1. Introduction
Voltage-gated sodium channels (VGSCs) are large pore-forming transmembrane-spanning proteins with four homologous domains (I-IV) that regulate the influx of Na
+ ions across neuronal membranes in response to local changes in voltage [
1]. This Na
+ influx is essential for action potential initiation and propagation in electrically excitable cells and crucial for the physiological function of neurons. In humans, nine different isoforms with relatively high sequence homology have been identified (Na
V1.1–Na
V1.9), each with discrete expression profiles and distinctive biophysical properties [
2]. While several VGSC isoforms expressed in peripheral sensory neurons, such as Na
V1.7, Na
V1.8, and Na
V1.9, have a well-established role in somatosensation and nociception, the contribution of Na
V1.3 to peripheral neuronal excitability is less well understood.
Na
V1.3 is a tetrodotoxin (TTX)-sensitive channel with fast-activating and fast-inactivating currents capable of rapid repriming and sustaining repetitive firing [
3]. Interestingly, Na
V1.3 is expressed at very low levels in adult rodent dorsal root ganglion (DRG) neurons but is upregulated in DRGs following nerve injury or streptozotocin-induced diabetes, suggesting the channel may contribute to the development and maintenance of neuropathic pain [
4,
5,
6]. Indeed, shRNA-mediated knockdown of Na
V1.3 in DRG neurons attenuates the development of mechanical allodynia in rodent models of spared nerve injury (SNI) and diabetic neuropathy [
7,
8], while global Na
V1.3 knockout has confounding results, with minimal effect on the development of mechanical allodynia following chronic constriction injury (CCI), spinal nerve transection (SNT), and spinal nerve ligation (SNL) [
9,
10]. Therefore, selective pharmacological modulators of Na
V1.3 are required to further elucidate the contribution of the channel to neuronal excitability in physiological and pathological pain states.
Venoms from scorpions, spiders, and cone snails are a rich source of novel bioactive peptides with activity at ion channels. Many venom-derived peptides have high potency and selectivity for VGSCs and have provided us with unique insights into VGSC gating, structure, and function [
11]. Scorpion peptides that act on VGSCs can be functionally distinguished as α- or β-scorpion toxins, depending on how they modulate channel opening or closing. α-Scorpion toxins cause a marked delay in fast inactivation, resulting in prolonged Na
+ influx; whereas, β-scorpion toxins shift the voltage dependence of activation to more hyperpolarized potentials, resulting in toxin-bound channels that open closer to the resting membrane potential [
12,
13]. Tf2 is a β-scorpion toxin originally identified from the venom of
Tityus fasciolatus that, at a concentration of 1 µM, selectively activates Na
V1.3, with no effect at the other Na
V subtypes (Na
V1.1, Na
V1.2, and Na
V1.4–Na
V1.8 tested) expressed in
Xenopus oocytes [
14]. We therefore hypothesized that Tf2 could be used as a pharmacological tool to assess the contribution of Na
V1.3 to the excitability of peripheral sensory neurons. The remaining sodium channel subtype Nav1.9 is historically hard to express, but recent progress showed that the chimera of the human Nav1.9 with the C-terminus of Nav1.4 (hNav1.9_C4) reveals reasonable currents in HEK293 cells [
8]. As Tf2 is not thought to interact with the C-terminus, this chimera is a valuable tool for testing its influences on Nav1.9 gating.
Here, we report the chemical synthesis of Tf2 and the mutant Tf2[S14R], along with previously undescribed β-scorpion activity of Tf2 at hNaV1.9_C4 in vitro. We show for the first time that Tf2 causes Ca2+ influx in small, medium, and large diameter mammalian sensory neurons and explore the activity of Tf2 on the peripheral somatosensory system in vivo.
2. Experimental Section
2.1. Peptide Synthesis
Tf2 was synthesized in two segments consisting of Tf2(1–26)-α-thioester and Tf2(27–62), which were linked by native chemical ligation. Tf2(1–26)-α-thioester was synthesized via Boc chemistry on trityl-associated mercaptopropionic acid lysine resin to yield the C-terminal α-thioester upon hydrogen fluoride (HF, BOC Australia, Sydney, NSW, Australia) cleavage. Tf2(27–62) was synthesized using Fmoc chemistry on Rink amide resin to yield the C-terminally amidated peptide segment after trifluoroacetic acid (TFA, Auspep, Melbourne, VIC, Australia) cleavage. The cleaved crude peptides were precipitated and washed twice with cold diethyl ether. The peptides were then dissolved in 50% acetonitrile, 0.1% TFA (v/v) in water and lyophilized. Peptides were purified by preparative reverse-phase (RP) HPLC on a Shimadzu Prominence system (Sydney, NSW, Australia). HPLC fractions containing the desired peptide (judged by HPLC/MS) of similar purity were pooled, lyophilized, and stored at −20 °C.
Tf2(1–26)-α-thioester: KEGYAMDHEGCKFSCFIRPSGFCDGY-[COS-CH2-CH2-CO]-Lys, expected MW (assuming average isotope composition): 3164.5 Da, observed MW: 3164.4 ± 0.5 Da. Tf2(27–62): CKTHLKASSGYCAWPACYCYGVPSNIKVWDYATNKC-NH2, expected MW (assuming average isotope composition): 4031.7 Da, observed MW: 4031.2 ± 0.5 Da.
