The miRNA Content of Exosomes Released from the Glioma Microenvironment Can Affect Malignant Progression

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Experimental Section
2.1. GASC Cell Culture, Supernatant Collection, and Exosomes Isolation
2.2. Nanoparticle Tracking Analysis
2.3. MACSPlex Analysis
2.4. Western Blot
2.5. miRNA Extraction, Library Preparation, and Sequencing
2.6. miRNA Targets Functional Enrichment Analysis
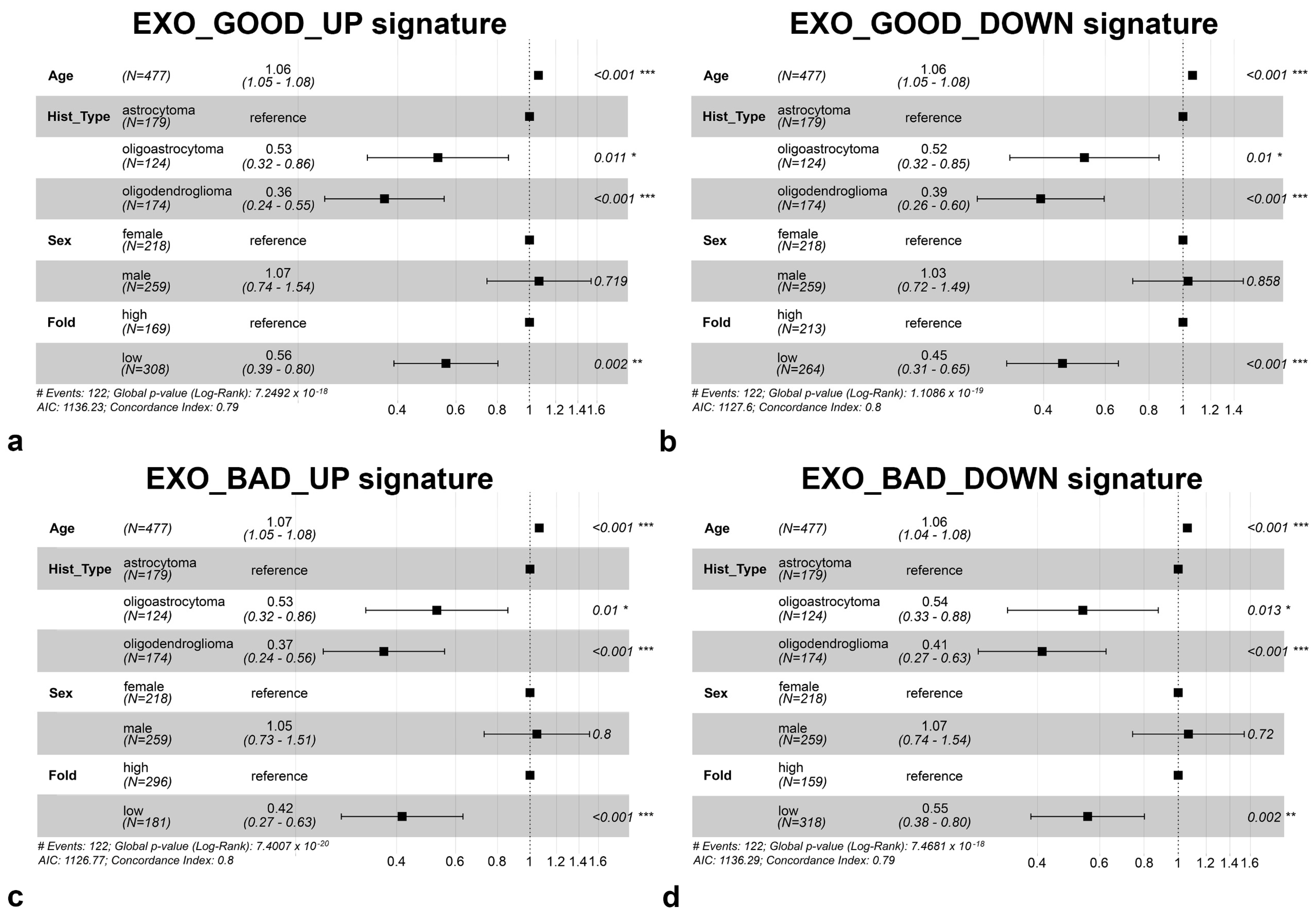
2.7. Survival Analysis
3. Results
3.1. Characterization of Exosomes Derived from LGG with Different Prognosis
3.2. EXO_GOOD and EXO_BAD Display Different miRNA Profiles
3.3. Most of Modulated miRNAs in EXO_GOOD and EXO_BAD Are Endowed with Prognostic Significance
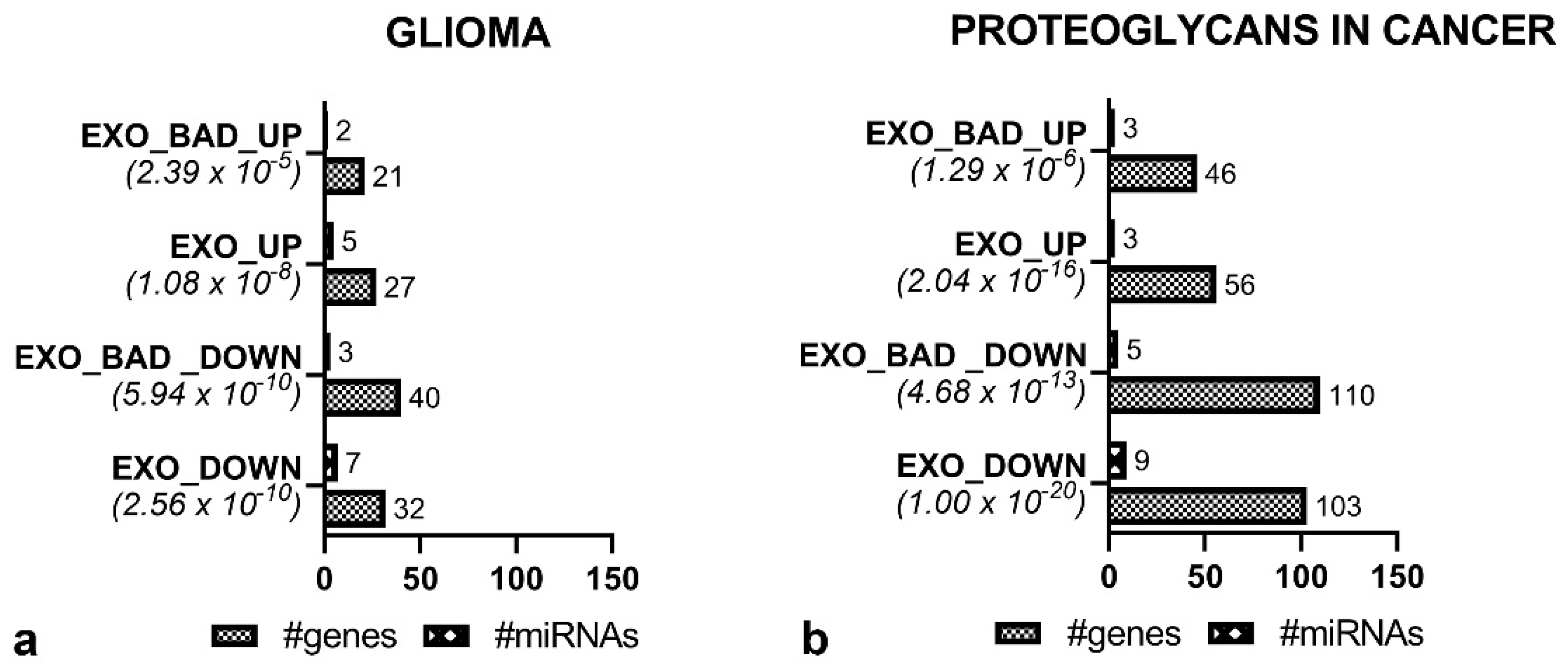
3.4. Functional Enrichment Analysis Reveals Common Pathways in EXO_GOOD and EXO_BAD
3.5. Specific miRNAs of EXO_BAD Could Provide Insight into the Poorer Outcome of Some LGG
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ruda, R.; Angileri, F.F.; Ius, T.; Silvani, A.; Sarubbo, S.; Solari, A.; Castellano, A.; Falini, A.; Pollo, B.; Del Basso De Caro, M.; et al. Italian consensus and recommendations on diagnosis and treatment of low-grade gliomas. An intersociety (SINch/AINO/SIN) document. J. Neurosurg. Sci. 2020, 64, 313–334. [Google Scholar] [CrossRef]
- Soffietti, R.; Baumert, B.G.; Bello, L.; von Deimling, A.; Duffau, H.; Frenay, M.; Grisold, W.; Grant, R.; Graus, F.; Hoang-Xuan, K.; et al. Guidelines on management of low-grade gliomas: Report of an EFNS-EANO Task Force. Eur. J. Neurol. 2010, 17, 1124–1133. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef]
- Cesselli, D.; Ius, T.; Isola, M.; Del Ben, F.; Da Col, G.; Bulfoni, M.; Turetta, M.; Pegolo, E.; Marzinotto, S.; Scott, C.A.; et al. Application of an Artificial Intelligence Algorithm to Prognostically Stratify Grade II Gliomas. Cancers (Basel) 2019, 12, 50. [Google Scholar] [CrossRef]
- Bai, H.; Harmanci, A.S.; Erson-Omay, E.Z.; Li, J.; Coskun, S.; Simon, M.; Krischek, B.; Ozduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Patel, S.; Ngounou Wetie, A.G.; Darie, C.C.; Clarkson, B.D. Cancer secretomes and their place in supplementing other hallmarks of cancer. Adv. Exp. Med. Biol. 2014, 806, 409–442. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Lathia, J.D.; Heddleston, J.M.; Venere, M.; Rich, J.N. Deadly teamwork: Neural cancer stem cells and the tumor microenvironment. Cell Stem Cell 2011, 8, 482–485. [Google Scholar] [CrossRef]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gercel-Taylor, C. Exosomes/microvesicles: Mediators of cancer-associated immunosuppressive microenvironments. Semin. Immunopathol. 2011, 33, 441–454. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes–vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef]
- Arita, T.; Ichikawa, D.; Konishi, H.; Komatsu, S.; Shiozaki, A.; Ogino, S.; Fujita, Y.; Hiramoto, H.; Hamada, J.; Shoda, K.; et al. Tumor exosome-mediated promotion of adhesion to mesothelial cells in gastric cancer cells. Oncotarget 2016, 7, 56855–56863. [Google Scholar] [CrossRef]
