Mutations and Copy Number Alterations in IDH Wild-Type Glioblastomas Are Shaped by Different Oncogenic Mechanisms

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tumor Samples
2.2. Pathology and Molecular Subsets
2.3. Whole Exome Sequencing, Pre-Processing and Variant Calling
2.4. Metrics
2.5. The Cancer Genome Atlas Pan-Glioma Data
2.6. Association of Somatically Mutated Genes with Metrics and Pathway Enrichment Analyses
3. Results
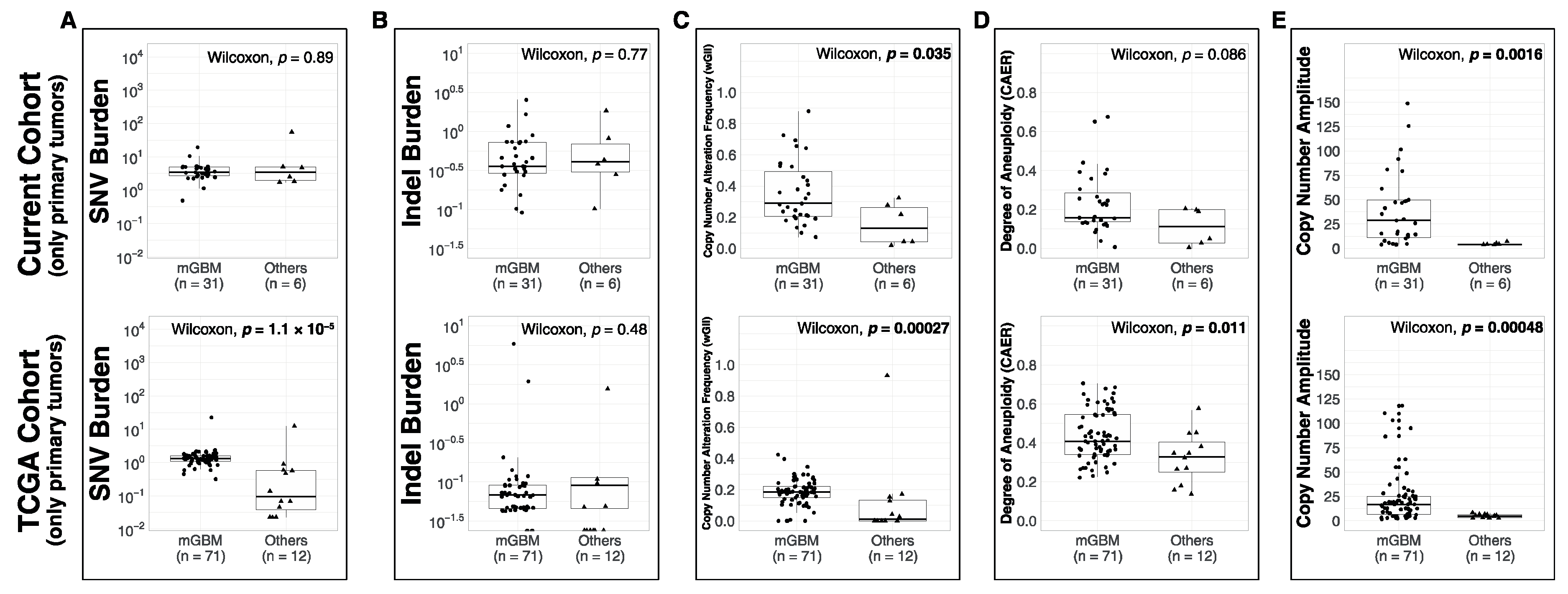
3.1. Comparison of Metrics between Molecular Subsets
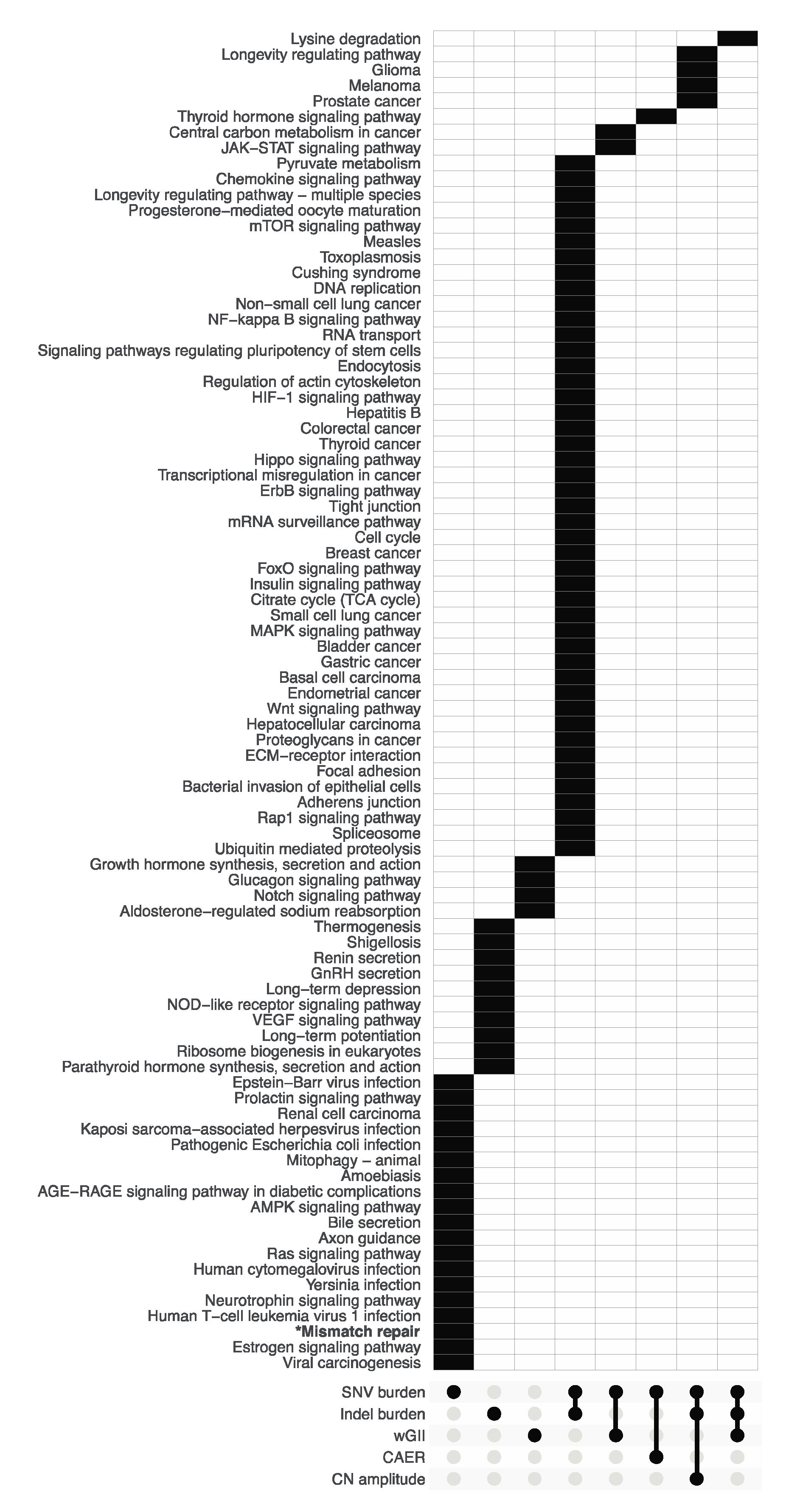
3.2. Pathway Enrichment Analysis of Metric-Associated Somatic Variants
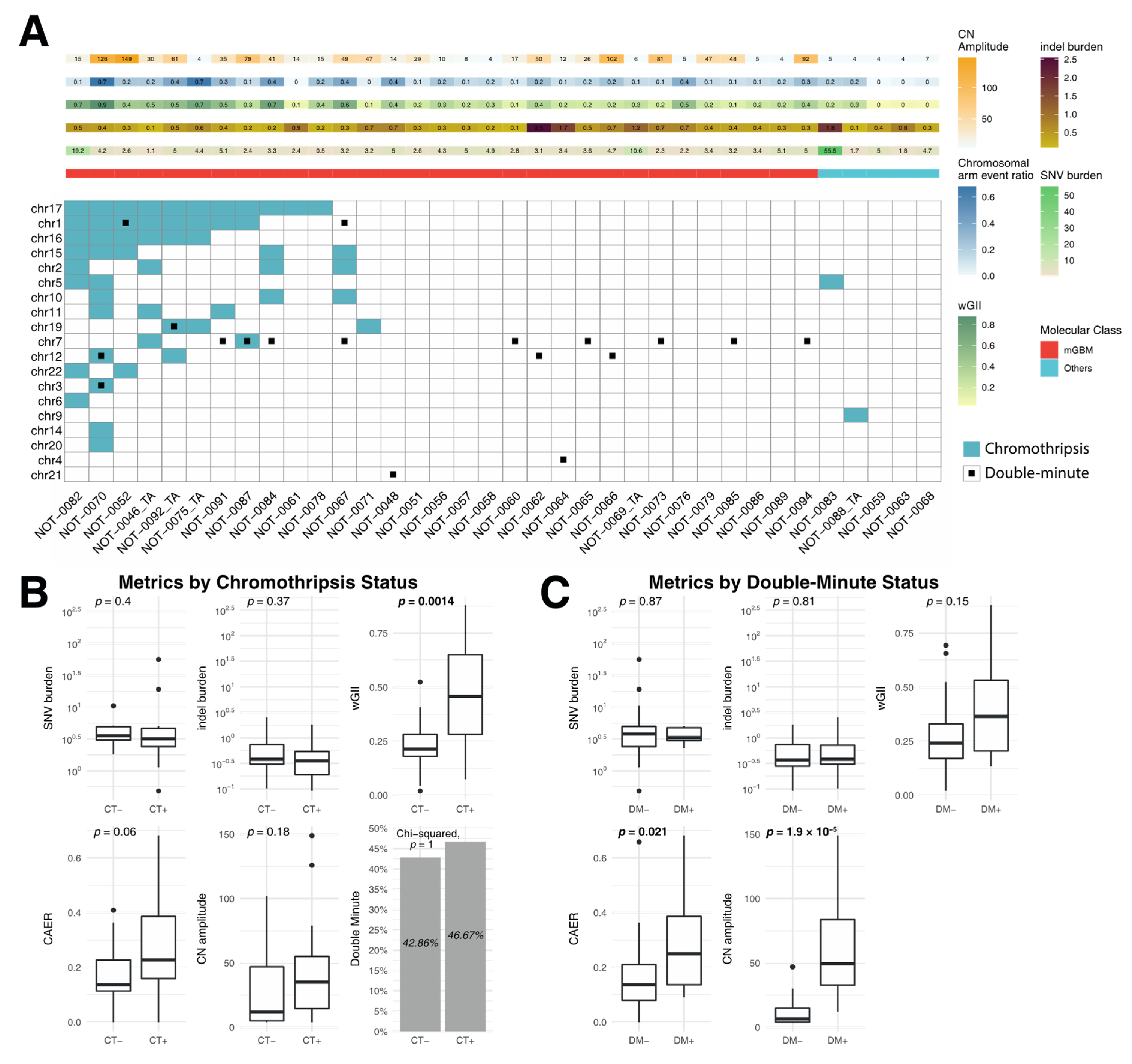
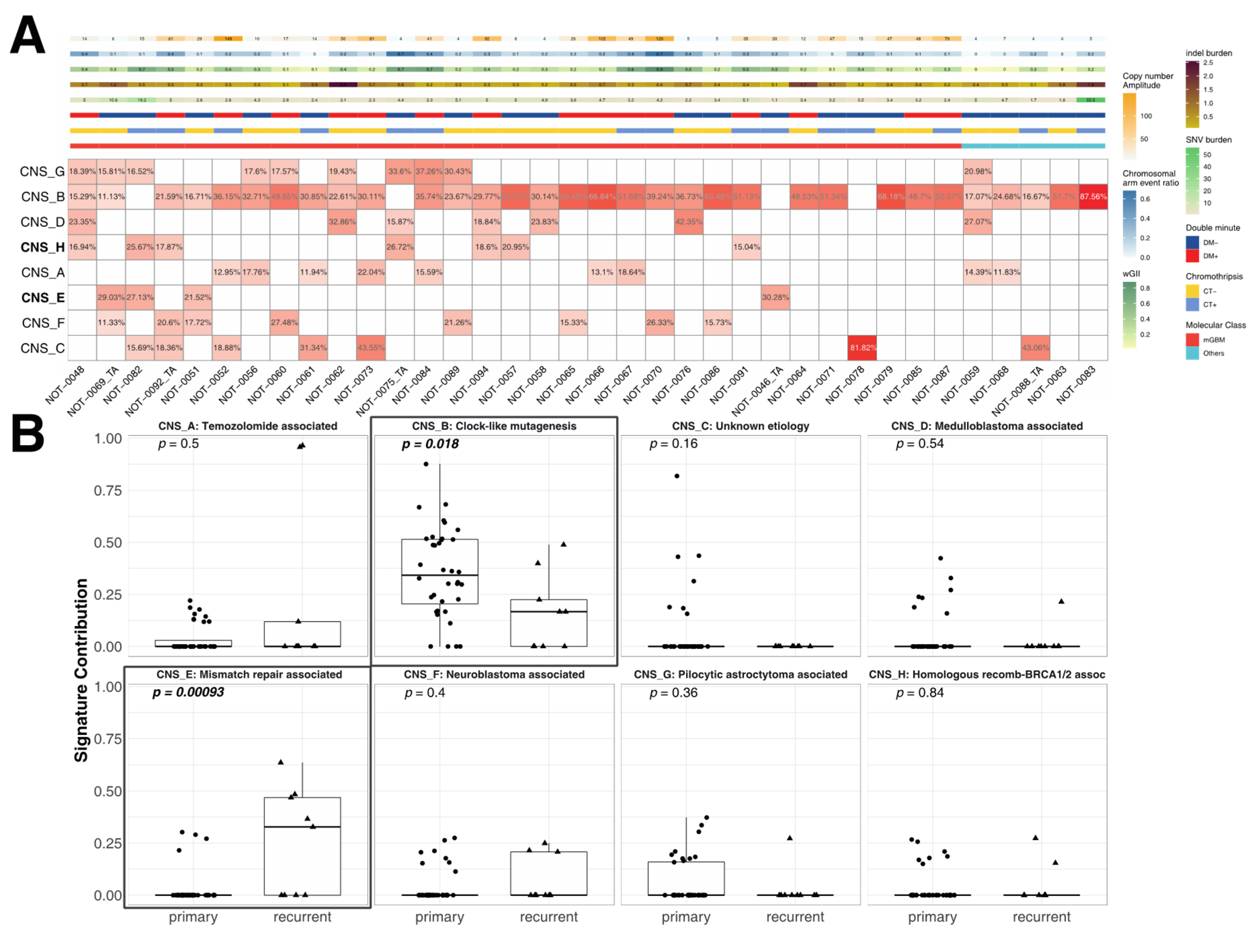
3.3. Correlation of Metrics with Chromothripsis and Double Minutes
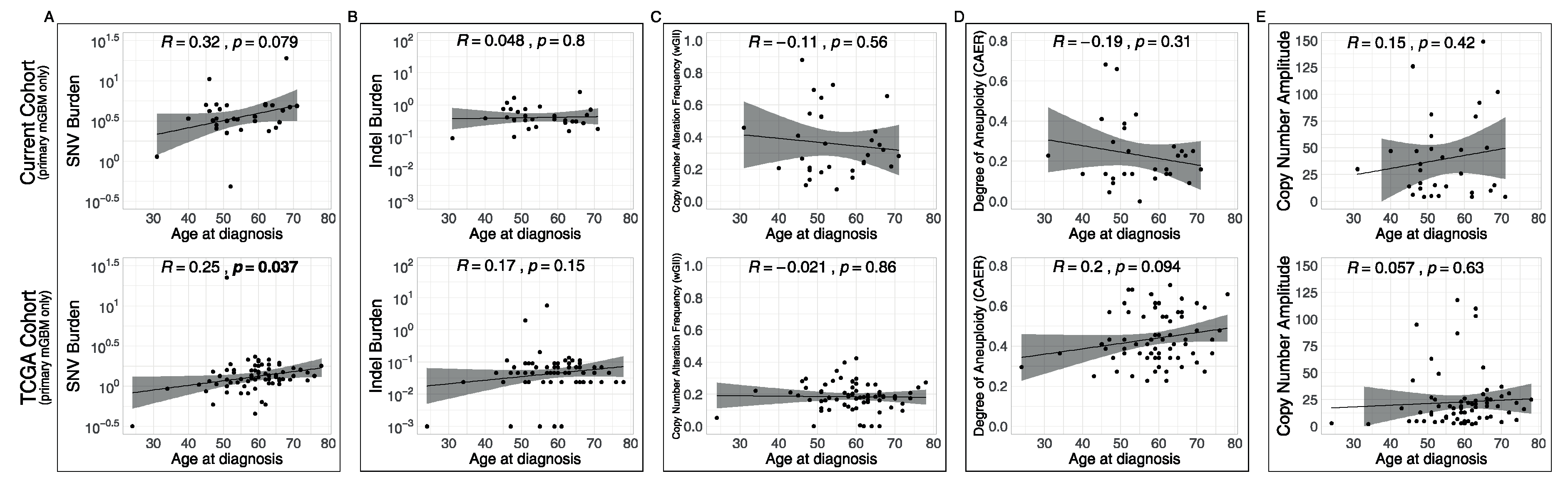
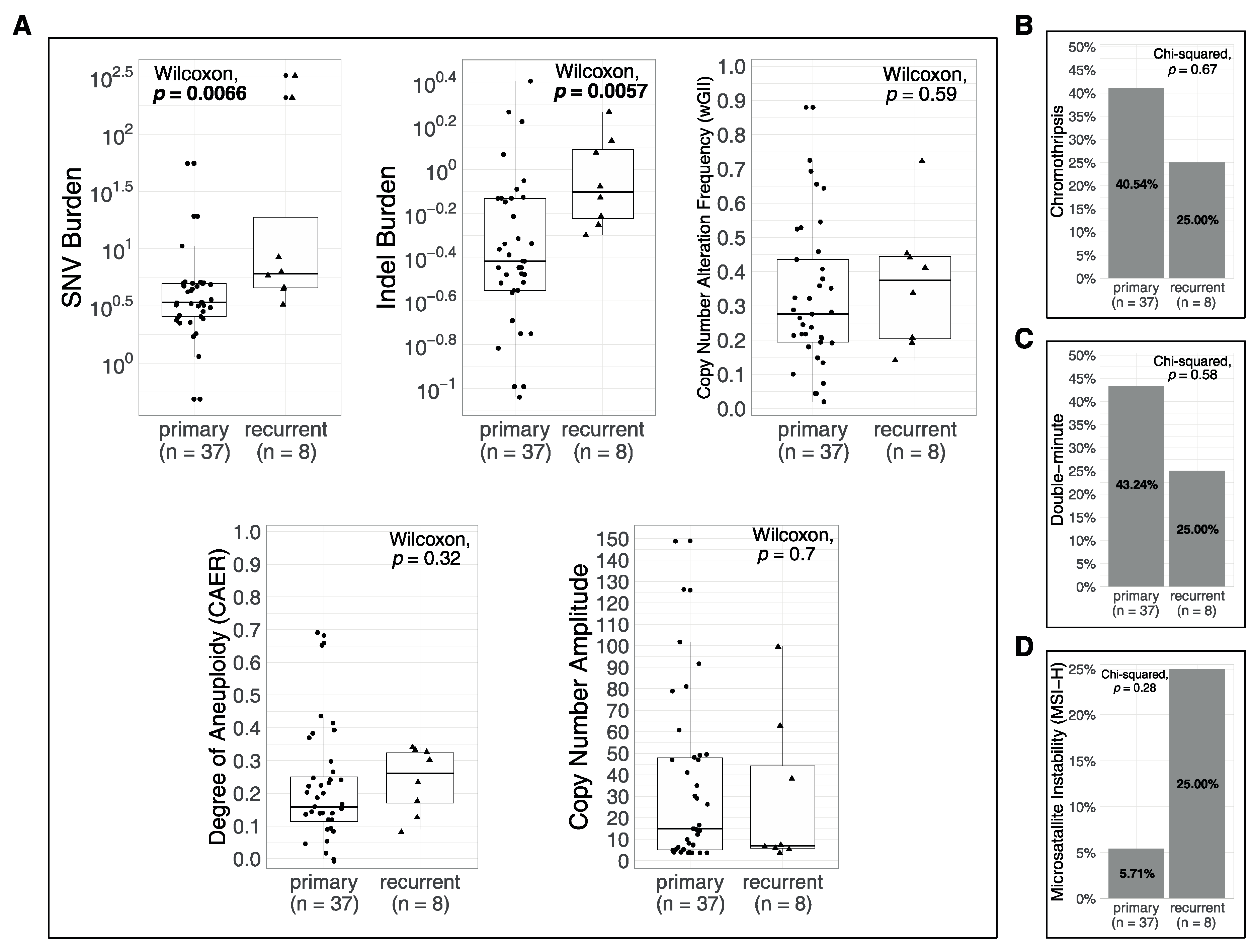
3.4. Associations of Metrics with Age and with Recurrences
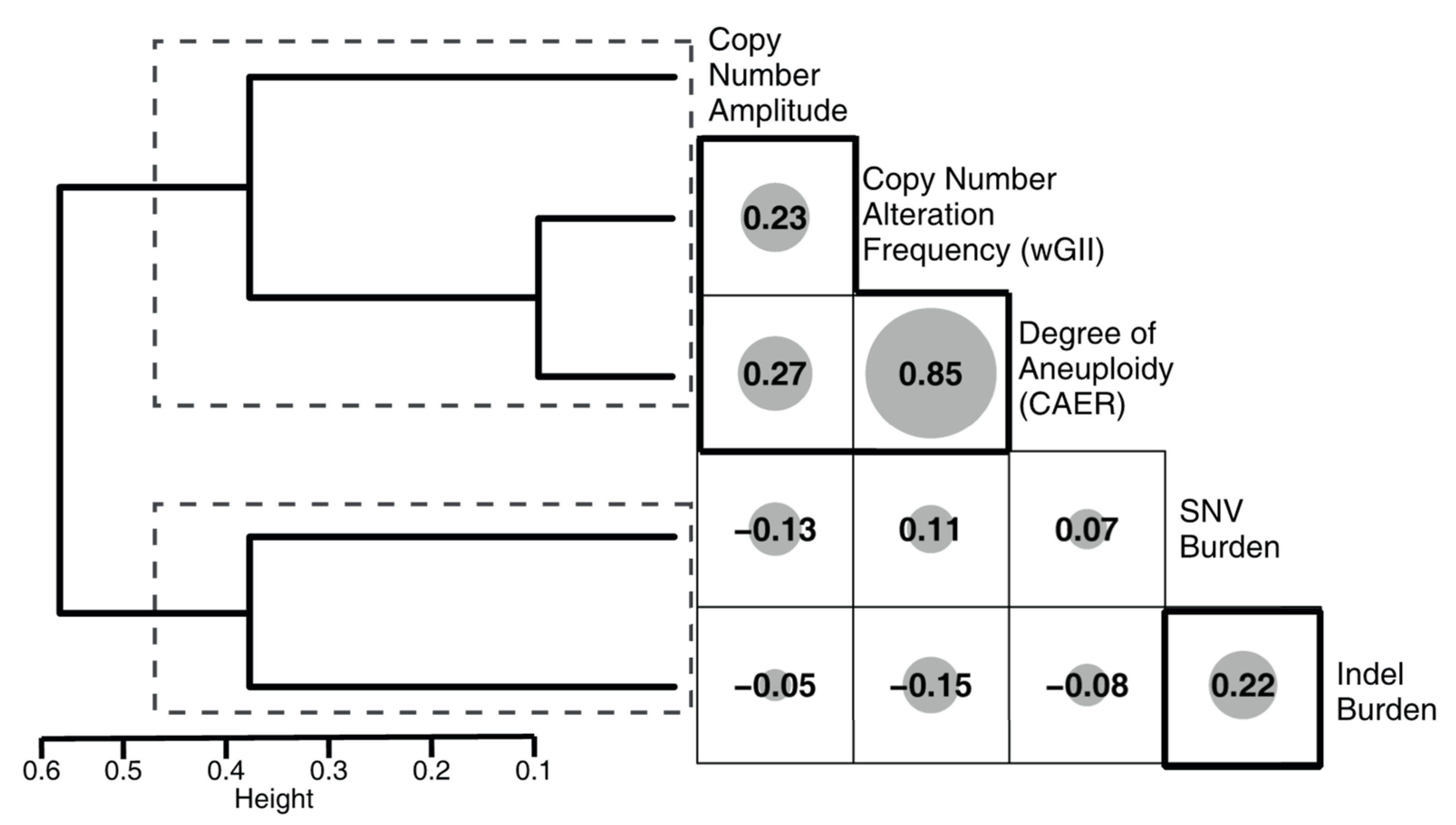
3.5. Correlation of the Metrics with Mutational Signatures
4. Discussion
4.1. Rationale for the Study
4.2. Molecular Subsets of IDH-WT Glioblastomas Differ in Genomic Alteration Burden
4.3. Chromothripsis and Double Minute Events May Be Drivers of Copy-Number Alterations in IDH-WT Glioblastoma
4.4. Mismatch Repair Deficiency Is Likely a Major Driver of Mutational Burden in Gliomas
4.5. Limitations and Future Prospects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Rogan, P.K.; Cancer Genome Atlas Research Network. Faculty Opinions recommendation of Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Turcan, Ş.; Cahill, D.P. Origin of Gliomas. Semin. Neurol. 2018, 38, 5–10. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Initiative, A.P.C.G.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; et al. Signatures of mutational processes in human cancer. Nat. Cell Biol. 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Ülgen, E.; Can, Ö.; Bilguvar, K.; Oktay, Y.; Akyerli, C.B.; Danyeli, A.E.; Yakıcıer, M.C.; Sezerman, O.U.; Pamir, M.N.; Özduman, K. Whole exome sequencing–based analysis to identify DNA damage repair deficiency as a major contributor to gliomagenesis in adult diffuse gliomas. J. Neurosurg. 2019, 132, 1435–1446. [Google Scholar] [CrossRef]
