An Exploratory Pilot Study with Plasma Protein Signatures Associated with Response of Patients with Depression to Antidepressant Treatment for 10 Weeks


 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Blood Collection and Plasma Preparation
2.3. Plasma Manipulation and Digestion
2.4. Nano-LC-ESI-MS/MS Analysis
2.5. Database Searching and Label-free Quantification
2.6. Analysis of Public Microarray Data
2.7. Batch Mean-Centering Correction, Missing Data Imputation, and Normalization
2.8. LC-ESI-MRM/MS Analysis
2.9. Statistical Analysis
2.10. Literature Search
3. Results
3.1. Demographic and Clinical Characteristics of Study Subjects
3.2. Plasma Sample Preparations and Development of LC-MS/MS
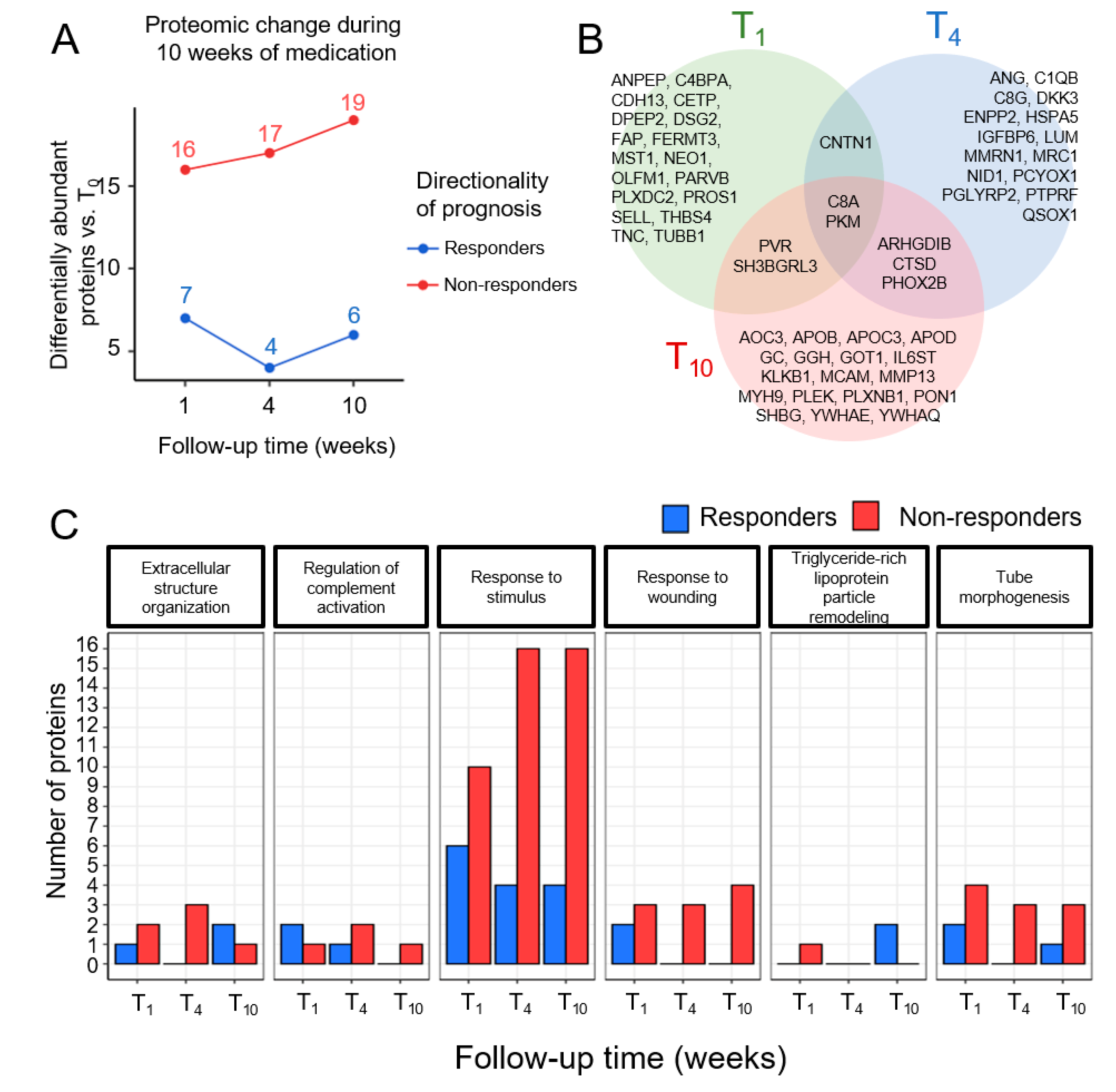
3.3. Time-Dependent Changes in Plasma Proteins in Responders and Non-Responders
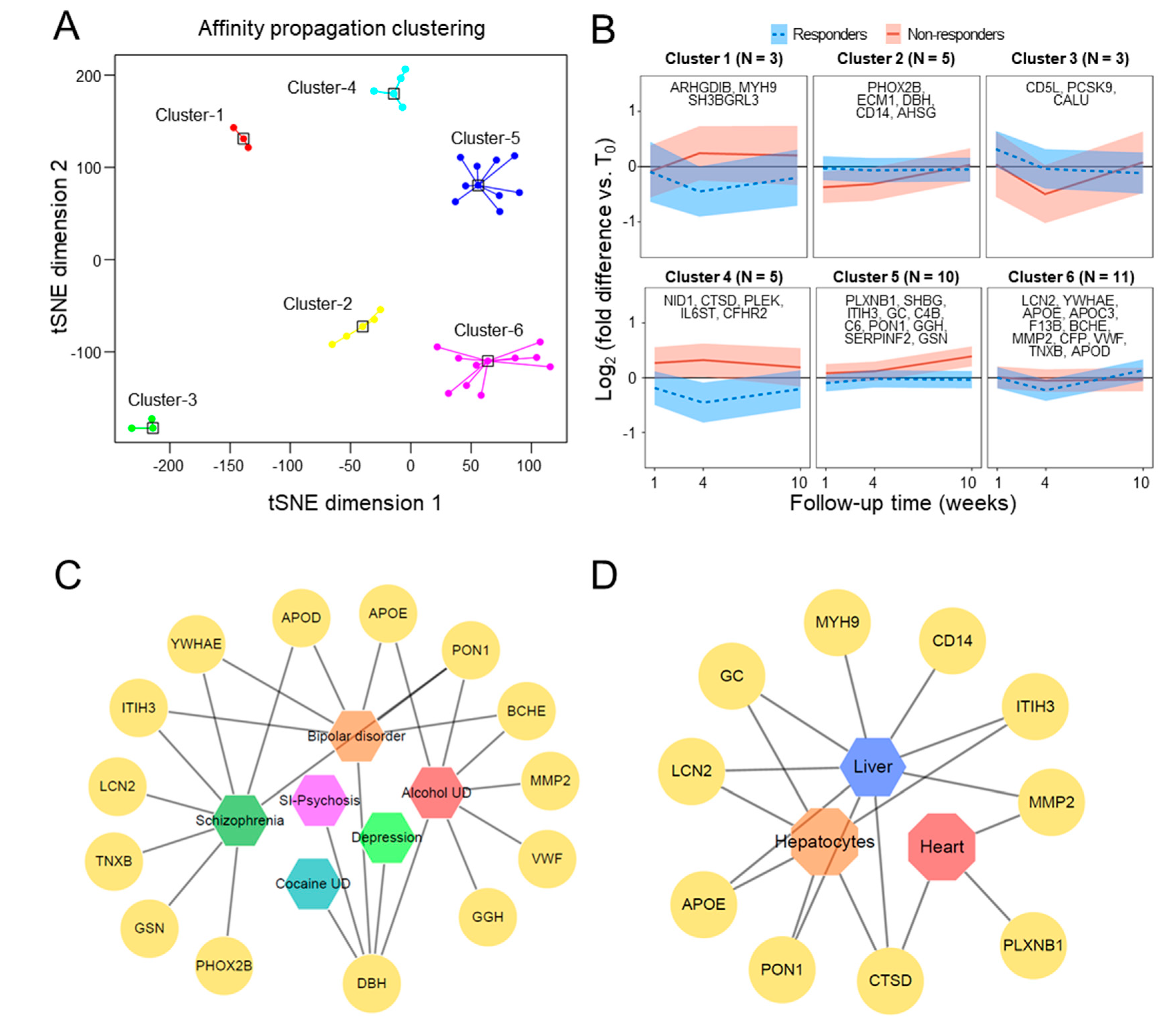
3.4. External Validation in Public Studies of mRNA Expression
3.5. LC-MRM/MS Validation of Candidate Plasma Proteins Predictive of Early Response
3.6. Relationship between Plasma Proteins and Psychiatric Morbidity Survey Scores
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDD | Major depressive disorder |
| SSRIs | Selective serotonin reuptake inhibitors |
| STAR*D | Sequenced Treatment Alternative to Relieve Depression |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| MEM | mixed-effect model |
| GEE | generalized estimating equation |
| LMM | Linear mixed model |
| LC-MRM/MS | Liquid chromatography-multiple reaction monitoring/mass spectrometry |
| LFQ | Label-free quantification |
| SD | Standard deviation |
| MADRS | Montgomery and Asberg Depression Rating Scale |
| CGI-S | Clinical Global Impression-Severity |
| BDI | Beck’s Depression Inventory |
| HAM-D | Hamilton Rating Scale for Depression |
| CUDOS | Clinically Useful Depression Outcome Scale |
| GO | Gene ontology |
| SGoF | Sequential goodness of fit metatest |
| t-SNE | t-stochastic neighbor embedding |
| PsyGeNet | Psychiatric disorders gene association network |
| PsychoQOL | Psychological quality of life |
| DBH | Dopamine beta-hydroxylase |
| NSF | Normalization scaling factor |
References
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V.; et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- Fava, M.; Kendler, K.S. Major depressive disorder. Neuron 2000, 28, 335–341. [Google Scholar] [CrossRef]
- Burcusa, S.L.; Iacono, W.G. Risk for recurrence in depression. Clin. Psychol. Rev. 2007, 27, 959–985. [Google Scholar] [CrossRef] [PubMed]
- Cain, R.A. Navigating the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study: Practical outcomes and implications for depression treatment in primary care. Prim. Care 2007, 34, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Stewart, J.W.; Nierenberg, A.A.; Thase, M.E.; Ritz, L.; Biggs, M.M.; Warden, D.; Luther, J.F.; et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N. Engl. J. Med. 2006, 354, 1231–1242. [Google Scholar] [CrossRef]
- Martins-de-Souza, D.; Harris, L.W.; Guest, P.C.; Turck, C.W.; Bahn, S. The role of proteomics in depression research. Eur. Arch. Psychiatry Clin. Neurosci. 2010, 260, 499–506. [Google Scholar] [CrossRef]
- Turck, C.W.; Guest, P.C.; Maccarrone, G.; Ising, M.; Kloiber, S.; Lucae, S.; Holsboer, F.; Martins-de-Souza, D. Proteomic Differences in Blood Plasma Associated with Antidepressant Treatment Response. Front. Mol. Neurosci. 2017, 10, 272. [Google Scholar] [CrossRef]