Native chemical ligation was done as previously described [
11]. Briefly, 8.7 μmol (35.3 mg) of peptide Tf2(27–62) and 8.7 μmol (27.6 mg) of Tf2(1–26)-α-thioester were dissolved in 8.7 mL of ligation buffer consisting of 6 M guanidine hydrochloride, 0.2 M Na
2HPO
4, 40 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and 50 mM 4-mercaptophenylacetic acid (MPAA), pH 7.25. The solution was stirred under nitrogen for 16 h and the reaction progress was monitored by HPLC-UV. The reduced full-length Tf2 (1–62) ligation product was isolated via RP-HPLC on an Agilent Zorbax 300SB, C3 column (9.4 × 250 mm, 5 μm Phenomenex Australia, Sydney, NSW, Australia). Yield: 42.5 mg; expected MW (assuming average isotope composition): 6963.0 Da, observed MW: 6962.8 ± 1.0 Da.
For oxidative folding of Tf2, 1.7 μmol (15 mg) of reduced Tf2 were dissolved in 3.75 mL of 6 M guanidine hydrochloride. Folding was initiated by diluting the peptide solution with 150 mL of folding buffer (0.1 M Tris, 0.5 M arginine, 1 M urea, 10 mM reduced glutathione, 1 mM oxidized glutathione, pH 8.0) and stirring for 72 h at 4 °C. Oxidized Tf2 was then purified by RP-HPLC on a Agilent Zorbax 300SB, C3 column (9.4 × 250 mm, 5 μm, Phenomenex Australia, Sydney, NSW, Australia). Yield: 2.2 mg. Calculated MW (assuming most abundant isotope composition): 6949.03 Da, observed MW: 6949.12 ± 0.20 Da. All reagents were obtained from Sigma-Aldrich (St Louis, MI, USA) unless otherwise stated.
2.2. Cell Culture
Human embryonic kidney (HEK) 293 cells that constitutively express the human VGSC channel α subunits NaV1.1–1.7/β1 (SB Drug Discovery, Glasgow, UK) were maintained in minimum essential media (MEM) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine, along with the selection antibiotics blasticidin, geneticin, and zeocin as recommended by the manufacturer. Chinese hamster ovary (CHO) cells expressing hNaV1.8/β3 via a tetracycline inducible system (ChanTest, Cleveland, OH, USA) were maintained in MEM, 10% FBS, and 2 mM l-glutamine. Tetracycline (1 µg mL−1) was added to culture media 48–72 h prior to assay to induce hNaV1.8 expression. Cells were passaged every 3–4 days after reaching 70–80% confluence with TrypLE Express (Thermo Fisher Scientific, Scoresby, VIC, Australia) and grown in an incubator at 37 °C with 5% CO2.
ND7/23 cells (Sigma-Aldrich, St. Louis, MO, USA) were maintained in high glucose Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) with a glucose level of 4.5 g/L and supplemented with 10% FBS (Biochrom AG, Berlin, Germany). ND7/23 cells were transiently transfected with 2.75 µg hNa
V1.9_C4 plasmid (chimera of human Na
V1.9 and the C-terminus of rat Na
V1.4; pCDNA3 vector [
8]), 0.25 μg of green fluorescent protein (GFP) DNA (Lonza, Basel, Switzerland), and 6 μL of JetPEI reagent (Polyplus Transfection, Illkirch, France). GFP was co-transfected to detect transfected cells via green fluorescence. Then, 24 h after transfection, cells were split either onto glass coverslips coated with poly-D-lysine (PDL) hydrobromide (Sigma-Aldrich) for treatment with 1 µM Tf2, or split into plastic petri dishes for control conditions. After recovering for 2–3 h, the cells were recorded with whole-cell patch clamp. Cells were passaged every 3–4 days after reaching 70–80% confluence with TrypLE Express (Thermo Fisher Scientific, Scoresby, VIC, Australia) in the case of HEK and CHO cells, and with accutase (Sigma-Aldrich) in the case of the ND7/23 cells. All cells were grown in an incubator at 37 °C with 5% CO
2.
2.3. Fluorescent Membrane Potential Assay
To rapidly assess the activity of Tf2 and Tf2[S14R] at hNaV1.1–1.7, we utilized the FLIPRTetra fluorescent imaging plate reader (Molecular Devices, Sunnyvale, CA, USA). Cells were plated in 384-well black-walled plates at a density of 10,000–15,000 per well in normal growth media 24–48 h before membrane potential experiments. Cells were loaded with red membrane potential dye (Molecular Devices) diluted in physiological salt solution (PSS; 140 mM NaCl, 11.5 mM glucose, 5.9 mM KCl, 1.4 mM MgCl2, 1.2 mM NaH2PO4, 5 mM NaHCO3, 1.8 mM CaCl2, 10 mM HEPES) and incubated for 30 min at 37 °C as per the manufacturer’s instructions. All synthetic toxins were diluted in PSS and 0.1% bovine serum albumin (BSA) to avoid adsorption to plastic surfaces. Changes in membrane potential (excitation/emission 510–545 nm/565–625 nm) were recorded each second for 5 min after the addition of toxin and the area under the curve (AUC) was computed using ScreenWorks (Molecular Devices, Version 3.2.0.14).