- Degasperi, A.; Amarante, T.D.; Czarnecki, J.; Shooter, S.; Zou, X.; Glodzik, D.; Morganella, S.; Nanda, A.S.; Badja, C.; Koh, G.; et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat. Cancer 2020, 1, 249–263. [Google Scholar] [CrossRef]
- Gerstung, M.; Van Loo, P.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; et al. The evolutionary history of 2658 cancers. Nat. Cell Biol. 2020, 578, 122–128. [Google Scholar] [CrossRef]
- Cortés-Ciriano, I.; Lee, J.J.-K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.-Z.; Park, P.J.; et al. Comprehensive analysis of chromothripsis in 2658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartkova, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nat. Cell Biol. 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nat. Cell Biol. 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt-DeMasters, B.K.; Levy, J.M.M. H3 K27M-mutant gliomas in adults vs. children share similar histological features and adverse prognosis. Clin. Neuropathol. 2018, 37, 53–63. [Google Scholar] [CrossRef]
- Schreck, K.C.; Ranjan, S.; Skorupan, N.; Bettegowda, C.; Eberhart, C.G.; Ames, H.M.; Holdhoff, M. Incidence and clinicopathologic features of H3 K27M mutations in adults with radiographically-determined midline gliomas. J. Neuro Oncol. 2019, 143, 87–93. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Bartels, U.; Bouffet, E.; Becher, O.; Hawkins, C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: Diagnostic and therapeutic implications. Acta Neuropathol. 2014, 128, 573–581. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH wild-type, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hänggi, D.; Wick, W.; et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Sathirapongsasuti, J.F.; Lee, H.; Horst, B.A.J.; Brunner, G.; Cochran, A.J.; Binder, S.; Quackenbush, J.; Nelson, S.F. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics 2011, 27, 2648–2654. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef]
- Pladsen, A.V.; Nilsen, G.; Rueda, O.M.; Aure, M.R.; Borgan, Ø.; Liestøl, K.; Vitelli, V.; Frigessi, A.; Langerød, A.; Mathelier, A.; et al. DNA copy number motifs are strong and independent predictors of survival in breast cancer. Commun. Biol. 2020, 3, 153–159. [Google Scholar] [CrossRef]
