Differential Roles of Dendritic Cells in Expanding CD4 T Cells in Sepsis

 ,
,  ,
, 
Abstract
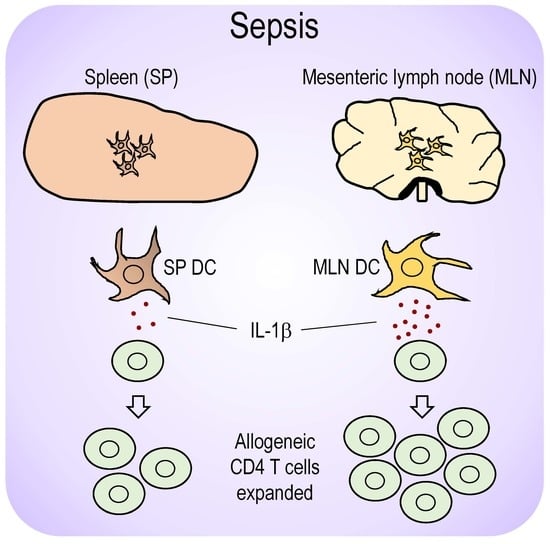
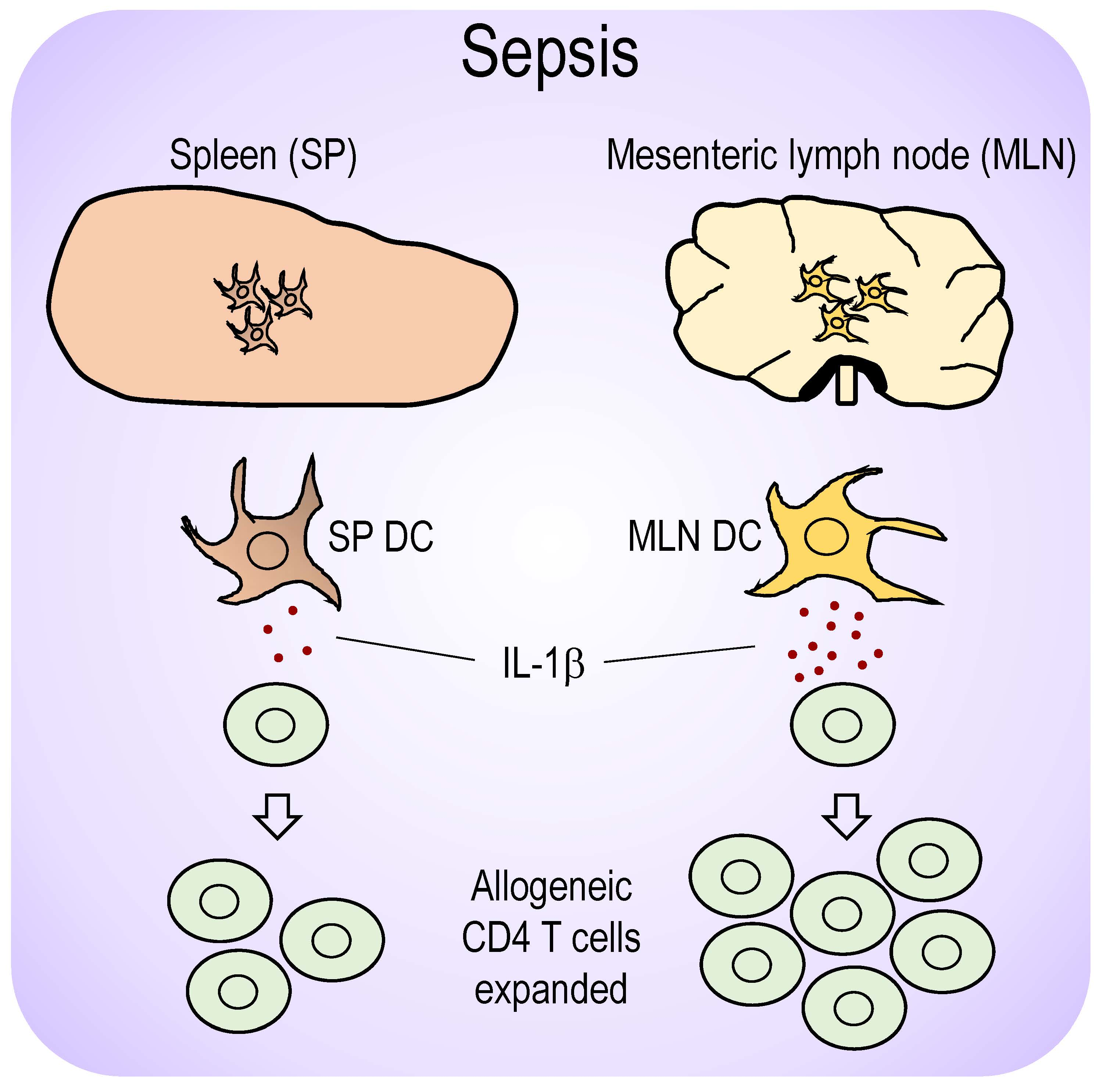{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
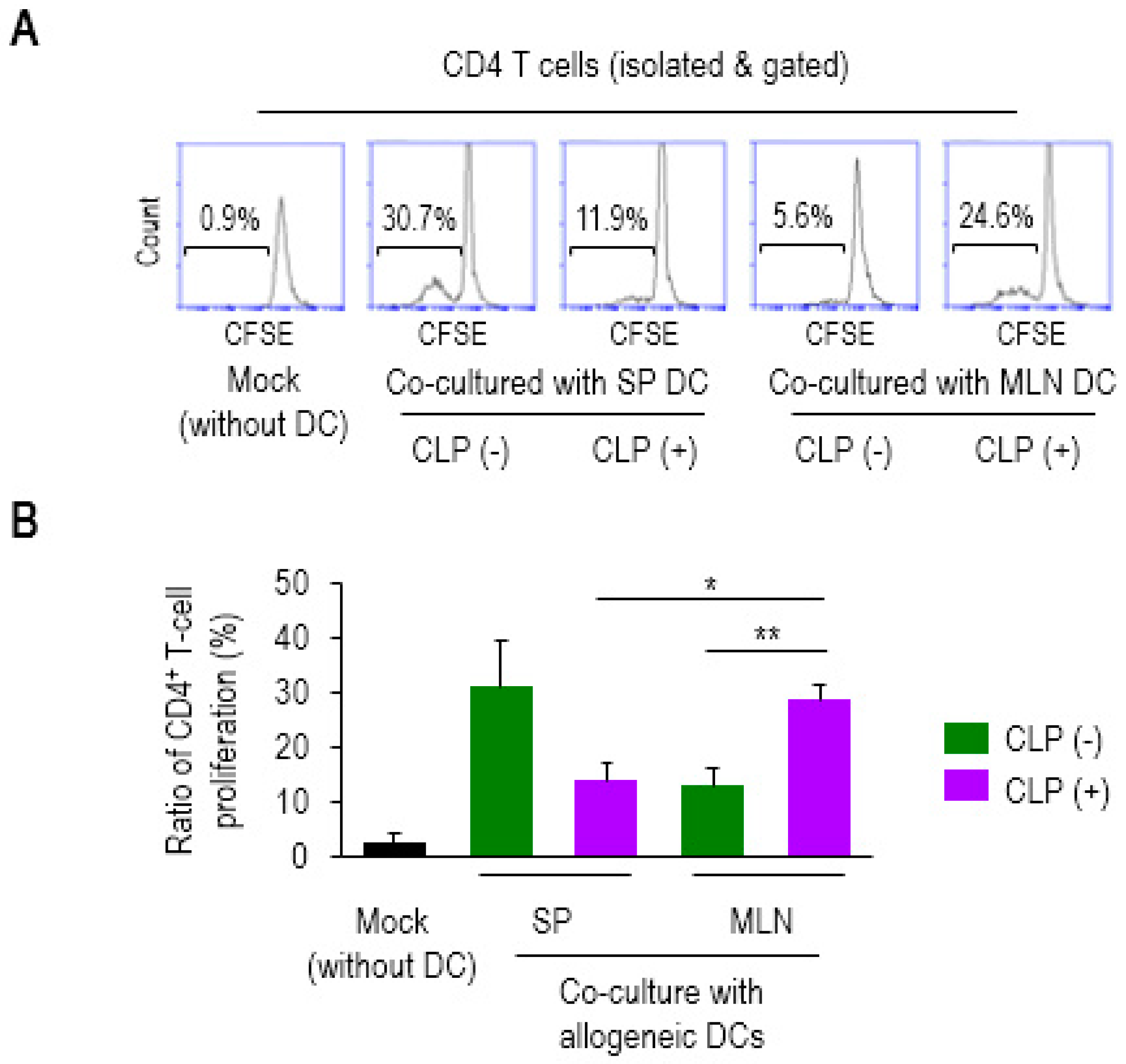
2.1. CD4 T-cell Proliferation is Augmented by Allogeneic MLN DCs in Sepsis
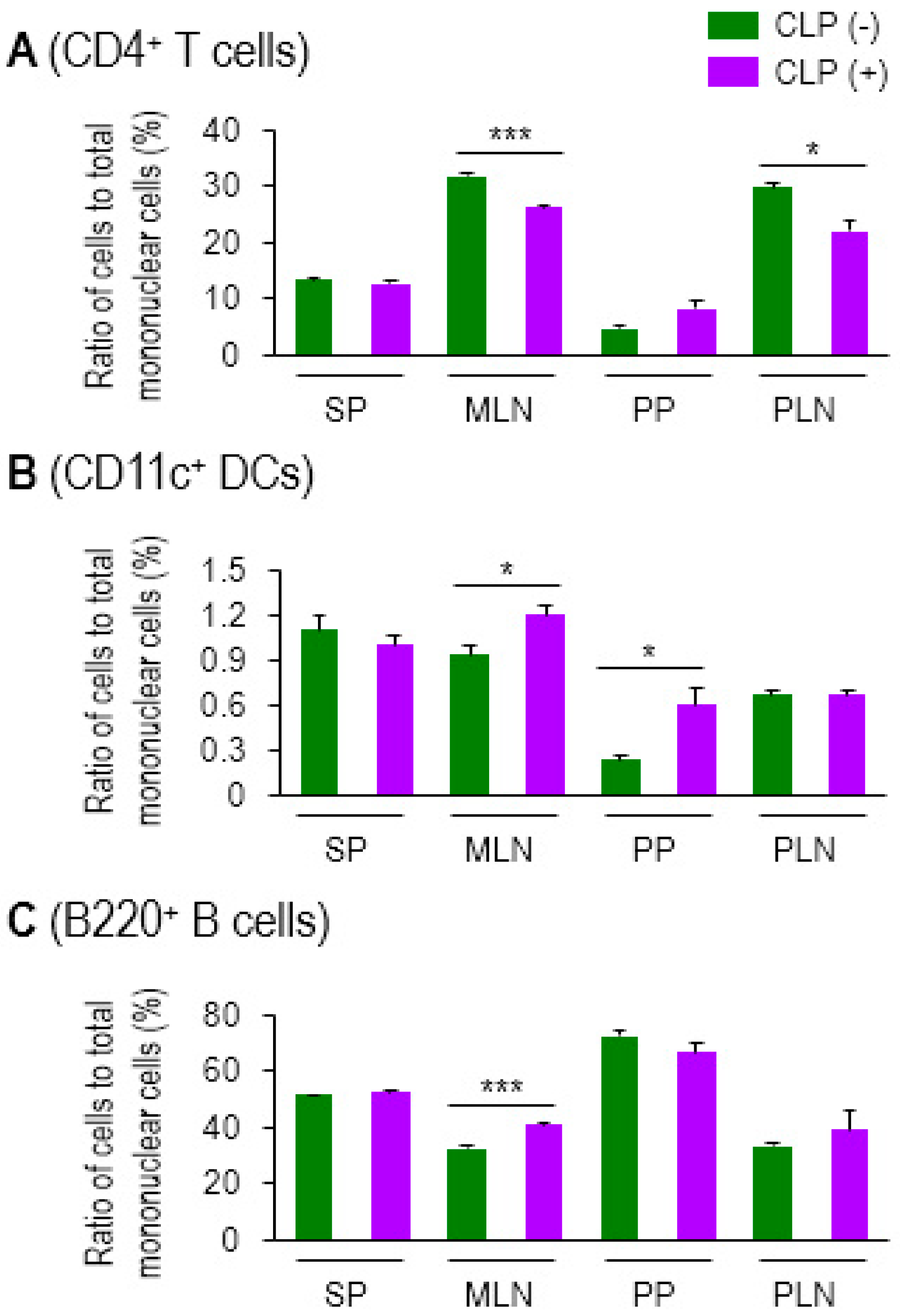
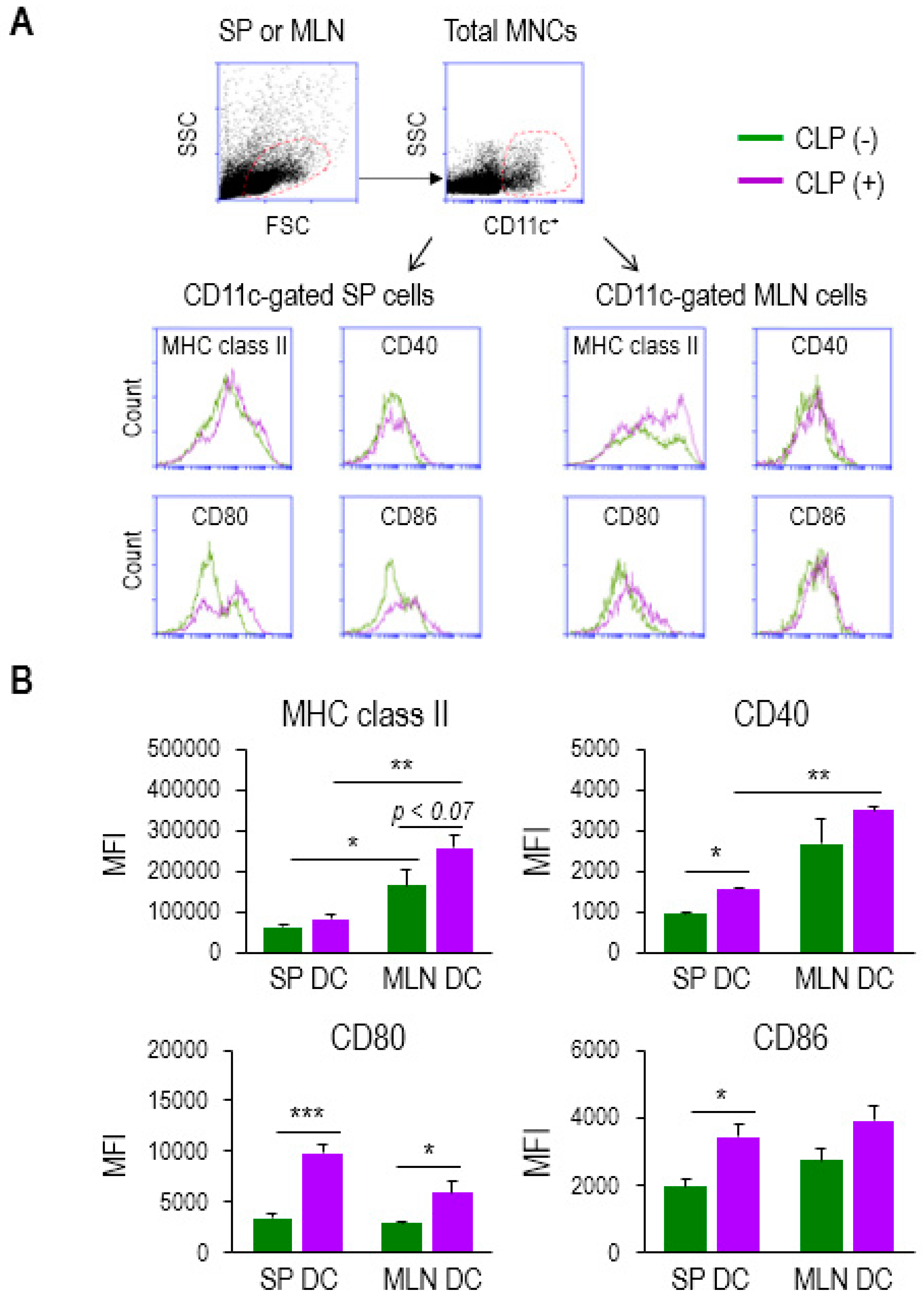
2.2. Some Activation Markers Are Highly Increased in MLN DCs in Sepsis