2.4. Whole Cell Patch-Clamp Electrophysiology
Whole cell patch-clamp experiments on hNaV1.3, hNaV1.7, and hNaV1.8 cell lines were performed using the QPatch-16 automated electrophysiology platform (Sophion, Ballerup, Denmark). This set-up utilizes 16-well planar chips plates (QPlates, Sophion) with a standard resistance of 2 ± 0.02 mΩ. Cells were expanded in T-175 flasks and maintained in 37 °C and 5% CO2 48–72 h prior to assay. On the day of experiment, cells were harvested with TrypLE Express and resuspended in DMEM with 25 mM HEPES, 100 U/mL penicillin-streptomycin, and 0.04 mg/mL trypsin inhibitor from Glycine max (soybean) and stirred for 30–60 min prior to use.
The external solution (ECS) for all automated VGSC recordings contained (in mM): NaCl (140), KCl (4), CaCl2 (2), MgCl2 (1), HEPES (10), TEA-Cl (20), and glucose (10) containing 0.1% BSA. The pH was adjusted to 7.4 with NaOH and osmolarity adjusted to 305 mOsm with sucrose. The intracellular solution (ICS) consisted of (in mM): CsF (140), EGTA/CsOH (1/5), HEPES (10), and NaCl (10) adjusted to pH 7.4 with CsOH and 320 mOsm with sucrose. The cell positioning pressure was set to −60 mbar, minimum seal resistance 0.1 GΩ, holding pressure −20 mbar, and currents were filtered at 25 kHz (8th order Bessel, cut off 5 kHz). A standard P/4 leak subtraction protocol was included, and as such, leak subtracted and non-leak subtracted currents were acquired in parallel.
After obtaining the stable whole-cell configuration, voltage clamp experiments proceeded as follows from a holding potential of −90 mV. All protocols included a brief (15 ms) pre-conditioning pulse to 0 mV followed by a 120 ms recovery to allow voltage-sensor trapping by β-scorpion toxins [
15]. Current–voltage (IV) relationships were determined using a series of 500 ms pulses ranging from −90 to +55 mV (in 5 mV steps), followed by a 20 ms pulse to −20 mV for hNa
V1.3 and hNa
V1.7 or +10 mV for hNa
V1.8 to assess the voltage dependence of fast inactivation. Peak current values were normalized to a buffer control. Toxins were diluted in ECS with 0.1% BSA and incubated for 5 min prior to recording.
Whole-cell patch-clamp experiments on ND7/23 cells transfected with hNav1.9_C4 were performed at room temperature, using an EPC-10 USB amplifier and PatchMaster software (HEKA Elektronik, Lambrecht, Germany) and analysis was done using Igor Pro 6.3 (WaveMetrics, Lake Oswego, OR, USA). Glass pipettes were prepared with a DMZ puller (Zeitz Instruments, Martinsried, Germany) to a resistance of 0.9 to 2.5 MΩ and filled with ICS. The ICS for hNav1.9_C4 recordings consisted of (in mM): CsF (140), EGTA (1), HEPES (10) and NaCl (10), sucrose (18) adjusted to pH 7.33 with CsOH, and 310 mOsm with additional sucrose. The ECS for hNav1.9_C4 recordings contained (in mM): NaCl (140), KCl (3), CaCl2 (1), MgCl2 (1), HEPES (10), glucose (20), and 0.1 % BSA. The pH was adjusted to 7.4 with NaOH and osmolarity adjusted to 305 mOsm with glucose. TTX (500 nM) was added to the ECS to block endogenous Na+ currents in ND7/23 cells. Tf2 (1 µM) was diluted in ECS with 0.1% BSA and incubated for 5 min prior to recording. Capacitive transients were cancelled and series resistance (≤6 MΩ) was compensated by at least 70%. Leak current was subtracted online using a P/4 procedure following each test pulse. Signals were sampled with 100 kHz and filtered at 10 kHz for activation and fast inactivation protocols and at 30 kHz for the deactivation protocol. Recording protocols were started 5 min after establishing the whole-cell configuration to allow for current stabilization. The holding potential for all recordings was −120 mV. The voltage dependence of activation was measured using a series of 40 ms pulses ranging from −120 mV to 30 mV in 10 mV steps at an interval of 5 s. The voltage dependence of steady-state fast inactivation was measured using a series of 500 ms pre-pulses ranging from −160 to −20 mV, in steps of 10 mV, followed by a 40 ms depolarizing test pulse at −40 mV. The voltage dependence of deactivation was measured using a series of 1.5 ms depolarizing pre-pulses to −20 mV. This was followed by a repolarization test pulse ranging from −130 to −20 mV, in 10 mV steps, at an interval of 5 s.