- Chan, M.K.; Cooper, J.D.; Bot, M.; Birkenhager, T.K.; Bergink, V.; Drexhage, H.A.; Steiner, J.; Rothermundt, M.; Penninx, B.W.; Bahn, S. Blood-based immune-endocrine biomarkers of treatment response in depression. J. Psychiatr. Res. 2016, 83, 249–259. [Google Scholar] [CrossRef]
- Feng, Z.; Diehr, P.; Peterson, A.; McLerran, D. Selected statistical issues in group randomized trials. Annu. Rev. Public Health 2001, 22, 167–187. [Google Scholar] [CrossRef]
- Gibbons, R.D.; Hedeker, D.; DuToit, S. Advances in analysis of longitudinal data. Annu. Rev. Clin. Psychol. 2010, 6, 79–107. [Google Scholar] [CrossRef]
- Laird, N.M.; Ware, J.H. Random-effects models for longitudinal data. Biometrics 1982, 38, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.Y.; Zeger, S.L. Longitudinal Data-Analysis Using Generalized Linear-Models. Biometrika 1986, 73, 13–22. [Google Scholar] [CrossRef]
- Pedroza, C.; Truong, V.T.T. Estimating relative risks in multicenter studies with a small number of centers—Which methods to use? A simulation study. Trials 2017, 18, 512. [Google Scholar] [CrossRef] [PubMed]
- Aktas Samur, A.; Coskunfirat, N.; Saka, O. Comparison of predictor approaches for longitudinal binary outcomes: Application to anesthesiology data. PeerJ 2014, 2, e648. [Google Scholar] [CrossRef] [PubMed]
- McNeish, D.M.; Harring, J.R. Clustered data with small sample sizes: Comparing the performance of model-based and design-based approaches. Commun. Stat-Simul. C 2017, 46, 855–869. [Google Scholar] [CrossRef]
- Zhou, C.; Simpson, K.L.; Lancashire, L.J.; Walker, M.J.; Dawson, M.J.; Unwin, R.D.; Rembielak, A.; Price, P.; West, C.; Dive, C.; et al. Statistical considerations of optimal study design for human plasma proteomics and biomarker discovery. J. Proteome Res. 2012, 11, 2103–2113. [Google Scholar] [CrossRef]
- Cairns, D.A.; Barrett, J.H.; Billingham, L.J.; Stanley, A.J.; Xinarianos, G.; Field, J.K.; Johnson, P.J.; Selby, P.J.; Banks, R.E. Sample size determination in clinical proteomic profiling experiments using mass spectrometry for class comparison. Proteomics 2009, 9, 74–86. [Google Scholar] [CrossRef]
- Wang, M.; Kong, L.; Li, Z.; Zhang, L. Covariance estimators for generalized estimating equations (GEE) in longitudinal analysis with small samples. Stat. Med. 2016, 35, 1706–1721. [Google Scholar] [CrossRef]
- Ren, J.; Zhao, G.; Sun, X.; Liu, H.; Jiang, P.; Chen, J.; Wu, Z.; Peng, D.; Fang, Y.; Zhang, C. Identification of plasma biomarkers for distinguishing bipolar depression from major depressive disorder by iTRAQ-coupled LC-MS/MS and bioinformatics analysis. Psychoneuroendocrinology 2017, 86, 17–24. [Google Scholar] [CrossRef]
- Park, D.I.; Stambuk, J.; Razdorov, G.; Pucic-Bakovic, M.; Martins-de-Souza, D.; Lauc, G.; Turck, C.W. Blood plasma/IgG N-glycome biosignatures associated with major depressive disorder symptom severity and the antidepressant response. Sci. Rep. 2018, 8, 179. [Google Scholar] [CrossRef]
- Lee, M.Y.; Kim, E.Y.; Kim, S.H.; Cho, K.C.; Ha, K.; Kim, K.P.; Ahn, Y.M. Discovery of serum protein biomarkers in drug-free patients with major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 69, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Fang, Z.; Xu, X.J.; Liu, M.L.; Du, X.; Zhang, X.; Wang, H.; Zhou, J.; Xie, P. Metabolite signature for diagnosing major depressive disorder in peripheral blood mononuclear cells. J. Affect Disord. 2016, 195, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Kim, S.H.; Lee, H.J.; Lee, N.Y.; Kim, H.Y.; Park, C.H.K.; Ahn, Y.M. A randomized, double-blind, 6-week prospective pilot study on the efficacy and safety of dose escalation in non-remitters in comparison to those of the standard dose of escitalopram for major depressive disorder. J. Affect. Disord. 2019, 259, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; Garcia-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef]