- Yang, J.; Liu, J.; Ouyang, L.; Chen, Y.; Liu, B.; Cai, H. CTLPScanner: A web server for chromothripsis-like pattern detection. Nucleic Acids Res. 2016, 44, W252–W258. [Google Scholar] [CrossRef]
- Sanborn, J.Z.; Salama, S.R.; Grifford, M.; Brennan, C.W.; Mikkelsen, T.; Jhanwar, S.; Katzman, S.; Chin, L.; Haussler, D. Double Minute Chromosomes in Glioblastoma Multiforme Are Revealed by Precise Reconstruction of Oncogenic Amplicons. Cancer Res. 2013, 73, 6036–6045. [Google Scholar] [CrossRef]
- Wang, C.; Liang, C. MSIpred: A python package for tumor microsatellite instability classification from tumor mutation annotation data using a support vector machine. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Ülgen, E.; Ozisik, O.; Sezerman, O.U. pathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data through Active Subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]
- Akyerli, C.B.; Yüksel, Ş.; Can, Ö.; Erson-Omay, E.Z.; Oktay, Y.; Coşgun, E.; Ülgen, E.; Erdemgil, Y.; Sav, A.; Von Deimling, A.; et al. Use of telomerase promoter mutations to mark specific molecular subsets with reciprocal clinical behavior in IDH mutant and IDH wild-type diffuse gliomas. J. Neurosurg. 2018, 128, 1102–1114. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.W.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, andTERTPromoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef]
- Melin, B.S.; GliomaScan, C.; Barnholtz-Sloan, J.S.; Wrensch, M.R.; Johansen, C.; Il’Yasova, D.; Kinnersley, B.; Ostrom, Q.T.; LaBreche, K.; Chen, Y.; et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 2017, 49, 789–794. [Google Scholar] [CrossRef]
- Oktay, Y.; Ülgen, E.; Can, Ö.; Akyerli, C.B.; Yüksel, Ş.; Erdemgil, Y.; Durası, İ.M.; Henegariu, O.I.; Nanni, P.; Selevsek, N.; et al. IDH-mutant glioma specific association of rs55705857 located at 8q24.21 involves MYC deregulation. Sci. Rep. 2016, 6, 27569. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Barker, P. Double minutes in human tumor cells. Cancer Genet. Cytogenet. 1982, 5, 81–94. [Google Scholar] [CrossRef]
- Bigner, S.H.; Mark, J.; Bigner, D.D. Cytogenetics of human brain tumors. Cancer Genet. Cytogenet. 1990, 47, 141–154. [Google Scholar] [CrossRef]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; De Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef]