- Khalyfa, A.; Almendros, I.; Gileles-Hillel, A.; Akbarpour, M.; Trzepizur, W.; Mokhlesi, B.; Huang, L.; Andrade, J.; Farre, R.; Gozal, D. Circulating exosomes potentiate tumor malignant properties in a mouse model of chronic sleep fragmentation. Oncotarget 2016, 7, 54676–54690. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringner, M.; Morgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef]
- Arscott, W.T.; Tandle, A.T.; Zhao, S.; Shabason, J.E.; Gordon, I.K.; Schlaff, C.D.; Zhang, G.; Tofilon, P.J.; Camphausen, K.A. Ionizing radiation and glioblastoma exosomes: Implications in tumor biology and cell migration. Transl. Oncol. 2013, 6, 638–648. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Mendell, J.T. MicroRNAs: Critical regulators of development, cellular physiology and malignancy. Cell Cycle 2005, 4, 1179–1184. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Fujita, Y.; Yoshioka, Y.; Ochiya, T. Extracellular vesicle transfer of cancer pathogenic components. Cancer Sci. 2016, 107, 385–390. [Google Scholar] [CrossRef]
- Mohammadi, S.; Yousefi, F.; Shabaninejad, Z.; Movahedpour, A.; Mahjoubin Tehran, M.; Shafiee, A.; Moradizarmehri, S.; Hajighadimi, S.; Savardashtaki, A.; Mirzaei, H. Exosomes and cancer: From oncogenic roles to therapeutic applications. IUBMB Life 2020, 72, 724–748. [Google Scholar] [CrossRef]
- Place, A.E.; Jin Huh, S.; Polyak, K. The microenvironment in breast cancer progression: Biology and implications for treatment. Breast Cancer Res. 2011, 13, 227. [Google Scholar] [CrossRef]
- Codrici, E.; Enciu, A.M.; Popescu, I.D.; Mihai, S.; Tanase, C. Glioma Stem Cells and Their Microenvironments: Providers of Challenging Therapeutic Targets. Stem Cells Int. 2016, 2016, 5728438. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Bourkoula, E.; Mangoni, D.; Ius, T.; Pucer, A.; Isola, M.; Musiello, D.; Marzinotto, S.; Toffoletto, B.; Sorrentino, M.; Palma, A.; et al. Glioma-associated stem cells: A novel class of tumor-supporting cells able to predict prognosis of human low-grade gliomas. Stem Cells 2014, 32, 1239–1253. [Google Scholar] [CrossRef]
- Ius, T.; Ciani, Y.; Ruaro, M.E.; Isola, M.; Sorrentino, M.; Bulfoni, M.; Candotti, V.; Correcig, C.; Bourkoula, E.; Manini, I.; et al. An NF-kappaB signature predicts low-grade glioma prognosis: A precision medicine approach based on patient-derived stem cells. Neuro Oncol. 2018, 20, 776–787. [Google Scholar] [CrossRef]
- Manini, I.; Caponnetto, F.; Dalla, E.; Ius, T.; Pepa, G.M.D.; Pegolo, E.; Bartolini, A.; Rocca, G.; Menna, G.; Loreto, C.D.; et al. Heterogeneity Matters: Different Regions of Glioblastoma Are Characterized by Distinctive Tumor-Supporting Pathways. Cancers (Basel) 2020, 12, 960. [Google Scholar] [CrossRef]
- Manini, I.; Ruaro, M.E.; Sgarra, R.; Bartolini, A.; Caponnetto, F.; Ius, T.; Skrap, M.; Di Loreto, C.; Beltrami, A.P.; Manfioletti, G.; et al. Semaphorin-7A on Exosomes: A Promigratory Signal in the Glioma Microenvironment. Cancers (Basel) 2019, 11, 758. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Sirko, S.; Behrendt, G.; Johansson, P.A.; Tripathi, P.; Costa, M.; Bek, S.; Heinrich, C.; Tiedt, S.; Colak, D.; Dichgans, M.; et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. [corrected]. Cell Stem Cell 2013, 12, 426–439. [Google Scholar] [CrossRef]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, Chapter 3, Unit 3.22. [Google Scholar] [CrossRef]