2.5. Ca2+ Imaging of Isolated Dorsal Root Ganglion Neurons
Primary cultures of mouse DRG neurons were prepared as previously described [
16,
17]. Briefly, DRG ganglia, removed from all spinal levels of 4-week-old male C57BL/6 mice, were incubated in DMEM (Gibco, Waltham, MD, USA) with 1 mg mL
−1 Collagenase IV (Gibco, Life Technologies, NY, USA) for 90 min at 37 °C, then triturated with fire-polished Pasteur pipettes and plated in 96-well plates coated with PDL (Corning, ME, USA). After 1 h, wells were flooded with DMEM supplemented with 10% FBS and pencillin/streptomycin (Gibco, Waltham, MD, USA) and maintained for 16–24 h before assay. Cells were loaded with Fluo-4 AM calcium indicator, according to the manufacturer’s instructions (ThermoFisher Scientific, MA, USA). After loading (1 h), the dye-containing solution was replaced with assay solution (1 x Hanks’ balanced salt solution, 20 mM HEPES). Fluorescence corresponding to [Ca
2+]
i of 100–200 DRG cells per experiment was monitored in parallel using a Nikon Ti-E Deconvolution inverted microscope (Nikon, Tokyo, Japan), equipped with a Lumencor Spectra LED Lightsource (Lumencor, Beavertown, OR, USA). Images were acquired at 10x objective at 1 frame/s (excitation 485 nm, emission 521 nm). For each experiment, baseline fluorescence was monitored for 30 s. At 30 s, 60 s, and 180 s, assay solution was replaced with fresh assay solution (as a negative control), Tf2 (1 µM in assay solution), and KCl (30 mM in assay solution), respectively. Experiments with TTX and Tf2[S14R] were performed in the same way. Cells responding to KCl and/or Tf2 were considered neuronal and grouped according to size: Large (>600 µm
2), medium (300–600 µm
2), and small diameter (<300 µm
2). Experiments involving the use of mouse tissue were approved by The University of Queensland Animal Ethics Committee. Data were derived from 2–3 independent experiments, and plotted using GraphPad Prism 8. Where differences were tested, unpaired
t-tests were used.
2.6. Animals
For behavioral assessment, we used adult male wildtype or homozygous NaV1.9-deficient (NaV1.9-/-) mice aged 6–8 weeks (~25g) on a C57BL/6J background. Homozygous Nav1.9 knockout mice on a C57BL/6J background were generated using CRISPR/Cas9 technology by the Queensland Facility for Advanced Genome Editing (The University of Queensland, Australia). In brief, two sgRNAs were designed to target exon 2 of the Scn11a gene. The deletion of exon 2 generated a frame shift mutation resulting in the introduction of stop codons in exon 3. The guide sgRNAs were microinjected together with Cas9 mRNA into fertilized eggs collected from C57BL/6J mice and transferred into pseudopregnant surrogate CD1 female mice. Offspring born to the foster mothers were genotyped by PCR (see below) and DNA sequencing. One F0 male mouse was bred to C57BL/6J female mice to generate F1 heterozygous mice, and F1 heterozygous mice were inbred to obtain homozygous mice. Genomic DNA was extracted from ear clippings and mice were genotyped by PCR using the following primers (forward: 5′-GCTGCTCAGACACTCACAGT-3′) and (reverse: 5′ATTCTGCCACCAGAGACTGC-3′) by the AEGRC genotyping and sequencing facility (The University of Queensland). Animals were housed in groups of 3 or 4 per cage, under 12-h light-dark cycles, and had standard rodent chow and water ad libitum. Age-matched controls were used for studies involving knockout animals. All behavioral assessment was performed by a blinded observer unaware of the genotype and/or treatments received.
Ethical approval for in vivo experiments was obtained from The University of Queensland Animal Ethics Committee prior to experimentation. All animals experiments were conducted in accordance with local and national regulations, including the International Association for the Study of Pain Guidelines for the Use of Animals in Research in agreement with the Animal Care and Protection Regulation Qld (2012), and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 8th edition (2013) (TRI/IMB/093/17, 31 March 2017; IMB/PACE/421/18, 18 October 2018).
2.7. Behavioral Assessment
First, Tf2 (1 µM), Tf2[S14R] (10 µM), or ICA-121431 (500 nM) were diluted in saline with 0.1%
w/
v BSA and administered either alone or in combination by intraplantar (i.pl) injection into the left hind paw in a volume of 40 µL under light isoflurane (3 %) anesthesia. Mice were then placed individually into polyvinyl boxes (10 × 10 × 10 cm) and recorded by video for 20 min post-injection. The number of spontaneous pain behaviors, including licks, shakes, and flinches, were counted later by a blinded observer. Once spontaneous pain behavior had ceased, mechanical thresholds were assessed using an electronic von Frey apparatus (MouseMet Electronic von Frey, TopCat Metrology, Ely, UK), heat thresholds were assessed using the thermal probe test (MouseMet Thermal), and weight bearing was assessed using the Catwalk XT (Noldus Information Technology, Wageningen, The Netherlands) as previously described [
18,
19]. To quantify weight bearing, the parameter ‘mean intensity of the 15 most intense pixels’ was used as a surrogate measure.
2.8. Paw Thickness
In a separate cohort of animals, paw thickness was measured along the distal–proximal axis at the metatarsal level using a digital vernier caliper (Kincrome, Vic, Australia) 10 min after i.pl. injection of Tf2 (1 µM) or Tf2[S14R] (10 µM).
2.9. Data Analysis
Data were plotted and analyzed by GraphPad Prism, versions 8.2.0 and 8.4.2. Statistical significance was defined as p < 0.05 and was determined by t-test or one-way ANOVA with Dunnett’s post-test, as indicated. In case of deactivation, multiple t-tests (one per row) were used without assuming equal standard deviation and with the Holm–Sidak method for correcting p-values for multiple comparisons. Concentration–response curves were fitted with a four-parameter Hill equation with a variable Hill coefficient. Conductance–voltage relationships for activation were calculated for each voltage step using where Vrev is the reversal potential. Resulting data were fitted using a Boltzmann equation: GNa =; GNa is the voltage-dependent sodium conductance, GNa,max is the maximal sodium conductance, Vm is the membrane potential, V1/2 is the membrane potential at half-maximal activation, and k is the slope factor. Current–voltage relationships for inactivation were fitted using a Boltzmann equation: INa = INa is the voltage-dependent sodium current, INa,max is the maximal sodium current, Vm is the membrane potential, V1/2 is the half-maximal inactivation, and k is the slope factor. The time course of current decay at deactivating voltages over 10 ms was fitted by the double-exponential equation,, where y0 is the current amplitude at steady state, amp1 and amp2 are the amplitude coefficient for the fast and slow time constants, and τ1 and τ2 are the fast and slow time constants. Data are expressed as the mean ± standard error of the mean (SEM).