- Barresi, V.; Simbolo, M.; Mafficini, A.; Piredda, M.L.; Caffo, M.; Cardali, S.M.; Germanò, A.; Cingarlini, S.; Ghimenton, C.; Scarpa, A. Ultra-Mutation in IDH Wild-Type Glioblastomas of Patients Younger than 55 Years is Associated with Defective Mismatch Repair, Microsatellite Instability, and Giant Cell Enrichment. Cancers 2019, 11, 1279. [Google Scholar] [CrossRef]
- Erson-Omay, E.Z.; Caglayan, A.O.; Schultz, N.; Weinhold, N.; Omay, S.B.; Özduman, K.; Köksal, Y.; Li, J.; Harmancı, A.S.; Clark, V.; et al. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol. 2015, 17, 1356–1364. [Google Scholar] [CrossRef]
- Erson-Omay, E.Z.; Henegariu, O.; Omay, S.B.; Harmancı, A.S.; Youngblood, M.W.; Mishra-Gorur, K.; Li, J.; Özduman, K.; Carrión-Grant, G.; Clark, V.E.; et al. Longitudinal analysis of treatment-induced genomic alterations in gliomas. Genome Med. 2017, 9, 12. [Google Scholar] [CrossRef][Green Version]
- Nguyen, S.A.; Stechishin, O.D.; Luchman, H.A.; Lun, X.Q.; Senger, D.L.; Robbins, S.M.; Cairncross, J.G.; Weiss, S. Novel MSH6 Mutations in Treatment-Naive Glioblastoma and Anaplastic Oligodendroglioma Contribute to Temozolomide Resistance Independently of MGMT Promoter Methylation. Clin. Cancer Res. 2014, 20, 4894–4903. [Google Scholar] [CrossRef]
- Van Thuijl, H.F.; Mazor, T.; Johnson, B.E.; Fouse, S.D.; Aihara, K.; Hong, C.; Malmström, A.; Hallbeck, M.; Heimans, J.J.; Kloezeman, J.J.; et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol. 2015, 129, 597–607. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nat. Cell Biol. 2020, 580, 517–523. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Analysis ID | Primary Status | Gender | Age at Initial Presentation | Predominant Localization | Sample Type | Pathological Diagnosis | Grade | ATRX | TERT | H3 | EGFR Amplification | 7+/10− | Other Putative Drivers | Molecular Subset |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NOT-0046 | NOT-0046_TA | primary | M | 31 | thalamic | FFPE | Glioblastoma, IDH wild-type | IV | MUT | WT | WT | amplification | FALSE | mGBM | |
| NOT-0046_TB | recurrent | M | 31 | thalamic | LiN2 | Glioblastoma, IDH wild-type | IV | MUT | WT | WT | amplification | TRUE | mGBM | ||