- Ahn, H.S.; Kim, J.H.; Jeong, H.; Yu, J.; Yeom, J.; Song, S.H.; Kim, S.S.; Kim, I.J.; Kim, K. Differential Urinary Proteome Analysis for Predicting Prognosis in Type 2 Diabetes Patients with and without Renal Dysfunction. Int. J. Mol. Sci. 2020, 21, 4236. [Google Scholar] [CrossRef]
- Belzeaux, R.; Gorgievski, V.; Fiori, L.M.; Lopez, J.P.; Grenier, J.; Lin, R.; Nagy, C.; Ibrahim, E.C.; Gascon, E.; Courtet, P.; et al. GPR56/ADGRG1 is associated with response to antidepressant treatment. Nat. Commun. 2020, 11, 1635. [Google Scholar] [CrossRef]
- Mehta, D.; Raison, C.L.; Woolwine, B.J.; Haroon, E.; Binder, E.B.; Miller, A.H.; Felger, J.C. Transcriptional signatures related to glucose and lipid metabolism predict treatment response to the tumor necrosis factor antagonist infliximab in patients with treatment-resistant depression. Brain Behav. Immun. 2013, 31, 205–215. [Google Scholar] [CrossRef]
- Goh, W.W.B.; Wang, W.; Wong, L. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol. 2017, 35, 498–507. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, S.; Su, P.F.; Li, C.I.; Ye, F.; Flynn, C.R.; Shyr, Y. Statistical strategies for microRNAseq batch effect reduction. Transl. Cancer Res. 2014, 3, 260–265. [Google Scholar] [CrossRef]
- Sims, A.H.; Smethurst, G.J.; Hey, Y.; Okoniewski, M.J.; Pepper, S.D.; Howell, A.; Miller, C.J.; Clarke, R.B. The removal of multiplicative, systematic bias allows integration of breast cancer gene expression—Improving meta-analysis and prediction of prognosis. BMC Med. Genom. 2008, 1, 42. [Google Scholar] [CrossRef]
- Kim, H.; Golub, G.H.; Park, H. Missing value estimation for DNA microarray gene expression data: Local least squares imputation. Bioinformatics 2005, 21, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Bodenhofer, U.; Kothmeier, A.; Hochreiter, S. APCluster: An R package for affinity propagation clustering. Bioinformatics 2011, 27, 2463–2464. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Sacristan, A.; Hernandez-Ferrer, C.; Gonzalez, J.R.; Furlong, L.I. psygenet2r: A R/Bioconductor package for the analysis of psychiatric disease genes. Bioinformatics 2017, 33, 4004–4006. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Rodriguez, A.; de Una-Alvarez, J.; Rolan-Alvarez, E. A new multitest correction (SGoF) that increases its statistical power when increasing the number of tests. BMC Bioinform. 2009, 10, 209. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Legendre, P. Comparison of permutation methods for the partial correlation and partial Mantel tests. J. Stat. Comput. Sim. 2000, 67, 37–73. [Google Scholar] [CrossRef]
- Wei, C.H.; Allot, A.; Leaman, R.; Lu, Z. PubTator central: Automated concept annotation for biomedical full text articles. Nucleic Acids Res. 2019, 47, W587–W593. [Google Scholar] [CrossRef]
- Ahn, H.S.; Sohn, T.S.; Kim, M.J.; Cho, B.K.; Kim, S.M.; Kim, S.T.; Yi, E.C.; Lee, C. SEPROGADIC—Serum protein-based gastric cancer prediction model for prognosis and selection of proper adjuvant therapy. Sci. Rep. 2018, 8, 16892. [Google Scholar] [CrossRef]
