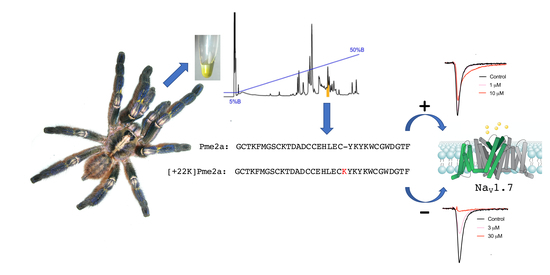
Addition of K22 Converts Spider Venom Peptide Pme2a from an Activator to an Inhibitor of NaV1.7

 ,
,  ,
,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Cell Culture
2.2. Venom Collection
2.3. Membrane Potential Assay at NaV1.8
2.4. Isolation of Pme1a and Pme2a
2.5. Peptide Synthesis
2.6. Calcium Responses in TRPV1-HEK Cells
2.7. Electrophysiology
2.8. Data Analysis
3. Results
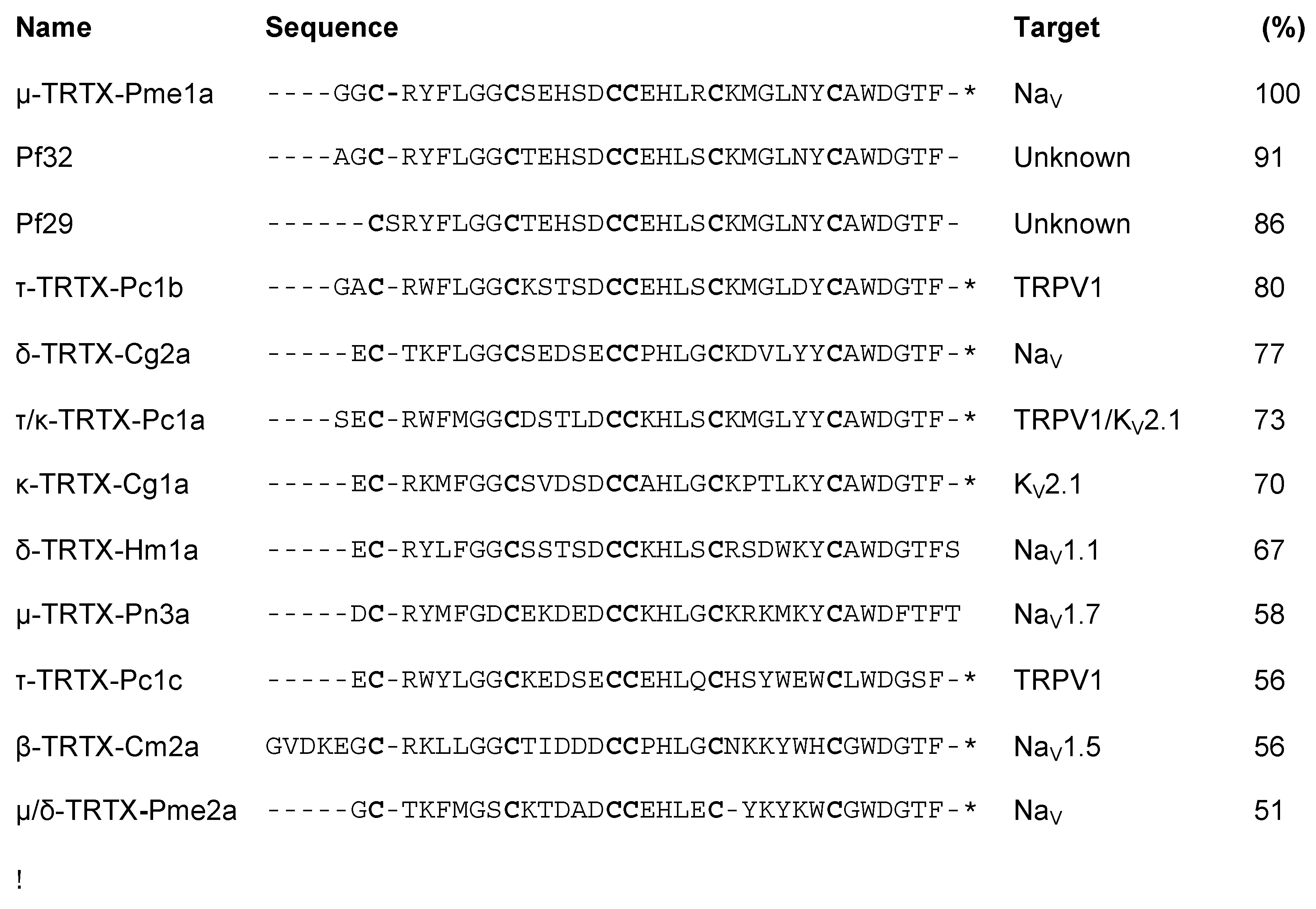
3.1. Isolation of the Novel Spider Venom Peptides μ-TRTX-Pme1a and μ/δ-TRTX-Pme2a from P. metallica
3.2. Pharmacological Activity of Pme1a
3.3. Pharmacological Activity of Pme2a
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.D.; Ahn, H.S.; Yang, Y.; Han, C.; Seal, R.P.; Wood, J.N.; Waxman, S.G.; Dib-Hajj, S.D. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain 2012, 153, 2017–2030. [Google Scholar] [CrossRef] [PubMed]
- Djouhri, L.; Fang, X.; Okuse, K.; Wood, J.N.; Berry, C.M.; Lawson, S.N. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): Expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J. Physiol. 2003, 550, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Frezel, N.; Dib-Hajj, S.D.; Waxman, S.G. Expression of NaV1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol. Pain 2012, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Lauria, G.; Merkies, I.S.; Cheng, X.; Han, C.; Ahn, H.S.; Persson, A.K.; Hoeijmakers, J.G.; Gerrits, M.M.; Pierro, T.; et al. Gain-of-function NaV1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Barbosa, C.; Pei, Z.; Xie, W.; Strong, J.A.; Zhang, J.M.; Cummins, T.R. Increased resurgent sodium currents in Nav1.8 contribute to nociceptive sensory neuron hyperexcitability associated with peripheral neuropathies. J. Neurosci. 2019, 39, 1539–1550. [Google Scholar] [CrossRef]
- Waxman, S.G.; Dib-Hajj, S.D. The two sides of NaV1.7: Painful and painless channelopathies. Neuron 2019, 101, 765–767. [Google Scholar] [CrossRef]
- Abrahamsen, B.; Zhao, J.; Asante, C.O.; Cendan, C.M.; Marsh, S.; Martinez-Barbera, J.P.; Nassar, M.A.; Dickenson, A.H.; Wood, J.N. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 2008, 321, 702–705. [Google Scholar] [CrossRef]
- Gingras, J.; Smith, S.; Matson, D.J.; Johnson, D.; Nye, K.; Couture, L.; Feric, E.; Yin, R.; Moyer, B.D.; Peterson, M.L.; et al. Global NaV1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS ONE 2014, 9, e105895. [Google Scholar] [CrossRef]