| NOT-0047 | NOT-0047 | recurrent | M | 49 | parietal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | WT | WT | amplification | TRUE | mGBM | |
| NOT-0048 | NOT-0048 | primary | F | 45 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | WT | WT | amplification | TRUE | mGBM | |
| NOT-0051 | NOT-0051 | primary | M | 48 | temporal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0052 | NOT-0052 | primary | F | 65 | hippocampus | FFPE | Anaplastic astrocytoma, IDH wild-type | III | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0054 | NOT-0054 | recurrent | F | 40 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C250 | WT | amplification | TRUE | mGBM | |
| NOT-0056 | NOT-0056 | primary | F | 67 | hippocampus | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0057 | NOT-0057 | primary | F | 62 | temporal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0058 | NOT-0058 | primary | F | 71 | frontal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0060 | NOT-0060 | primary | M | 48 | hippocampus | FFPE | Glioblastoma, IDH wild-type | IV | WT | C250 | WT | amplification | TRUE | mGBM | |
| NOT-0061 | NOT-0061 | primary | M | 55 | occipital | FFPE | Glioblastoma, IDH wild-type | IV | WT | C250 | WT | copy neutral | FALSE | mGBM | |
| NOT-0062 | NOT-0062 | primary | M | 66 | frontal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C250 | WT | amplification | TRUE | mGBM | |
| NOT-0064 | NOT-0064 | primary | F | 48 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0065 | NOT-0065 | primary | M | 59 | occipital | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | FALSE | mGBM | |
| NOT-0066 | NOT-0066 | primary | F | 69 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0067 | NOT-0067 | primary | F | 51 | parietal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | WT | WT | amplification | TRUE | mGBM | |
| NOT-0069 | NOT-0069_TA | primary | M | 46 | parietal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0069_TB | recurrent | M | 46 | parietal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | ||
| NOT-0070 | NOT-0070 | primary | M | 46 | temporal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C250 | WT | copy neutral | TRUE | mGBM | |