- Pigati, L.; Yaddanapudi, S.C.; Iyengar, R.; Kim, D.J.; Hearn, S.A.; Danforth, D.; Hastings, M.L.; Duelli, D.M. Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS ONE 2010, 5, e13515. [Google Scholar] [CrossRef]
- Guduric-Fuchs, J.; O’Connor, A.; Camp, B.; O’Neill, C.L.; Medina, R.J.; Simpson, D.A. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genom. 2012, 13, 357. [Google Scholar] [CrossRef]
- Jenjaroenpun, P.; Kremenska, Y.; Nair, V.M.; Kremenskoy, M.; Joseph, B.; Kurochkin, I.V. Characterization of RNA in exosomes secreted by human breast cancer cell lines using next-generation sequencing. PeerJ 2013, 1, e201. [Google Scholar] [CrossRef]
- Batagov, A.O.; Kurochkin, I.V. Exosomes secreted by human cells transport largely mRNA fragments that are enriched in the 3′-untranslated regions. Biol. Direct. 2013, 8, 12. [Google Scholar] [CrossRef]
- Bolukbasi, M.F.; Mizrak, A.; Ozdener, G.B.; Madlener, S.; Strobel, T.; Erkan, E.P.; Fan, J.B.; Breakefield, X.O.; Saydam, O. miR-1289 and “Zipcode”-like Sequence Enrich mRNAs in Microvesicles. Mol. Ther. Nucleic Acids 2012, 1, e10. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef]
- Wang, X.R.; Luo, H.; Li, H.L.; Cao, L.; Wang, X.F.; Yan, W.; Wang, Y.Y.; Zhang, J.X.; Jiang, T.; Kang, C.S.; et al. Overexpressed let-7a inhibits glioma cell malignancy by directly targeting K-ras, independently of PTEN. Neuro-Oncology 2013, 15, 1491–1501. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Oh, H.J.; Im, W.S.; Lim, J.Y.; Kim, S.K.; Park, C.K.; Jung, K.H.; Lee, S.K.; Kim, M.; et al. Let-7 microRNA inhibits the proliferation of human glioblastoma cells. J. Neurooncol. 2011, 102, 19–24. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Efe, I.E.; Guneykaya, D.; Ivanov, A.; Huang, Y.; Orlowski, E.; Kruger, C.; Deisz, R.A.; Markovic, D.; Fluh, C.; et al. let-7 MicroRNAs Regulate Microglial Function and Suppress Glioma Growth through Toll-Like Receptor 7. Cell Rep. 2019, 29, 3460–3471. [Google Scholar] [CrossRef]
- Zhou, X.; Ren, Y.; Moore, L.; Mei, M.; You, Y.; Xu, P.; Wang, B.; Wang, G.; Jia, Z.; Pu, P.; et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab. Investig. 2010, 90, 144–155. [Google Scholar] [CrossRef]
- Sayed, D.; He, M.; Hong, C.; Gao, S.; Rane, S.; Yang, Z.; Abdellatif, M. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J. Biol. Chem. 2010, 285, 20281–20290. [Google Scholar] [CrossRef]
- Zhang, C.; Kang, C.; You, Y.; Pu, P.; Yang, W.; Zhao, P.; Wang, G.; Zhang, A.; Jia, Z.; Han, L.; et al. Co-suppression of miR-221/222 cluster suppresses human glioma cell growth by targeting p27kip1 in vitro and in vivo. Int. J. Oncol. 2009, 34, 1653–1660. [Google Scholar] [CrossRef]
- Zhang, J.; Han, L.; Ge, Y.; Zhou, X.; Zhang, A.; Zhang, C.; Zhong, Y.; You, Y.; Pu, P.; Kang, C. miR-221/222 promote malignant progression of glioma through activation of the Akt pathway. Int. J. Oncol. 2010, 36, 913–920. [Google Scholar] [CrossRef]
- Gal, H.; Pandi, G.; Kanner, A.A.; Ram, Z.; Lithwick-Yanai, G.; Amariglio, N.; Rechavi, G.; Givol, D. MIR-451 and Imatinib mesylate inhibit tumor growth of Glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2008, 376, 86–90. [Google Scholar] [CrossRef]
- Nan, Y.; Han, L.; Zhang, A.; Wang, G.; Jia, Z.; Yang, Y.; Yue, X.; Pu, P.; Zhong, Y.; Kang, C. MiRNA-451 plays a role as tumor suppressor in human glioma cells. Brain Res. 2010, 1359, 14–21. [Google Scholar] [CrossRef]