- Martinez-Aguilar, J.; Molloy, M.P. Label-free selected reaction monitoring enables multiplexed quantitation of S100 protein isoforms in cancer cells. J. Proteome Res. 2013, 12, 3679–3688. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.M.; Omenn, G.S.; Sun, Z.; Campbell, D.S.; Baker, M.S.; Overall, C.M.; Aebersold, R.; Moritz, R.L.; Deutsch, E.W. The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J. Proteome Res. 2017, 16, 4299–4310. [Google Scholar] [CrossRef] [PubMed]
- Karpievitch, Y.V.; Dabney, A.R.; Smith, R.D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinform. 2012, 13 (Suppl. 16), S5. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.J.; Abbatiello, S.E.; Kim, K.; Yan, P.; Whiteaker, J.R.; Lin, C.; Kim, J.S.; Zhang, Y.; Wang, X.; Ivey, R.G.; et al. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat. Methods 2014, 11, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, Y.; Lee, M.Y.; Shin, J.; Han, J.M.; Yang, E.G.; Yu, M.H.; Kim, S.; Hwang, D.; Lee, C. Multiple reaction monitoring of multiple low-abundance transcription factors in whole lung cancer cell lysates. J. Proteome Res. 2013, 12, 2582–2596. [Google Scholar] [CrossRef]
- Bhowmick, P.; Mohammed, Y.; Borchers, C.H. MRMAssayDB: An integrated resource for validated targeted proteomics assays. Bioinformatics 2018, 34, 3566–3571. [Google Scholar] [CrossRef]
- Muth, C.; Bales, K.L.; Hinde, K.; Maninger, N.; Mendoza, S.P.; Ferrer, E. Alternative Models for Small Samples in Psychological Research: Applying Linear Mixed Effects Models and Generalized Estimating Equations to Repeated Measures Data. Educ. Psychol. Meas. 2016, 76, 64–87. [Google Scholar] [CrossRef]
- Wang, M.; Long, Q. Modified robust variance estimator for generalized estimating equations with improved small-sample performance. Stat. Med. 2011, 30, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- McNeish, D.; Stapleton, L.M. Modeling Clustered Data with Very Few Clusters. Multivar Behav. Res. 2016, 51, 495–518. [Google Scholar] [CrossRef]
- Lopez-Vilchez, I.; Diaz-Ricart, M.; Navarro, V.; Torramade, S.; Zamorano-Leon, J.; Lopez-Farre, A.; Galan, A.M.; Gasto, C.; Escolar, G. Endothelial damage in major depression patients is modulated by SSRI treatment, as demonstrated by circulating biomarkers and an in vitro cell model. Transl. Psychiatry 2016, 6, e886. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C.; Corponi, F.; Albani, D.; Raimondi, I.; Forloni, G.; Schruers, K.; Kasper, S.; Kautzky, A.; Zohar, J.; Souery, D.; et al. Pleiotropic genes in psychiatry: Calcium channels and the stress-related FKBP5 gene in antidepressant resistance. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Bobinska, K.; Szemraj, J.; Galecki, P.; Talarowska, M. The role of MMP genes in recurrent depressive disorders and cognitive functions. Acta Neuropsychiatr. 2016, 28, 221–231. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, P.; Raza, M.U.; Szebeni, A.; Szebeni, K.; Ordway, G.A.; Stockmeier, C.A.; Zhu, M.Y. Altered Expression of Phox2 Transcription Factors in the Locus Coeruleus in Major Depressive Disorder Mimicked by Chronic Stress and Corticosterone Treatment In Vivo and In Vitro. Neuroscience 2018, 393, 123–137. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, H.X.; Li, Z.Q.; Li, T.; Li, J.Y.; Wang, T.; Li, Y.; Feng, G.Y.; Shi, Y.Y.; He, L. The YWHAE gene confers risk to major depressive disorder in the male group of Chinese Han population. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 77, 172–177. [Google Scholar] [CrossRef]
- Pope, H.G., Jr.; Kouri, E.M.; Hudson, J.I. Effects of supraphysiologic doses of testosterone on mood and aggression in normal men: A randomized controlled trial. Arch. Gen. Psychiatry 2000, 57, 133–140. [Google Scholar] [CrossRef]