| NOT-0071 | NOT-0071 | primary | M | 47 | multifocal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | WT | WT | amplification | FALSE | mGBM | |
| NOT-0073 | NOT-0073 | primary | M | 51 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0076 | NOT-0076 | primary | M | 51 | thalamus | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0078 | NOT-0078 | primary | F | 52 | frontal | LiN2 | Diffuse astrocytoma, WHO grade II, IDH wild-type | II | MUT | WT | WT | amplification | FALSE | mGBM | |
| NOT-0079 | NOT-0079 | primary | M | 40 | parietal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0082 | NOT-0082 | primary | M | 68 | gliomatosis | FFPE | Anaplastic astrocytoma, WHO grade III, IDH wild-type | III | WT | C228 | WT | amplification | FALSE | mGBM | |
| NOT-0084 | NOT-0084 | primary | M | 54 | parietal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | WT | WT | amplification | FALSE | mGBM | |
| NOT-0085 | NOT-0085 | primary | M | 59 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C250 | WT | amplification | TRUE | mGBM | |
| NOT-0086 | NOT-0086 | primary | M | 53 | temporal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C250 | WT | amplification | TRUE | mGBM | |
| NOT-0087 | NOT-0087 | primary | M | 63 | frontal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C250 | WT | amplification | TRUE | mGBM | |
| NOT-0089 | NOT-0089 | primary | M | 62 | parietal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | copy neutral | FALSE | mGBM | |
| NOT-0091 | NOT-0091 | primary | F | 48 | frontal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0092 | NOT-0092_TA | primary | M | 51 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | WT | WT | WT | amplification | FALSE | mGBM | |
| NOT-0092_TB | recurrent | M | 51 | frontal | FFPE | Glioblastoma, IDH wild-type | IV | WT | WT | WT | amplification | FALSE | mGBM | ||
| NOT-0094 | NOT-0094 | primary | M | 64 | frontal | FFPE | Glioblastoma, IDH wild-type | IV | WT | C228 | WT | amplification | TRUE | mGBM | |
| NOT-0075 | NOT-0075_TA | primary | F | 49 | cerebellar | LiN2 | Anaplastic astrocytoma, IDH wild-type | III | MUT | WT | WT | amplification | TRUE | mGBM | |