- Sakr, M.; Takino, T.; Sabit, H.; Nakada, M.; Li, Z.; Sato, H. miR-150-5p and miR-133a suppress glioma cell proliferation and migration through targeting membrane-type-1 matrix metalloproteinase. Gene 2016, 587, 155–162. [Google Scholar] [CrossRef]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-kappaB pathways. Oncogene 2020, 39, 428–442. [Google Scholar] [CrossRef]
- Cui, T.; Bell, E.H.; McElroy, J.; Becker, A.P.; Gulati, P.M.; Geurts, M.; Mladkova, N.; Gray, A.; Liu, K.; Yang, L.; et al. miR-4516 predicts poor prognosis and functions as a novel oncogene via targeting PTPN14 in human glioblastoma. Oncogene 2019, 38, 2923–2936. [Google Scholar] [CrossRef]
- Lv, Q.L.; Zhu, H.T.; Li, H.M.; Cheng, X.H.; Zhou, H.H.; Chen, S.H. Down-regulation of miRNA-320c promotes tumor growth and metastasis and predicts poor prognosis in human glioma. Brain Res. Bull. 2018, 139, 125–132. [Google Scholar] [CrossRef]
- Pan, C.; Gao, H.; Zheng, N.; Gao, Q.; Si, Y.; Zhao, Y. MiR-320 inhibits the growth of glioma cells through downregulating PBX3. Biol. Res. 2017, 50, 31. [Google Scholar] [CrossRef]
- Li, T.; Ma, J.; Han, X.; Jia, Y.; Yuan, H.; Shui, S.; Guo, D. MicroRNA-320 Enhances Radiosensitivity of Glioma Through Down-Regulation of Sirtuin Type 1 by Directly Targeting Forkhead Box Protein M1. Transl. Oncol. 2018, 11, 205–212. [Google Scholar] [CrossRef]
- Han, L.; Li, Z.; Jiang, Y.; Jiang, Z.; Tang, L. SNHG29 regulates miR-223-3p/CTNND1 axis to promote glioblastoma progression via Wnt/beta-catenin signaling pathway. Cancer Cell Int. 2019, 19, 345. [Google Scholar] [CrossRef]
- Ji, Y.; Wei, Y.; Wang, J.; Gong, K.; Zhang, Y.; Zuo, H. Correlation of microRNA-10b upregulation and poor prognosis in human gliomas. Tumour Biol. 2015, 36, 6249–6254. [Google Scholar] [CrossRef]
- Li, W.; Li, C.; Xiong, Q.; Tian, X.; Ru, Q. MicroRNA-10b-5p downregulation inhibits the invasion of glioma cells via modulating homeobox B3 expression. Exp. Ther. Med. 2019, 17, 4577–4585. [Google Scholar] [CrossRef]
- Xue, J.; Zhou, A.; Wu, Y.; Morris, S.A.; Lin, K.; Amin, S.; Verhaak, R.; Fuller, G.; Xie, K.; Heimberger, A.B.; et al. miR-182-5p Induced by STAT3 Activation Promotes Glioma Tumorigenesis. Cancer Res. 2016, 76, 4293–4304. [Google Scholar] [CrossRef]
- Li, J.; Yuan, H.; Xu, H.; Zhao, H.; Xiong, N. Hypoxic Cancer-Secreted Exosomal miR-182-5p Promotes Glioblastoma Angiogenesis by Targeting Kruppel-like Factor 2 and 4. Mol. Cancer Res. 2020, 18, 1218–1231. [Google Scholar] [CrossRef]
- Yu, H.; Zheng, J.; Liu, X.; Xue, Y.; Shen, S.; Zhao, L.; Li, Z.; Liu, Y. Transcription Factor NFAT5 Promotes Glioblastoma Cell-driven Angiogenesis via SBF2-AS1/miR-338-3p-Mediated EGFL7 Expression Change. Front. Mol. Neurosci. 2017, 10, 301. [Google Scholar] [CrossRef]