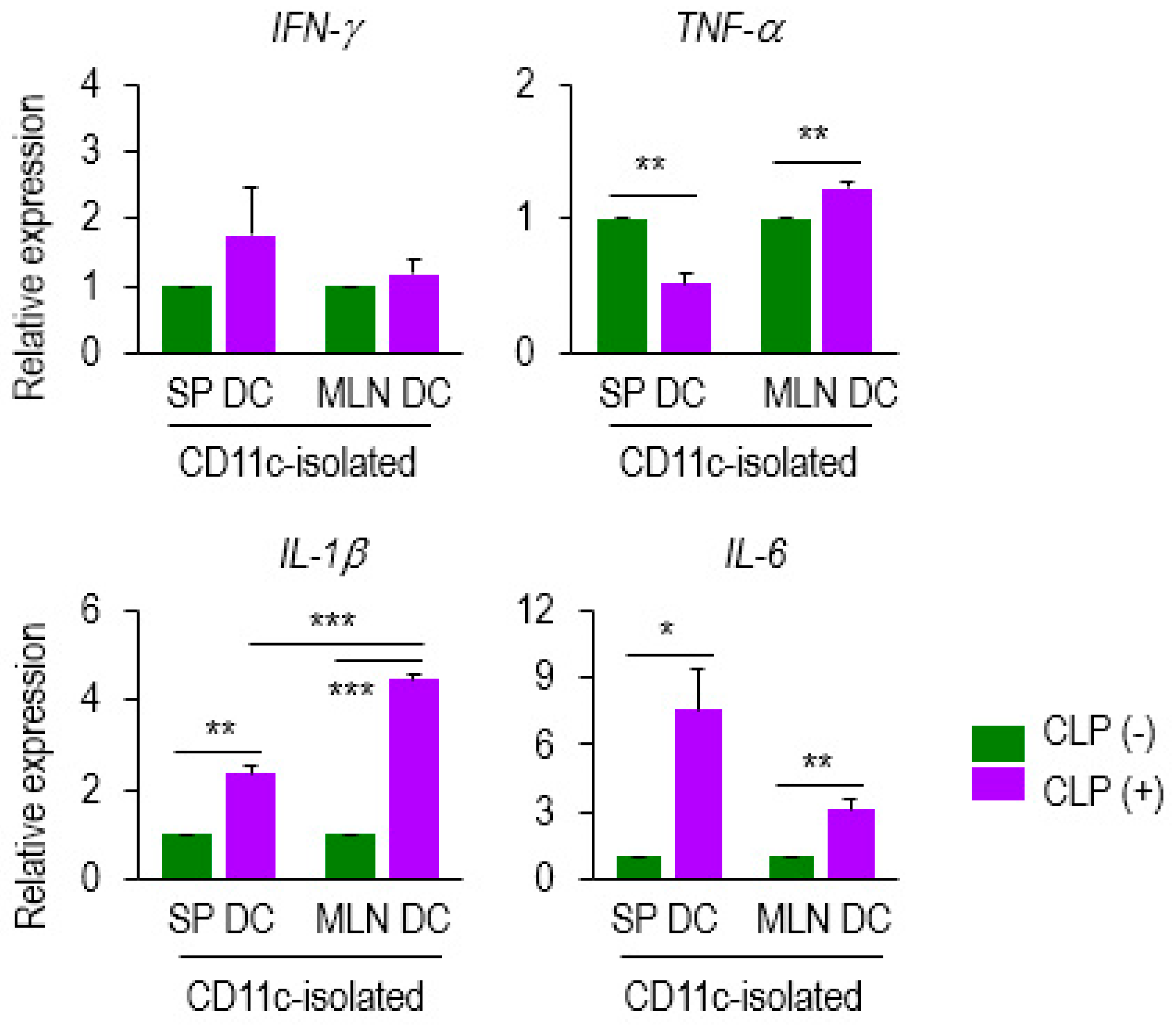
2.3. Level of IL-1β mRNA is Markedly Increased in Septic MLN DCs
2.4. CD4 T-Cell Proliferation is Augmented by IL-1β Treatment in a Dose-Dependent Manner
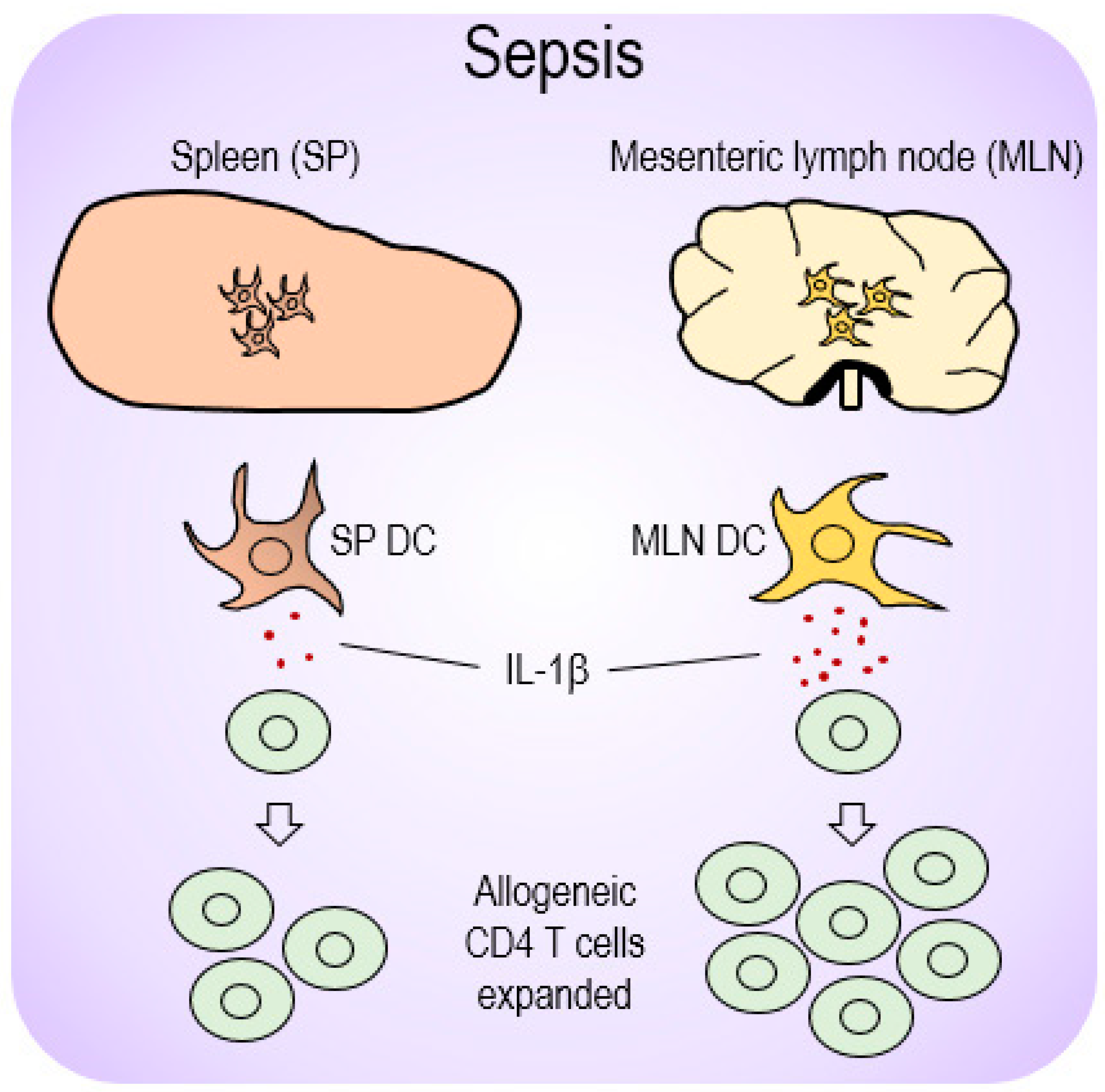
3. Discussions
4. Methods
4.1. Mice
4.2. Polymicrobial Sepsis
4.3. Isolation of CD4+ T Cells and CD11c+ Dendritic Cells (DCs)
4.4. Mixed Lymphocyte Reaction (MLR) Culture and CD4 T-Cell Proliferation Assay
4.5. Flow Cytometry Analysis