| NOT-0075_TB | recurrent | F | 49 | cerebellar | LiN2 | Glioblastoma, IDH wild-type | IV | MUT | WT | WT | copy neutral | FALSE | mGBM | ||
| NOT-0059 | NOT-0059 | primary | F | 76 | frontal | FFPE | Glioblastoma, IDH wild-type | IV | WT | WT | WT | copy neutral | FALSE | SETD2 (Y2523) | Others |
| NOT-0063 | NOT-0063 | primary | M | 37 | gliomatosis | LiN2 | Glioblastoma, IDH wild-type | IV | MUT | WT | G34R | copy neutral | FALSE | Others | |
| NOT-0068 | NOT-0068 | primary | M | 64 | gliomatosis | LiN2 | Diffuse astrocytoma, IDH wild-type | II | MUT | WT | WT | copy neutral | FALSE | SETD2 (A2553T) | Others |
| NOT-0083 | NOT-0083 | primary | F | 41 | corpus callosum | FFPE | Glioblastoma, IDH wild-type | IV | MUT | WT | WT | copy neutral | FALSE | SETD2 (R2040*; R2510H) | Others |
| NOT-0088 | NOT-0088_TA | primary | F | 34 | cerebellar | LiN2 | Anaplastic astrocytoma with piloid features, IDH wild-type* | III | WT | WT | WT | copy neutral | TRUE | FGFR1 (K567E; V742M) | Others |
| NOT-0088_TB | recurrent | F | 34 | cerebellar | LiN2 | Glioblastoma, IDH wild-type | IV | WT | WT | WT | copy neutral | FALSE | FGFR1 (K567E; V742M) | Others | |
| NOT-0090 | NOT-0090_TA | primary | M | 28 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | MUT | WT | WT | copy neutral | FALSE | SETD2 (KQ583fs) | Others |
| NOT-0090_TB | recurrent | M | 28 | frontal | LiN2 | Glioblastoma, IDH wild-type | IV | MUT | WT | WT | copy neutral | FALSE | Others |
| Metric | Assessed Genomic Alteration | Possible Mechanisms |
|---|---|---|
| SNV Burden | Frequency of single nucleotide variations (SNV) | DNA damage repair deficiency, Polymerase errors, APOBEC mutagenesis in hypermutated/ultra-mutated tumors |
| Indel Burden | Frequency of short insertion deletions (indels) | Polymerase slippage, Non-homologous End Joining (NHEJ), hairpin loops |
| Weighted Genome Instability Index (wGII) | Frequency of copy number variation (CNV) events | Double stand breaks, NHEJ, Mitotic nondisjunction, Chromosomal instability, Break-Fusion Bridge (BFB) cycles, Chromothripsis |
| Chromosomal Arm Event Ratio (CAER) | Number of chromosomal arm amplifications or deletions (excludes X, Y): Aneuploidy | Mitotic nondisjunction, Chromothripsis |