- Wang, W.Y.; Lu, W.C. Reduced Expression of hsa-miR-338-3p Contributes to the Development of Glioma Cells by Targeting Mitochondrial 3-Oxoacyl-ACP Synthase (OXSM) in Glioblastoma (GBM). OncoTargets Ther. 2020, 13, 9513–9523. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Liu, F.; Zhang, J.; Liu, L. Decreased Expression of MiRNA-204-5p Contributes to Glioma Progression and Promotes Glioma Cell Growth, Migration and Invasion. PLoS ONE 2015, 10, e0132399. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Ge, P.; Ma, C. MiR-126 Regulates the ERK Pathway via Targeting KRAS to Inhibit the Glioma Cell Proliferation and Invasion. Mol. Neurobiol. 2017, 54, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Ahn, J.H.; Ryu, D.R.; Lee, J.; Cho, M.S.; Choi, Y.H. Effect of Necrosis on the miRNA-mRNA Regulatory Network in CRT-MG Human Astroglioma Cells. Cancer Res. Treat. 2018, 50, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Ames, H.M.; Yuan, M.; Vizcaino, M.A.; Yu, W.; Rodriguez, F.J. MicroRNA profiling of low-grade glial and glioneuronal tumors shows an independent role for cluster 14q32.31 member miR-487b. Mod. Pathol. 2017, 30, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Wade, A.; Robinson, A.E.; Engler, J.R.; Petritsch, C.; James, C.D.; Phillips, J.J. Proteoglycans and their roles in brain cancer. FEBS J. 2013, 280, 2399–2417. [Google Scholar] [CrossRef]
- Steck, P.A.; Moser, R.P.; Bruner, J.M.; Liang, L.; Freidman, A.N.; Hwang, T.L.; Yung, W.K. Altered expression and distribution of heparan sulfate proteoglycans in human gliomas. Cancer Res. 1989, 49, 2096–2103. [Google Scholar]
- Atukeren, P.; Kunbaz, A.; Turk, O.; Kemerdere, R.; Ulu, M.O.; Turkmen Inanir, N.; Tanriverdi, T. Expressions of Endocan in Patients with Meningiomas and Gliomas. Dis. Markers 2016, 2016, 7157039. [Google Scholar] [CrossRef]
- Yoshida, T.; Matsuda, Y.; Naito, Z.; Ishiwata, T. CD44 in human glioma correlates with histopathological grade and cell migration. Pathol. Int. 2012, 62, 463–470. [Google Scholar] [CrossRef]
- Song, L.; Liu, L.; Wu, Z.; Li, Y.; Ying, Z.; Lin, C.; Wu, J.; Hu, B.; Cheng, S.Y.; Li, M.; et al. TGF-beta induces miR-182 to sustain NF-kappaB activation in glioma subsets. J. Clin. Investig. 2012, 122, 3563–3578. [Google Scholar] [CrossRef]
- Huang, B.S.; Luo, Q.Z.; Han, Y.; Li, X.B.; Cao, L.J.; Wu, L.X. microRNA-223 promotes the growth and invasion of glioblastoma cells by targeting tumor suppressor PAX6. Oncol. Rep. 2013, 30, 2263–2269. [Google Scholar] [CrossRef]
- Villain, G.; Poissonnier, L.; Noueihed, B.; Bonfils, G.; Rivera, J.C.; Chemtob, S.; Soncin, F.; Mattot, V. miR-126-5p promotes retinal endothelial cell survival through SetD5 regulation in neurons. Development 2018, 145. [Google Scholar] [CrossRef]
- Gao, H.; Zhao, H.; Xiang, W. Expression level of human miR-34a correlates with glioma grade and prognosis. J. Neurooncol. 2013, 113, 221–228. [Google Scholar] [CrossRef]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef]
- Duan, J.; Zhou, K.; Tang, X.; Duan, J.; Zhao, L. MicroRNA-34a inhibits cell proliferation and induces cell apoptosis of glioma cells via targeting of Bcl-2. Mol. Med. Rep. 2016, 14, 432–438. [Google Scholar] [CrossRef]
- Li, Q.; Wang, C.; Cai, L.; Lu, J.; Zhu, Z.; Wang, C.; Su, Z.; Lu, X. miR34a derived from mesenchymal stem cells stimulates senescence in glioma cells by inducing DNA damage. Mol. Med. Rep. 2019, 19, 1849–1857. [Google Scholar] [CrossRef]
- Feng, F.; Kuai, D.; Wang, H.; Li, T.; Miao, W.; Liu, Y.; Fan, Y. Reduced expression of microRNA-497 is associated with greater angiogenesis and poor prognosis in human gliomas. Hum. Pathol. 2016, 58, 47–53. [Google Scholar] [CrossRef]