4. Discussion
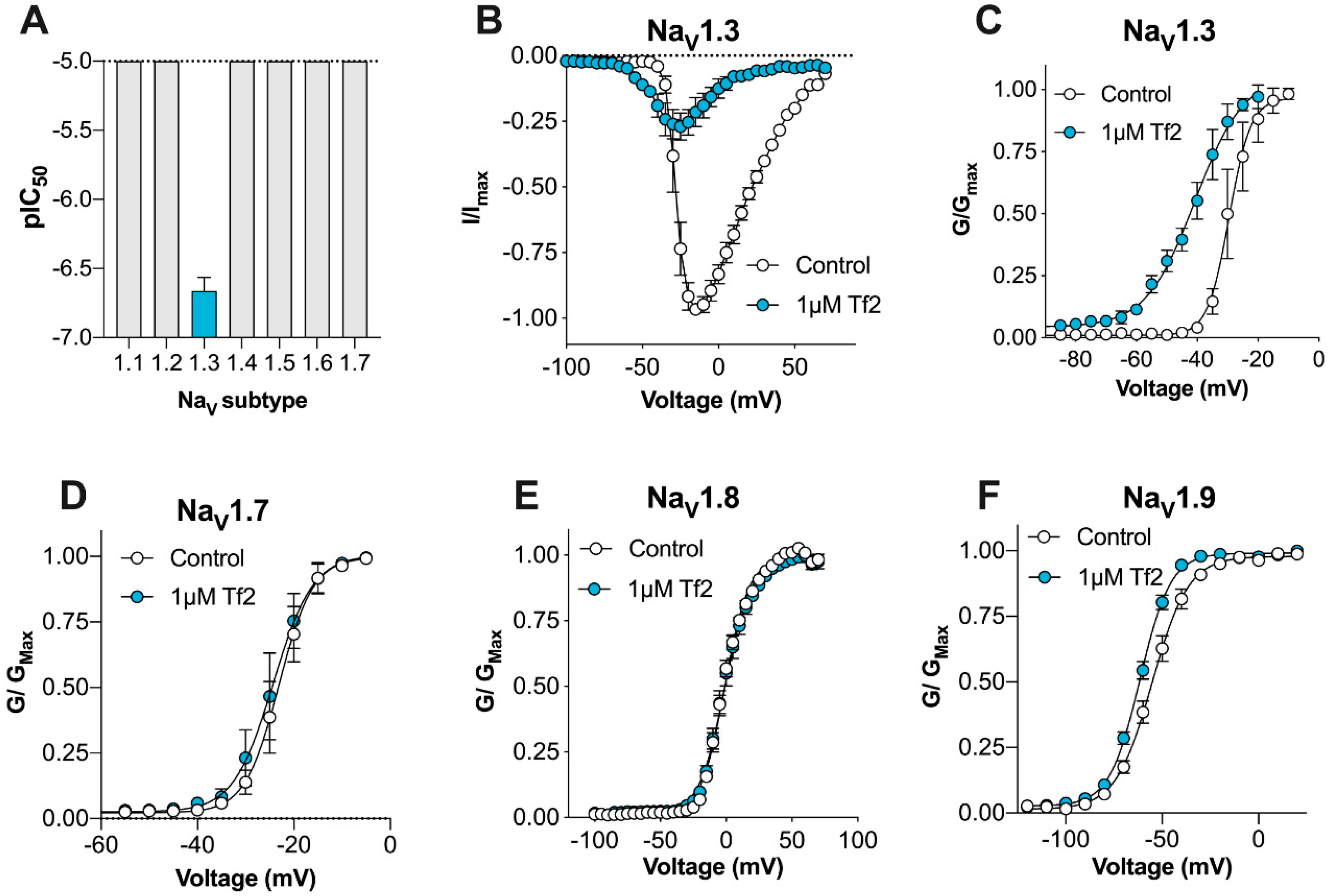
Here, we describe for the first time the activity of synthetic β-scorpion toxin Tf2 on the full panel of human VGSCs (Na
V1.1–1.9) constitutively expressed in mammalian cells, on the calcium influx in primary sensory DRG neurons, and on nociceptive behaviors in mice in vivo, with the overall aim to investigate its suitability as a selective Na
V1.3 pharmacological probe. Modern chemical peptide synthesis strategies applied to α- and β-long chain scorpion toxins have allowed the production of these molecules on the multi-milligram scale and have allowed structural and functional studies that are difficult to perform with the small quantities usually isolated from scorpion venom [
22,
23,
24,
25]. Synthetic Tf2 faithfully reproduced the activity and mode-of-action of venom-derived Tf2 reported previously, strongly suggesting that both samples are also structurally identical. Whilst previously described as a selective Na
V1.3 activator [
14], we found that Tf2 had additional activity at hNa
V1.9_C4, which is a TTX-resistant isoform preferentially expressed on small diameter DRG neurons that is associated with both ‘painless’ and ‘painful’ channelopathies [
26,
27].