- Asselmann, E.; Kische, H.; Haring, R.; Hertel, J.; Schmidt, C.O.; Nauck, M.; Beesdo-Baum, K.; Grabe, H.J.; Pane-Farre, C.A. Prospective associations of androgens and sex hormone-binding globulin with 12-month, lifetime and incident anxiety and depressive disorders in men and women from the general population. J. Affect. Disord. 2019, 245, 905–911. [Google Scholar] [CrossRef]
- Whalley, L.J.; Kutcher, S.; Blackwood, D.H.; Bennie, J.; Dick, H.; Fink, G. Increased plasma LH in manic-depressive illness: Evidence of a state-independent abnormality. Br. J. Psychiatry 1987, 150, 682–684. [Google Scholar] [CrossRef]
- Cakici, N.; Bot, M.; Lamers, F.; Janssen, T.; van der Spek, P.J.; de Haan, L.; Bahn, S.; Penninx, B.; van Beveren, N.J.M. Increased serum levels of leptin and insulin in both schizophrenia and major depressive disorder: A cross-disorder proteomics analysis. Eur. Neuropsychopharmacol. 2019, 29, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Cho, H.W.; Carroll, B.J.; Russo, P. Serum dopamine-beta-hydroxylase activity in the affective psychoses and schizophrenia. Decreased activity in unipolar psychotically depressed patients. Arch. Gen. Psychiatry 1976, 33, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Paclt, I.; Koudelova, J.; Pacltova, D.; Kopeckova, M. Dopamine beta hydroxylase (DBH) plasma activity in childhood mental disorders. Neuro Endocrinol. Lett. 2009, 30, 604–609. [Google Scholar] [PubMed]
- Puzynski, S.; Hauptmann, M.; Rode, A.; Kalinowski, A.; Bidzinska, E.; Beresewicz, M.; Bidzinski, A. Blood MAO/DBH index and the results of the treatment of endogenous depression. Psychiatr. Pol. 1990, 24, 202–208. [Google Scholar] [PubMed]
- Yu, P.H.; O’Sullivan, K.S.; Keegan, D.; Boulton, A.A. Dopamine-beta-hydroxylase and its apparent endogenous inhibitory activity in the plasma of some psychiatric patients. Psychiatry Res. 1980, 3, 205–210. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; He, Y.; Zhou, J.; Xi, Q.; Song, X.; Ye, Y.; Ying, B. Association between dopamine beta-hydroxylase 19-bp insertion/deletion polymorphism and major depressive disorder. J. Mol. Neurosci. 2015, 55, 367–371. [Google Scholar] [CrossRef]
- Zhan, Y.; Yang, Y.T.; You, H.M.; Cao, D.; Liu, C.Y.; Zhou, C.J.; Wang, Z.Y.; Bai, S.J.; Mu, J.; Wu, B.; et al. Plasma-based proteomics reveals lipid metabolic and immunoregulatory dysregulation in post-stroke depression. Eur. Psychiatry 2014, 29, 307–315. [Google Scholar] [CrossRef]
- Wang, Q.; Su, X.; Jiang, X.; Dong, X.; Fan, Y.; Zhang, J.; Yu, C.; Gao, W.; Shi, S.; Jiang, J.; et al. iTRAQ technology-based identification of human peripheral serum proteins associated with depression. Neuroscience 2016, 330, 291–325. [Google Scholar] [CrossRef]
- Stelzhammer, V.; Haenisch, F.; Chan, M.K.; Cooper, J.D.; Steiner, J.; Steeb, H.; Martins-de-Souza, D.; Rahmoune, H.; Guest, P.C.; Bahn, S. Proteomic changes in serum of first onset, antidepressant drug-naive major depression patients. Int. J. Neuropsychopharmacol. 2014, 17, 1599–1608. [Google Scholar] [CrossRef]
- Ramsey, J.M.; Cooper, J.D.; Bot, M.; Guest, P.C.; Lamers, F.; Weickert, C.S.; Penninx, B.W.; Bahn, S. Sex Differences in Serum Markers of Major Depressive Disorder in the Netherlands Study of Depression and Anxiety (NESDA). PLoS ONE 2016, 11, e0156624. [Google Scholar] [CrossRef]