- Lu, F.; Ye, Y.; Zhang, H.; He, X.; Sun, X.; Yao, C.; Mao, H.; He, X.; Qian, C.; Wang, B.; et al. miR-497/Wnt3a/c-jun feedback loop regulates growth and epithelial-to-mesenchymal transition phenotype in glioma cells. Int. J. Biol. Macromol. 2018, 120, 985–991. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Young, A. Re-education of the Tumor Microenvironment With Targeted Therapies and Immunotherapies. Front. Immunol. 2020, 11, 1633. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UPREGULATED miRNAs | DOWNREGULATED miRNAs | |
|---|---|---|
| EXO_GOOD | hsa-miR-320c, hsa-miR-4532, hsa-miR-122-5p, hsa-miR-1246, hsa-miR-126-5p, hsa-miR-142-3p, hsa-miR-142-5p, hsa-miR-144-3p, hsa-miR-144-5p, hsa-miR-150-5p, hsa-miR-378c, hsa-miR-378d, hsa-miR-4516, hsa-miR-451a, hsa-miR-486-5p | hsa-let-7d-5p, hsa-let-7g-5p, hsa-miR-100-3p, hsa-miR-1185-1-3p, hsa-miR-125b-5p, hsa-miR-143-5p, hsa-miR-145-3p, hsa-miR-145-5p, hsa-miR-148b-3p, hsa-miR-22-3p, hsa-miR-24-2-5p, hsa-miR-2682-5p, hsa-miR-374a-5p, hsa-miR-377-3p, hsa-miR-381-3p, hsa-miR-424-5p, hsa-miR-487a-3p, hsa-miR-539-3p, hsa-miR-98-5p, hsa-let-7a-5p, hsa-let-7e-5p, hsa-let-7f-5p, hsa-miR-140-5p, hsa-miR-152-5p, hsa-miR-193a-3p, hsa-miR-193a-5p, hsa-miR-21-3p, hsa-miR-221-5p, hsa-miR-23a-5p, hsa-miR-29a-3p, hsa-miR-337-3p, hsa-miR-34c-5p, hsa-miR-374a-3p, hsa-miR-411-3p, hsa-miR-454-3p, hsa-miR-455-3p, hsa-miR-7974 |
| EXO_BAD | hsa-miR-223-3p, hsa-miR-223-5p, hsa-miR-338-3p, hsa-miR-4492, hsa-miR-4508, hsa-miR-7704, hsa-miR-4488, hsa-miR-10b-5p, hsa-miR-204-5p, hsa-miR-151b, hsa-miR-182-5p, hsa-miR-126-3p, hsa-miR-122-5p, hsa-miR-1246, hsa-miR-126-5p, hsa-miR-142-5p, hsa-miR-144-3p, hsa-miR-144-5p, hsa-miR-150-5p, hsa-miR-378c, hsa-miR-378d, hsa-miR-4516, hsa-miR-451a, hsa-miR-486-5p | hsa-miR-335-3p, hsa-miR-497-5p, hsa-miR-29c-3p, hsa-miR-34a-5p, hsa-let-7a-3p, hsa-miR-582-3p, hsa-miR-29b-3p, hsa-miR-137, hsa-miR-378a-5p, hsa-miR-539-5p, hsa-miR-3607-3p, hsa-miR-29b-1-5p, hsa-miR-31-3p, hsa-miR-1304-3p, hsa-miR-190a-5p, hsa-let-7a-5p, hsa-let-7e-5p, hsa-let-7f-5p, hsa-miR-140-5p, hsa-miR-152-5p, hsa-miR-193a-3p, hsa-miR-193a-5p, hsa-miR-21-3p, hsa-miR-221-5p, hsa-miR-23a-5p, hsa-miR-29a-3p, hsa-miR-337-3p, hsa-miR-34c-5p, hsa-miR-374a-3p, hsa-miR-411-3p, hsa-miR-454-3p, hsa-miR-455-3p, hsa-miR-7974 |
| EXO_GOOD_UP | EXO_GOOD_DOWN | ||||||
|---|---|---|---|---|---|---|---|
| KEGG Pathway | p-Value | #Genes | #miRNAs | KEGG Pathway | p-Value | #Genes | #miRNAs |
| Proteoglycans in cancer | 1.50 × 10−9 | 56 | 3 | Prion diseases | 0 | 11 | 4 |
| Viral carcinogenesis | 8.92 × 10−9 | 59 | 6 | Fatty acid biosynthesis | 0 | 5 | 8 |
| Glioma | 2.52 × 10−6 | 30 | 6 | Steroid biosynthesis | 0 | 9 | 9 |
| Chronic myeloid leukemia | 5.74 × 10−6 | 34 | 4 | Fatty acid metabolism | 0 | 19 | 9 |
| MicroRNAs in cancer | 1.56 × 10−4 | 26 | 4 | Viral carcinogenesis | 0 | 119 | 11 |
| Non-small cell lung cancer | 3.37 × 10−4 | 21 | 2 | Hippo signaling pathway | 0 | 91 | 15 |
| Renal cell carcinoma | 4.01 × 10−3 | 23 | 3 | ECM-receptor interaction | 0 | 39 | 17 |
| Prostate cancer | 4.31 × 10−3 | 33 | 3 | Proteoglycans in cancer | 0 | 132 | 17 |
| Adherens junction | 4.44 × 10−3 | 19 | 4 | Cell cycle | 3.33 × 10−16 | 79 | 10 |
| Central carbon metabolism in cancer | 4.74 × 10−3 | 19 | 3 | Adherens junction | 8.88 × 10−16 | 58 | 14 |