The pharmacology of scorpion toxins at Na
V1.9 remains largely unexplored, partly due to difficulties in the functional expression of Na
V1.9 in heterologous systems. However, the β-scorpion toxin Ts1 (also known as TsVII), which has 73.4% sequence identity to Tf2, is also active on Na
V1.9 [
28]. This suggests that activity at Na
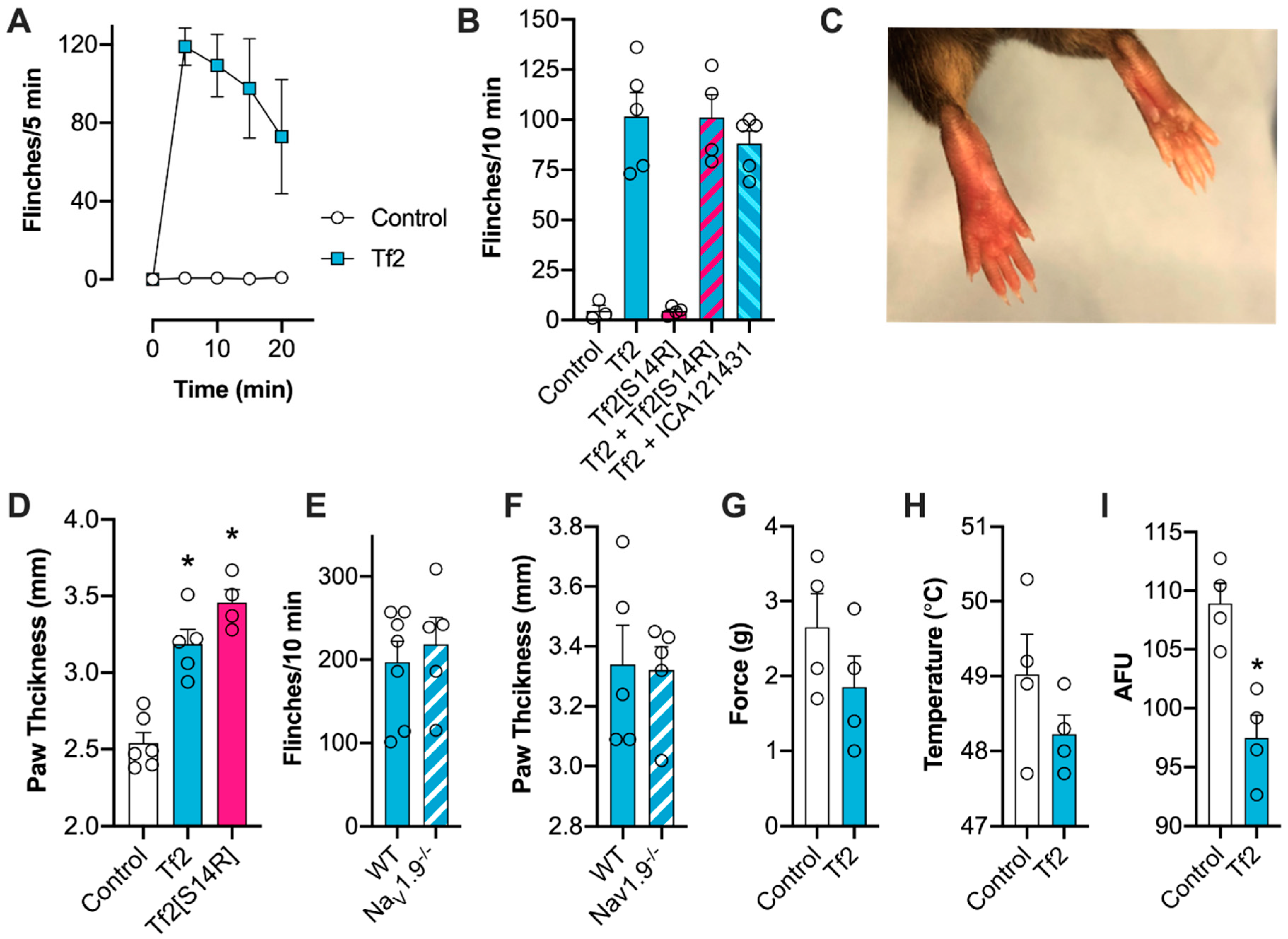
V1.9 may be broadly conserved amongst other related β-scorpion toxins, but this remains to be assessed. Despite shifting the voltage dependence of activation at Na
V1.9 (Δ −6.3 mV), Tf2-induced spontaneous pain and swelling developed normally in Na
V1.9
-/- mice, indicating that this isoform had a minimal contribution to in vivo activity. This result was somewhat surprising, given the mutation L1158P in human Na
V1.9 causes a similar hyperpolarizing shift in activation (Δ −6.7 mV), and has been linked to increased excitability of DRG neurons and painful peripheral neuropathy [
29]. However, this mutation also slows deactivation, which was a channel parameter unaffected by Tf2, suggesting that changes in Na
V1.9 deactivation may be important for neuronal excitability.