- Moreira, E.G.; Correia, D.G.; Bonifacio, K.L.; Moraes, J.B.; Cavicchioli, F.L.; Nunes, C.S.; Nunes, S.O.V.; Vargas, H.O.; Barbosa, D.S.; Maes, M. Lowered PON1 activities are strongly associated with depression and bipolar disorder, recurrence of (hypo)mania and depression, increased disability and lowered quality of life. World J. Biol. Psychiatry 2019, 20, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Ullas Kamath, S.; Chaturvedi, A.; Bhaskar Yerrapragada, D.; Kundapura, N.; Amin, N.; Devaramane, V. Increased Levels of Acetylcholinesterase, Paraoxonase 1, and Copper in Patients with Moderate Depression- a Preliminary Study. Rep. Biochem. Mol. Biol. 2019, 7, 174–180. [Google Scholar] [PubMed]
- Siwek, M.; Sowa-Kucma, M.; Styczen, K.; Misztak, P.; Nowak, R.J.; Szewczyk, B.; Dudek, D.; Rybakowski, J.K.; Nowak, G.; Maes, M. Associations of Serum Cytokine Receptor Levels with Melancholia, Staging of Illness, Depressive and Manic Phases, and Severity of Depression in Bipolar Disorder. Mol. Neurobiol. 2017, 54, 5883–5893. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.A.; Freitas, T.H.; Maes, M.; de Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef]
- Zhou, R.; Lu, Y.; Han, Y.; Li, X.; Lou, H.; Zhu, L.; Zhen, X.; Duan, S. Mice heterozygous for cathepsin D deficiency exhibit mania-related behavior and stress-induced depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 63, 110–118. [Google Scholar] [CrossRef]
- Heun, R.; Ptok, U.; Kolsch, H.; Maier, W.; Jessen, F. Contribution of apolipoprotein E and cathepsin D genotypes to the familial aggregation of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2004, 18, 151–158. [Google Scholar] [CrossRef]
- Zhao, F.; Yue, Y.; Jiang, H.; Yuan, Y. Shared genetic risk factors for depression and stroke. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 93, 55–70. [Google Scholar] [CrossRef]
- Tsang, R.S.; Mather, K.A.; Sachdev, P.S.; Reppermund, S. Systematic review and meta-analysis of genetic studies of late-life depression. Neurosci. Biobehav. Rev. 2017, 75, 129–139. [Google Scholar] [CrossRef]
- Okada, M.; Nadanaka, S.; Shoji, N.; Tamura, J.; Kitagawa, H. Biosynthesis of heparan sulfate in EXT1-deficient cells. Biochem. J. 2010, 428, 463–471. [Google Scholar] [CrossRef]
- Irie, F.; Badie-Mahdavi, H.; Yamaguchi, Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc. Natl. Acad. Sci. USA 2012, 109, 5052–5056. [Google Scholar] [CrossRef]
- Tulke, S.; Williams, P.; Hellysaz, A.; Ilegems, E.; Wendel, M.; Broberger, C. Nucleobindin 1 (NUCB1) is a Golgi-resident marker of neurons. Neuroscience 2016, 314, 179–188. [Google Scholar] [CrossRef]
- Gupta, R.; Kapoor, N.; Raleigh, D.P.; Sakmar, T.P. Nucleobindin 1 caps human islet amyloid polypeptide protofibrils to prevent amyloid fibril formation. J. Mol. Biol. 2012, 421, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Han, D.; Park, J.; Choi, H.; Park, J.C.; Cha, M.Y.; Woo, J.; Byun, M.S.; Lee, D.Y.; Kim, Y.; et al. Deep proteome profiling of the hippocampus in the 5XFAD mouse model reveals biological process alterations and a novel biomarker of Alzheimer’s disease. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.I.; Lim, S.W.; Myung, W.; Kim, D.K.; Lee, S.Y. Differentially expressed genes related to major depressive disorder and antidepressant response: Genome-wide gene expression analysis. Exp. Mol. Med. 2018, 50, 92. [Google Scholar] [CrossRef] [PubMed]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395 e386. [Google Scholar] [CrossRef]
- Paz, J.; Yao, H.; Lim, H.S.; Lu, X.Y.; Zhang, W. The neuroprotective role of attractin in neurodegeneration. Neurobiol. Aging 2007, 28, 1446–1456. [Google Scholar] [CrossRef]