| Other types of O-glycan biosynthesis | 5.85 × 10−3 | 9 | 2 | Lysine degradation | 2.89 × 10−15 | 26 | 11 |
| Pancreatic cancer | 7.21 × 10−3 | 27 | 3 | Pathways in cancer | 1.69 × 10−13 | 197 | 12 |
| Lysine degradation | 9.84 × 10−3 | 12 | 3 | Hepatitis B | 8.91 × 10−12 | 77 | 9 |
| TGF-beta signaling pathway | 1.04 × 10−2 | 25 | 3 | Glioma | 1.70 × 10−10 | 43 | 14 |
| FoxO signaling pathway | 1.10 × 10−2 | 38 | 2 | TGF-beta signaling pathway | 1.13 × 10−9 | 56 | 12 |
| EXO_BAD_UP | EXO_BAD_DOWN | ||||||
|---|---|---|---|---|---|---|---|
| KEGG Pathway | p-Value | #Genes | #miRNAs | KEGG Pathway | p-Value | #Genes | #miRNAs |
| Fatty acid biosynthesis | 0 | 3 | 3 | Prion diseases | 0 | 7 | 2 |
| Viral carcinogenesis | 2.22 × 10−16 | 87 | 9 | Fatty acid biosynthesis | 0 | 4 | 8 |
| Proteoglycans in cancer | 2.72 × 10−11 | 80 | 6 | Fatty acid metabolism | 0 | 22 | 9 |
| Prion diseases | 1.08 × 10−10 | 1 | 1 | Viral carcinogenesis | 0 | 121 | 10 |
| Chronic myeloid leukemia | 1.00 × 10−8 | 48 | 7 | Hippo signaling pathway | 0 | 83 | 14 |
| Glioma | 6.75 × 10−6 | 33 | 7 | Proteoglycans in cancer | 0 | 137 | 14 |
| Adherens junction | 1.59 × 10−5 | 29 | 5 | ECM-receptor interaction | 0 | 43 | 19 |
| Non-small cell lung cancer | 3.00 × 10−5 | 31 | 5 | Steroid biosynthesis | 1.11 × 10−16 | 6 | 9 |
| Prostate cancer | 2.55 × 10−4 | 45 | 5 | Adherens junction | 7.77 × 10−16 | 57 | 12 |
| Pancreatic cancer | 4.83 × 10−4 | 38 | 5 | Lysine degradation | 2.00 × 10−15 | 23 | 9 |
| Renal cell carcinoma | 2.50 × 10−3 | 32 | 5 | p53 signaling pathway | 1.12 × 10−11 | 51 | 10 |
| Other types of O-glycan biosynthesis | 2.70 × 10−3 | 11 | 3 | Glioma | 2.96 × 10−11 | 47 | 10 |
| Hepatitis B | 3.16 × 10−3 | 47 | 4 | Cell cycle | 1.02 × 10−10 | 85 | 6 |
| Pathways in cancer | 3.39 × 10−3 | 125 | 4 | Hepatitis B | 9.02 × 10−10 | 90 | 8 |
| Endometrial cancer | 8.10 × 10−3 | 21 | 4 | Pathways in cancer | 9.96 × 10−9 | 225 | 9 |
| GLIOMA | |
|---|---|
| UPREGULATED | BAD-SPECIFIC: hsa-miR-126-3p, hsa-miR-182-5p EXO-SHARED: hsa-miR-122-5p, hsa-miR-1246, hsa-miR-126-5p, hsa-miR-142-5p, hsa-miR-378d |
| DOWNREGULATED | BAD-SPECIFIC: hsa-miR-29b-3p, hsa-miR-34a-5p, hsa-miR-497-5p EXO-SHARED: hsa-let-7a-5p, hsa-let-7e-5p, hsa-let-7f-5p, hsa-miR-193a-3p, hsa-miR-29a-3p, hsa-miR-34c-5p, hsa-miR-454-3p |
| PROTEOGLYCANS IN CANCER | |
|---|---|
| UPREGULATED | BAD-SPECIFIC: hsa-miR-126-3p, hsa-miR-182-5p, hsa-miR-223-3p EXO-SHARED: hsa-miR-122-5p, hsa-miR-126-5p, hsa-miR-142-3p |
| DOWNREGULATED | BAD-SPECIFIC: hsa-miR-29b-3p, hsa-miR-29c-3p, hsa-miR-34a-5p, hsa-miR-497-5p, hsa-miR-582-3p EXO-SHARED: hsa-let-7a-5p, hsa-let-7e-5p, hsa-let-7f-5p, hsa-miR-140-5p, hsa-miR-193a-3p, hsa-miR-21-3p, hsa-miR-29a-3p, hsa-miR-374a-3p, hsa-miR-454-3p |
| miRNAs | VALIDATED TARGETS | |
|---|---|---|
| UPREGULATED | “BAD-SPECIFIC” only | AKT3, CDKN1A, NRAS, PIK3R1, RB1, SHC1 |
| COMMON TARGETS | AKT1, CALM1, CCND1, CDK4, CDK6, E2F1, E2F3, IGF1R, MAPK1, PIK3CA, PIK3CB, PIK3CD, PIK3R2, SOS1, TP53 | |
| “EXO-SHARED” only | CALM2, E2F2, EGFR, GRB2, KRAS, MDM2, MTOR, PIK3CG, PLCG1, PTEN, RAF1, SOS2 | |
| DOWNREGULATED | “BAD-SPECIFIC” only | AKT1, AKT3, ARAF, CDKN2A, E2F2, GRB2, IGF1, MAP2K1, MAP2K2, MTOR, PIK3CA, PRKCB, RAF1, SOS2, TGFA |