Selective VGSC modulators remain useful tool compounds for delineating the contribution of particular isoforms to the excitability of neurons in both physiological and pathological states [
17,
30]. Despite being implicated in neuropathic pain and epilepsy, there is a lack of highly potent and selective inhibitors of Na
V1.3 available. The small molecule ICA-121431 is the most potent Na
V1.3 inhibitor described to the date, but off-target activity at Na
V1.1, which is also expressed on peripheral and central neurons, limits its use as a research tool in vivo [
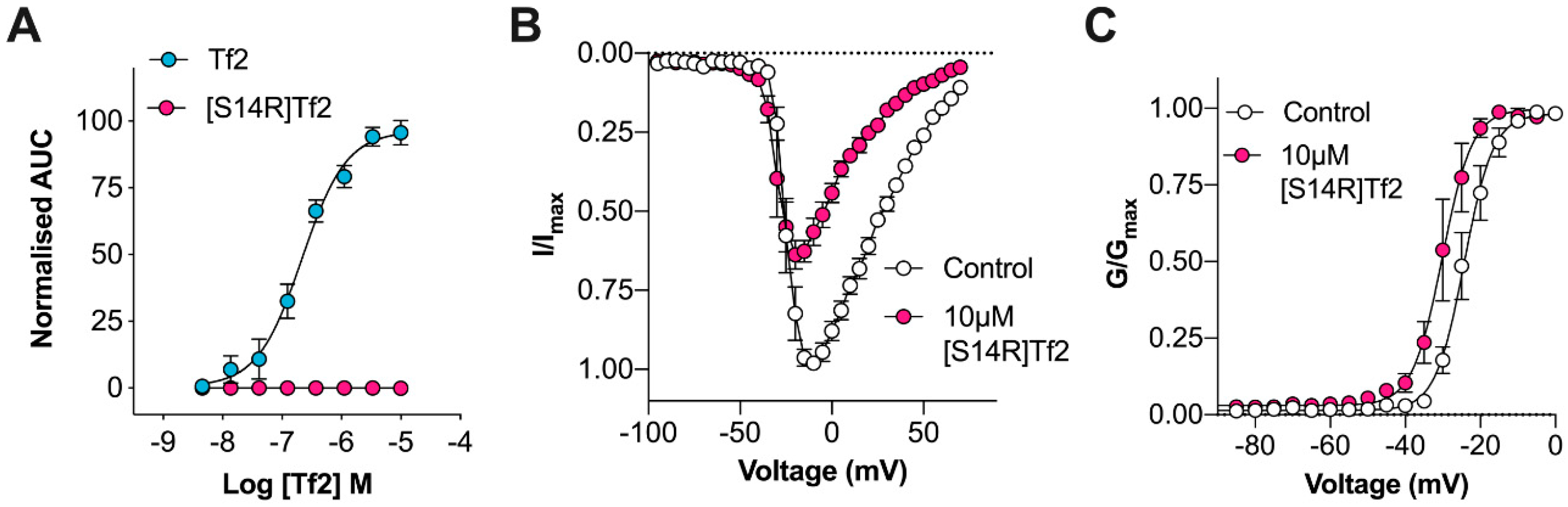
31]. In an attempt to rationally design a selective Na
V1.3 inhibitor, we synthesized Tf2[S14R], where we mutated the serine at position 14 to a positively charged arginine. In the homologous β-scorpion toxin Ts1, mutation of S14 to an arginine resulted in a toxin that could still bind to Na
V1.4 but no longer shifted the voltage dependence of activation [
32]. In line with this observation, Tf2[S14R] inhibited the peak current of Na
V1.3 without causing the same early channel opening as the native Tf2; however, it was unable to reverse Tf2-induced spontaneous pain behaviors in vivo. This is possibly due to the mutation causing a loss in potency, as the related toxin Cn2[E15R] required application at a concentration 200 times higher than the native to inhibit Na
V1.6 in vivo [
11]. Unfortunately, because the potency of Tf2 is already relatively weak, and Tf2[S14R] caused paw swelling at 10 µM, higher concentrations could not be assessed.