- English, J.A.; Lopez, L.M.; O’Gorman, A.; Focking, M.; Hryniewiecka, M.; Scaife, C.; Sabherwal, S.; Wynne, K.; Dicker, P.; Rutten, B.P.F.; et al. Blood-Based Protein Changes in Childhood Are Associated With Increased Risk for Later Psychotic Disorder: Evidence From a Nested Case-Control Study of the ALSPAC Longitudinal Birth Cohort. Schizophr. Bull. 2018, 44, 297–306. [Google Scholar] [CrossRef]
- Surinova, S.; Schiess, R.; Huttenhain, R.; Cerciello, F.; Wollscheid, B.; Aebersold, R. On the development of plasma protein biomarkers. J. Proteome Res. 2011, 10, 5–16. [Google Scholar] [CrossRef]
- Ahn, H.S.; Park, S.J.; Jung, H.G.; Woo, S.J.; Lee, C. Quantification of protein markers monitoring the pre-analytical effect of blood storage time before plasma isolation using (15) N metabolically labeled recombinant proteins. J. Mass Spectrom. 2018, 53, 1189–1197. [Google Scholar] [CrossRef]
- Kaisar, M.; van Dullemen, L.F.A.; Thezenas, M.L.; Zeeshan Akhtar, M.; Huang, H.; Rendel, S.; Charles, P.D.; Fischer, R.; Ploeg, R.J.; Kessler, B.M. Plasma degradome affected by variable storage of human blood. Clin. Proteom. 2016, 13, 26. [Google Scholar] [CrossRef]
- Rai, A.J.; Vitzthum, F. Effects of preanalytical variables on peptide and protein measurements in human serum and plasma: Implications for clinical proteomics. Expert Rev. Proteom. 2006, 3, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Pasella, S.; Baralla, A.; Canu, E.; Pinna, S.; Vaupel, J.; Deiana, M.; Franceschi, C.; Baggio, G.; Zinellu, A.; Sotgia, S.; et al. Pre-analytical stability of the plasma proteomes based on the storage temperature. Proteome Sci. 2013, 11, 10. [Google Scholar] [CrossRef]
- Ferguson, R.E.; Hochstrasser, D.F.; Banks, R.E. Impact of preanalytical variables on the analysis of biological fluids in proteomic studies. Proteom. Clin. Appl. 2007, 1, 739–746. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Responders (N = 5) | Non-Responders (N = 5) | p-Value |
|---|---|---|---|
| Age (SD) | 44.2 (14.2) | 42.8 (16.4) | 0.841 |
| Male (%) | 1 (20) | 1 (20) | 1.000 |
| Age at onset (SD) | 41.8 (11.9) | 33.4 (9.8) | 0.093 |
| Body mass index (kg/m2) (SD) | 23.1 (3.7) | 24.7 (4.6) | 0.309 |
| Clinical characteristics at baseline | |||
| Montgomery and Asberg Depression Rating Scale (SD) | 31.0 (4.6) | 28.8 (2.5) | 0.599 |
| Clinical Global Impression-Severity (SD) | 5.0 (0.7) | 4.2 (1.3) | 0.344 |
| Beck’s Depression Inventory (SD) | 32.6 (7.3) | 26.8 (3.6) | 0.206 |
| Hamilton Rating Scale for Depression (SD) | 21.6 (3.4) | 21.0 (2.9) | 1.000 |
| Clinically Useful Depression Outcome Scale (SD) | 38.4 (12.8) | 40.4 (3.0) | 0.917 |
| World Health Organization Quality of Life abbreviated version | |||
| Physical quality of life (SD) | 8.8 (1.7) | 8.5 (0.9) | 0.831 |
| Psychological quality of life (SD) | 8.0 (0.8) | 8.3 (1.7) | 0.827 |
| Social quality of life (SD) | 10.1 (1.5) | 11.7 (2.4) | 0.193 |
| Environmental quality of life (SD) | 10.1 (1.7) | 10.1 (1.1) | 0.914 |
| UNIPROT Accession | Adjusted p-Value | Gene Name | Protein Name | Cluster No. | COR a |
|---|---|---|---|---|---|
| P04278 | 2.70 × 10-3 | SHBG | Sex hormone-binding globulin | 5 | 0.12 |
| P05090 | 2.95 × 10-3 | APOD | Apolipoprotein D | 6 | −0.34 b |
| Q06033 | 4.01 × 10-3 | ITIH3 | Inter-alpha-trypsin inhibitor heavy chain H3 | 5 | −0.21 |
| P08567 | 4.36 × 10-3 | PLEK | Pleckstrin | 4 | 0.04 |
| P04275 | 4.69 × 10-3 | VWF | von Willebrand factor | 6 | −0.19 |
| P52566 | 5.11 × 10-3 | ARHGDIB | Rho GDP-dissociation inhibitor 2 | 1 | −0.15 |
| P02656 | 6.89 × 10-3 | APOC3 | Apolipoprotein C-III | 6 | −0.06 |