- Rogart, R.B.; Cribbs, L.L.; Muglia, L.K.; Kephart, D.D.; Kaiser, M.W. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc. Natl. Acad. Sci. USA 1989, 86, 8170–8174. [Google Scholar] [CrossRef]
- Whitaker, W.R.; Faull, R.L.; Waldvogel, H.J.; Plumpton, C.J.; Emson, P.C.; Clare, J.J. Comparative distribution of voltage-gated sodium channel proteins in human brain. Brain Res. Mol. Brain Res. 2001, 88, 37–53. [Google Scholar] [CrossRef]
- Vetter, I.; Deuis, J.R.; Mueller, A.; Israel, M.R.; Starobova, H.; Zhang, A.; Rash, L.D.; Mobli, M. NaV1.7 as a pain target—From gene to pharmacology. Pharmacol. Ther. 2017, 172, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.R.; Morgan, M.; Tay, B.; Deuis, J.R. Toxins as tools: Fingerprinting neuronal pharmacology. Neurosci. Lett. 2018, 679, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.R.; Tay, B.; Deuis, J.R.; Vetter, I. Sodium channels and venom peptide pharmacology. Adv. Pharmacol. 2017, 79, 67–116. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, J.; Bosmans, F. Animal toxins can alter the function of NaV1.8 and NaV1.9. Toxins (Basel) 2012, 4, 620–632. [Google Scholar] [CrossRef]
- Vetter, I.; Wyse, B.D.; Monteith, G.R.; Roberts-Thomson, S.J.; Cabot, P.J. The μ opioid agonist morphine modulates potentiation of capsaicin-evoked TRPV1 responses through a cyclic AMP-dependent protein kinase a pathway. Mol. Pain 2006, 2, 22. [Google Scholar] [CrossRef]
- Herzig, V.; Hodgson, W.C. Neurotoxic and insecticidal properties of venom from the Australian theraphosid spider Selenotholus foelschei. Neurotoxicology 2008, 29, 471–475. [Google Scholar] [CrossRef]
- Deuis, J.R.; Dekan, Z.; Inserra, M.C.; Lee, T.H.; Aguilar, M.I.; Craik, D.J.; Lewis, R.J.; Alewood, P.F.; Mobli, M.; Schroeder, C.I.; et al. Development of a μO-Conotoxin analogue with improved lipid membrane interactions and potency for the analgesic sodium channel NaV1.8. J. Biol. Chem. 2016, 291, 11829–11842. [Google Scholar] [CrossRef]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E.A.; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar] [CrossRef]
- Pineda, S.S.; Chaumeil, P.A.; Kunert, A.; Kaas, Q.; Thang, M.W.C.; Le, L.; Nuhn, M.; Herzig, V.; Saez, N.J.; Cristofori-Armstrong, B.; et al. ArachnoServer 3.0: An online resource for automated discovery, analysis and annotation of spider toxins. Bioinformatics 2018, 34, 1074–1076. [Google Scholar] [CrossRef] [PubMed]