| COMMON TARGETS | AKT2, BRAF, CALM1, CALM2, CALM3, CCND1, CDK4, CDK6, CDKN1A, E2F1, E2F3, EGFR, IGF1R, MAPK1, MDM2, NRAS, PDGFRA, PDGFRB, PIK3R1, PIK3R2, PIK3R3, PLCG1, PTEN, SOS1, TP53 | |
| “EXO-SHARED” only | CAMK2D, KRAS, PDGFA, PDGFB, PIK3CB, PRKCA, RB1 | |
| miRNAs | VALIDATED TARGETS | |
|---|---|---|
| UPREGULATED | “BAD-SPECIFIC” only | AKT3, CDC42, CDKN1A, EIF4B, ELK1, ESR1, FGF2, FZD6, HIF1A, ITPR3, MET, MMP2, MMP9, MSN, NRAS, PIK3CB, PIK3R1, PPP1R1, PRKACB, RPS6KB2, SDC4, SOS1, STAT3, THBS1 |
| COMMON TARGETS | AKT1, CBL, CCND1, CTNNB1, CTTN, FRS2, FZD3, IGF1R, IQGAP1, ITGB1, MAPK1, MTOR, PDPK1, PIK3CA, PIK3CD, PIK3R2, RAC1, RDX, ROCK2, SDC2, TP53, VEGFA | |
| “EXO-SHARED” only | ACTB, AKT2, ANK1, ARHGEF12, CD44, DCN, EGFR, FGFR1, FLNB, FZD1, FZD4, FZD7, GAB1, GRB2, HCLS1, ITGAV, KRAS, MAPK13, MDM2, MYC, PAK1, PIK3CG, PIK3R5, PLCG1, PTCH1, PTPN11, RAF1, RPS6KB1, SLC9A1, SMAD2, SOS2, VAV2, VMP1, WNT5A | |
| DOWNREGULATED | “BAD-SPECIFIC” only | AKT1, AKT2, ANK3, ARAF, CBLC, CD44, CD63, COL21A1, CTTN, ELK1, FGF2, FZD1, GAB1, GPC1, GRB2, HGF, HPSE, IGF1, ITGA5, ITPR1, MAP2K1, MAP2K2, MAPK12, MMP9, PIK3CA, PPP1R12B, PRKACA, PRKACB, PRKCB, PRKX, PTCH1, RAF1, ROCK2, TFAP4 |
| COMMON TARGETS | ACTB, ACTG1, AKT3, ARHGEF12, BRAF, CAV1, CAV2, CCND1, CDC42, CDKN1A, CTNNB1, DDX5, EGFR, EZR, FGFR1, FLNB, FN1, FRS2, FZD5, FZD6, GPC3, HIF1A, HSPG2, IGF1R, IGF2, IQGAP1, ITGAV, ITGB1, ITGB5, ITPR3, MAPK1, MAPK13, MDM2, MET, MMP2, MSN, MTOR, MYC, NRAS, NUDT16L1, PDCD4, PIK3R1, PIK3R2, PIK3R3, PLAU, PLAUR, PLCG1, PPP1CA, PPP1CB, PPP1CC, PPP1R12A, PPP1R12C, PRKCA, PTPN11, PXN, RDX, RPS6, RRAS, SDC1, SDC2, SDC4, SLC9A1, SMAD2, SMO, SOS1, SOS2, SRC, STAT3, TGFB1, TGFB2, THBS1, TP53, VAV2, VEGFA, WNT5A, WNT9A | |
| “EXO-SHARED” only | ARHGEF1, CAMK2D, CASP3, CBL, DROSHA, EIF4B, ESR1, FAS, FLNA, FLNC, FZD2, FZD3, FZD4, HOXD10, ITGA2, ITPR2, KDR, KRAS, PIK3CB, PTK2, PTPN6, RAC1, RPS6KB2, TLR4, VMP1, WNT3A, WNT5B | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caponnetto, F.; Dalla, E.; Mangoni, D.; Piazza, S.; Radovic, S.; Ius, T.; Skrap, M.; Di Loreto, C.; Beltrami, A.P.; Manini, I.; et al. The miRNA Content of Exosomes Released from the Glioma Microenvironment Can Affect Malignant Progression. Biomedicines 2020, 8, 564. https://doi.org/10.3390/biomedicines8120564
Caponnetto F, Dalla E, Mangoni D, Piazza S, Radovic S, Ius T, Skrap M, Di Loreto C, Beltrami AP, Manini I, et al. The miRNA Content of Exosomes Released from the Glioma Microenvironment Can Affect Malignant Progression. Biomedicines. 2020; 8(12):564. https://doi.org/10.3390/biomedicines8120564
Chicago/Turabian StyleCaponnetto, Federica, Emiliano Dalla, Damiano Mangoni, Silvano Piazza, Slobodanka Radovic, Tamara Ius, Miran Skrap, Carla Di Loreto, Antonio Paolo Beltrami, Ivana Manini, and et al. 2020. "The miRNA Content of Exosomes Released from the Glioma Microenvironment Can Affect Malignant Progression" Biomedicines 8, no. 12: 564. https://doi.org/10.3390/biomedicines8120564
APA StyleCaponnetto, F., Dalla, E., Mangoni, D., Piazza, S., Radovic, S., Ius, T., Skrap, M., Di Loreto, C., Beltrami, A. P., Manini, I., & Cesselli, D. (2020). The miRNA Content of Exosomes Released from the Glioma Microenvironment Can Affect Malignant Progression. Biomedicines, 8(12), 564. https://doi.org/10.3390/biomedicines8120564