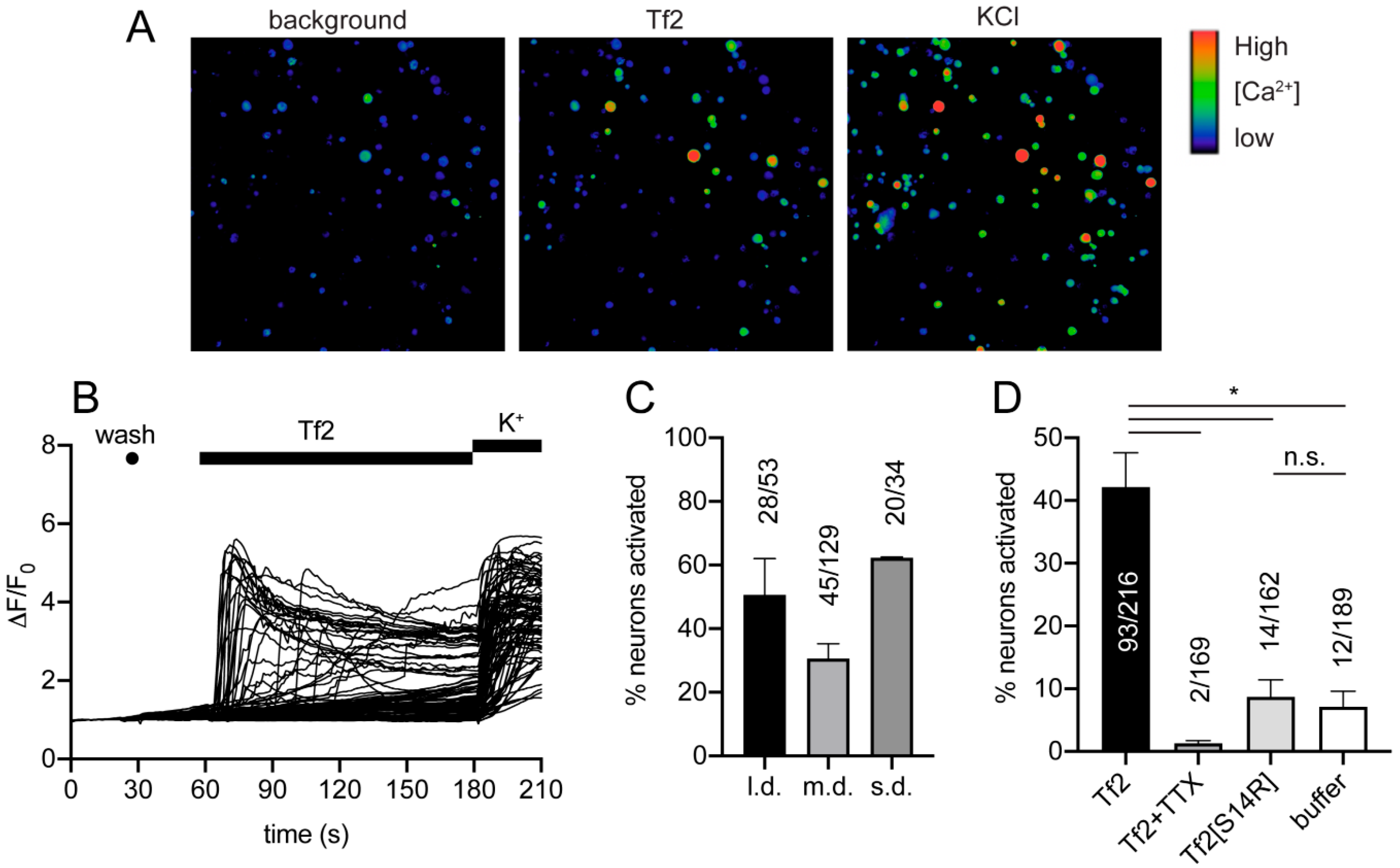
Tf2 caused Ca
2+ influx in both small and large mouse adult DRG neurons, which was surprising given Na
V1.3 is only expressed at significant levels in embryonic sensory neurons or in adult neurons after axotomy [
4]. Interestingly, colonic neurons isolated from mouse thoracolumbar (T10-L1) and lumbosacral (L5-S2) DRGs express Na
V1.3 mRNA transcripts at similar levels to Na
V1.1 and Na
V1.2 [
33]. As DRG neurons in this study were isolated from all spinal levels, we cannot rule out the presence of these Na
V1.3-expressing neurons in culture. However, given the definitive lack of proteomic evidence for Na
V1.3 expression in adult uninjured DRG neurons, and that Tf2 activated a large proportion of all DRG neurons, it is more likely that the observed effect on Ca
2+ influx is due to activity at other neuronal targets. This is consistent with Tf2-induced spontaneous flinching not being reversed by the Na
V1.1/1.3 inhibitor ICA-121431 in vivo. Indeed, it is not uncommon for scorpion toxins to have promiscuous activity on both VGSCs and voltage-gated potassium (K
V) channels, and blockade of a K
V channel could also result in Ca
2+ influx [
34,
35]. Furthermore, there are scorpion toxins that modulate members of the family of voltage-gated calcium channels (Ca
V), such as kurtoxin from
Parabuthus transvaalicus [
36]; however, the activity of Tf2 at voltage-gated potassium and calcium channels in heterologous expression systems remains to be tested. Inhibition of Tf2-induced Ca
2+ influx by TTX could also be explained by a global depression of neuronal excitability rather than direct opposition of Tf2-induced Na
V1.3 activation. The observation that Tf2[S14R], which lost activity at Na
V1.3, also lost excitatory activity in DRG neurons suggests that the pharmacophore of Tf2 at this other neuronal target(s) overlaps at least partially with that of Na
V1.3. In addition, the observation that Tf2[S14R], which lost excitatory activity in DRG neurons, still caused swelling in vivo suggests that Tf2-induced inflammation is mediated by an additional non-neuronal target. Scorpion venom- or toxin-induced inflammatory responses have been described previously and have been linked to the activation of innate immune cells via interaction with pattern recognition receptors [
37]. The use of global and sensory neuron-specific Na
V1.3 knockout mice would be required to tease out what contribution, if any, Na
V1.3 has to Tf2-induced pain and inflammation.
In summary, Tf2 does not appear to be a suitable tool to study the effects of Nav1.3 channel activation in sensory neurons. This is due to additional activity at NaV1.9 and the probability of other neuronal and non-neuronal targets. Results from in vivo experiments that use Tf2 should be interpreted accordingly. There is, however, scope to use Tf2 in studying the activity of NaV1.9 in vitro, particularly in heterologous expression systems. Indeed, very few toxins to date are known to modulate the NaV1.9 isoform. Finally, the somewhat surprising finding that Tf2 and Tf2[S14R] cause inflammation in vivo could be explored further to understand how scorpion toxin envenomation affects inflammatory processes in humans.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}