- Oldrati, V.; Koua, D.; Allard, P.M.; Hulo, N.; Arrell, M.; Nentwig, W.; Lisacek, F.; Wolfender, J.L.; Kuhn-Nentwig, L.; Stocklin, R. Peptidomic and transcriptomic profiling of four distinct spider venoms. PLoS ONE 2017, 12, e0172966. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Smith, J.J.; Vetter, I.; Rupasinghe, D.B.; Er, S.Y.; Senff, S.; Herzig, V.; Mobli, M.; Lewis, R.J.; Bosmans, F.; et al. Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 2015, 172, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef]
- Deuis, J.R.; Dekan, Z.; Wingerd, J.S.; Smith, J.J.; Munasinghe, N.R.; Bhola, R.F.; Imlach, W.L.; Herzig, V.; Armstrong, D.A.; Rosengren, K.J.; et al. Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci. Rep. 2017, 7, 40883. [Google Scholar] [CrossRef]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the NaV1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef]
- Lin King, J.V.; Emrick, J.J.; Kelly, M.J.S.; Herzig, V.; King, G.F.; Medzihradszky, K.F.; Julius, D. A Cell-penetrating scorpion toxin enables mode-specific modulation of TRPA1 and pain. Cell 2019, 178, 1362–1374. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, K.; Deuis, J.R.; Dekan, Z.; Jin, A.-H.; Alewood, P.F.; King, G.F.; Herzig, V.; Vetter, I. Addition of K22 Converts Spider Venom Peptide Pme2a from an Activator to an Inhibitor of NaV1.7. Biomedicines 2020, 8, 37. https://doi.org/10.3390/biomedicines8020037
Yin K, Deuis JR, Dekan Z, Jin A-H, Alewood PF, King GF, Herzig V, Vetter I. Addition of K22 Converts Spider Venom Peptide Pme2a from an Activator to an Inhibitor of NaV1.7. Biomedicines. 2020; 8(2):37. https://doi.org/10.3390/biomedicines8020037
Chicago/Turabian StyleYin, Kathleen, Jennifer R. Deuis, Zoltan Dekan, Ai-Hua Jin, Paul F. Alewood, Glenn F. King, Volker Herzig, and Irina Vetter. 2020. "Addition of K22 Converts Spider Venom Peptide Pme2a from an Activator to an Inhibitor of NaV1.7" Biomedicines 8, no. 2: 37. https://doi.org/10.3390/biomedicines8020037
APA StyleYin, K., Deuis, J. R., Dekan, Z., Jin, A.-H., Alewood, P. F., King, G. F., Herzig, V., & Vetter, I. (2020). Addition of K22 Converts Spider Venom Peptide Pme2a from an Activator to an Inhibitor of NaV1.7. Biomedicines, 8(2), 37. https://doi.org/10.3390/biomedicines8020037