| Copy number amplitude | Maximum number of amplifications | Extrachromosomal minutes and chromothripsis in cases with high level amplification (>10) |
| Chromothripsis | Massive, clustered, single chromosomal rearrangements | Mitotic nondisjunction |
| Double Minutes | circularization of double-stranded DNA, resulting in highly amplified genes | Chromothripsis, gene amplification, NHEJ |
| Microsatellite instability (MSI) | Length variation in microsatellite repeats | Mismatch Repair (MMR) deficiency |
| Mutational Signatures | Mechanisms underlying single nucleotide variations | CNS_A: Temozolomide-associated CNS_B: Clock-like mutagenesis CNS_C: Unknown etiology CNS_D: Medulloblastoma-associated CNS_E: Mismatch-repair-associated CNS_F: Neuroblastoma-associated CNS_G: Pilocytic astrocytoma-associated CNS_H: Homologous recombination-BRCA1/2-associated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ülgen, E.; Karacan, S.; Gerlevik, U.; Can, Ö.; Bilguvar, K.; Oktay, Y.; B. Akyerli, C.; K. Yüksel, Ş.; E. Danyeli, A.; Tihan, T.; et al. Mutations and Copy Number Alterations in IDH Wild-Type Glioblastomas Are Shaped by Different Oncogenic Mechanisms. Biomedicines 2020, 8, 574. https://doi.org/10.3390/biomedicines8120574
Ülgen E, Karacan S, Gerlevik U, Can Ö, Bilguvar K, Oktay Y, B. Akyerli C, K. Yüksel Ş, E. Danyeli A, Tihan T, et al. Mutations and Copy Number Alterations in IDH Wild-Type Glioblastomas Are Shaped by Different Oncogenic Mechanisms. Biomedicines. 2020; 8(12):574. https://doi.org/10.3390/biomedicines8120574
Chicago/Turabian StyleÜlgen, Ege, Sıla Karacan, Umut Gerlevik, Özge Can, Kaya Bilguvar, Yavuz Oktay, Cemaliye B. Akyerli, Şirin K. Yüksel, Ayça E. Danyeli, Tarık Tihan, and et al. 2020. "Mutations and Copy Number Alterations in IDH Wild-Type Glioblastomas Are Shaped by Different Oncogenic Mechanisms" Biomedicines 8, no. 12: 574. https://doi.org/10.3390/biomedicines8120574
APA StyleÜlgen, E., Karacan, S., Gerlevik, U., Can, Ö., Bilguvar, K., Oktay, Y., B. Akyerli, C., K. Yüksel, Ş., E. Danyeli, A., Tihan, T., Sezerman, O. U., Yakıcıer, M. C., Pamir, M. N., & Özduman, K. (2020). Mutations and Copy Number Alterations in IDH Wild-Type Glioblastomas Are Shaped by Different Oncogenic Mechanisms. Biomedicines, 8(12), 574. https://doi.org/10.3390/biomedicines8120574