| P06276 | 7.13 × 10-3 | BCHE | Cholinesterase | 6 | −0.11 |
| P27169 | 8.89 × 10-3 | PON1 | Serum paraoxonase/arylesterase 1 | 5 | −0.08 |
| P22105-4 | 9.85 × 10-3 | TNXB | Tenascin-X | 6 | −0.25 |
| P02774-3 | 1.09 × 10-2 | GC | Vitamin D-binding protein | 5 | 0.04 |
| P0C0L5 | 1.10 × 10-2 | C4B | Complement C4-B | 5 | −0.21 |
| P02649 | 1.15 × 10-2 | APOE | Apolipoprotein E | 6 | −0.04 |
| P07339 | 1.45 × 10-2 | CTSD | Cathepsin D | 4 | 0.02 |
| Q92820 | 1.50 × 10-2 | GGH | Gamma-glutamyl hydrolase | 5 | −0.01 |
| P09172 | 1.69 × 10-2 | DBH | Dopamine beta-hydroxylase | 2 | 0.03 |
| P40189 | 1.75 × 10-2 | IL6ST | Interleukin-6 receptor subunit beta | 4 | 0.15 |
| Q8NBP7 | 1.81 × 10-2 | PCSK9 | Proprotein convertase subtilisin/kexin type 9 | 3 | −0.14 |
| Q16610 | 1.83 × 10-2 | ECM1 | Extracellular matrix protein 1 | 2 | 0.02 |
| P62258 | 1.83 × 10-2 | YWHAE | 14-3-3 protein epsilon | 6 | 0.18 |
| P80188 | 1.83 × 10-2 | LCN2 | Neutrophil gelatinase-associated lipocalin | 6 | −0.11 |
| Q9H299 | 1.99 × 10-2 | SH3BGRL3 | SH3 domain-binding glutamic acid-rich-like protein 3 | 1 | 0.13 |
| P27918 | 2.05 × 10-2 | CFP | Properdin | 6 | 0.08 |
| P08571 | 2.12 × 10-2 | CD14 | Monocyte differentiation antigen CD14 | 2 | 0.24 |
| P08697 | 2.33 × 10-2 | SERPINF2 | Alpha-2-antiplasmin | 5 | 0.22 |
| P36980 | 2.34 × 10-2 | CFHR2 | Complement factor H-related protein 2 | 4 | 0.16 |
| P08253 | 2.57 × 10-2 | MMP2 | 72 kDa type IV collagenase | 6 | 0.13 |
| P13671 | 2.65 × 10-2 | C6 | Complement component C6 | 5 | 0.13 |
| O43852-3 | 2.80 × 10-2 | CALU | Calumenin | 3 | 0.10 |
| P14543 | 2.93 × 10-2 | NID1 | Nidogen-1 | 4 | −0.05 |
| P35579 | 2.95 × 10-2 | MYH9 | Myosin-9 | 1 | 0.05 |
| P05160 | 3.70 × 10-2 | F13B | Coagulation factor XIII B chain | 6 | −0.17 |
| P02765 | 3.75 × 10-2 | AHSG | Alpha-2-HS-glycoprotein | 2 | 0.20 |
| Q99453 | 3.85 × 10-2 | PHOX2B | Paired mesoderm homeobox protein 2B | 2 | −0.12 |
| O43157 | 4.01 × 10-2 | PLXNB1 | Plexin-B1 | 5 | −0.01 |
| P06396 | 4.26 × 10-2 | GSN | Gelsolin | 6 | 0.04 |
| O43866 | 4.41 × 10-2 | CD5L | CD5 antigen-like | 3 | −0.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.Y.; Ahn, H.-S.; Lee, M.Y.; Yu, J.; Yeom, J.; Jeong, H.; Min, H.; Lee, H.J.; Kim, K.; Ahn, Y.M. An Exploratory Pilot Study with Plasma Protein Signatures Associated with Response of Patients with Depression to Antidepressant Treatment for 10 Weeks. Biomedicines 2020, 8, 455. https://doi.org/10.3390/biomedicines8110455
Kim EY, Ahn H-S, Lee MY, Yu J, Yeom J, Jeong H, Min H, Lee HJ, Kim K, Ahn YM. An Exploratory Pilot Study with Plasma Protein Signatures Associated with Response of Patients with Depression to Antidepressant Treatment for 10 Weeks. Biomedicines. 2020; 8(11):455. https://doi.org/10.3390/biomedicines8110455
Chicago/Turabian StyleKim, Eun Young, Hee-Sung Ahn, Min Young Lee, Jiyoung Yu, Jeonghun Yeom, Hwangkyo Jeong, Hophil Min, Hyun Jeong Lee, Kyunggon Kim, and Yong Min Ahn. 2020. "An Exploratory Pilot Study with Plasma Protein Signatures Associated with Response of Patients with Depression to Antidepressant Treatment for 10 Weeks" Biomedicines 8, no. 11: 455. https://doi.org/10.3390/biomedicines8110455
APA StyleKim, E. Y., Ahn, H.-S., Lee, M. Y., Yu, J., Yeom, J., Jeong, H., Min, H., Lee, H. J., Kim, K., & Ahn, Y. M. (2020). An Exploratory Pilot Study with Plasma Protein Signatures Associated with Response of Patients with Depression to Antidepressant Treatment for 10 Weeks. Biomedicines, 8(11), 455. https://doi.org/10.3390/biomedicines8110455