4.6. Reverse Transcription (RT) and Quantitative PCR (qPCR)
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | allophycocyanin |
| CFSE | 5-(and -6)-carboxyfluorescein diacetate succinimidyl ester |
| CLP | cecal ligation and puncture |
| cDC | classical dendritic cell |
| DC | dendritic cell |
| ED50 | median effective dose |
| EDTA | ethylenediaminetetraacetic acid |
| FITC | fluorescein isothiocyanate |
| FoxP3 | forkhead box P3 |
| FSC | forward scatter |
| HMGB1 | high mobility group box 1 |
| IFN-γ | interferon-γ |
| IL-1β | interleukin-1β |
| LPS | lipopolysaccharide |
| mAb | monoclonal antibody |
| MFI | mean fluorescence intensity |
| MHC | major histocompatibility complex |
| MLN | mesenteric lymph node |
| MLR | mixed lymphocyte reaction |
| MNC | mononuclear cell |
| pDC | plasmacytoid dendritic cell |
| PE | phycoerythrin |
| PerCP | peridinin-chlorophyll-protein |
| PLN | peripheral lymph node |
| PP | Peyer’s patch |
| qPCR | quantitative polymerase chain reaction |
| RT | reverse transcription |
| SEM | standard error of the mean |
| SSC | side scatter |
| TLR | toll-like receptor |
| TNF-α | tumor necrosis factor-α |
References
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D., 2nd; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Park, E.J. Advances in understanding sepsis. Eur. J. Anaesthesiol. Suppl. 2008, 42, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Delano, M.J.; Ward, P.A. Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J. Clin. Investig. 2016, 126, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Scicluna, B.P.; Arts, R.J.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.; Cremer, O.L.; et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Nicholson, D.W. Apoptosis and caspases regulate death and inflammation in sepsis. Nat. Rev. Immunol. 2006, 6, 813–822. [Google Scholar] [CrossRef]
- Steinman, R.M. Linking innate to adaptive immunity through dendritic cells. Novartis Found. Symp. 2006, 279, 101–109. [Google Scholar]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Dzionek, A.; Inagaki, Y.; Okawa, K.; Nagafune, J.; Rock, J.; Sohma, Y.; Winkels, G.; Zysk, M.; Yamaguchi, Y.; Schmitz, J. Plasmacytoid dendritic cells: From specific surface markers to specific cellular functions. Hum. Immunol. 2002, 63, 1133–1148. [Google Scholar] [CrossRef]
- Shigematsu, H.; Reizis, B.; Iwasaki, H.; Mizuno, S.; Hu, D.; Traver, D.; Leder, P.; Sakaguchi, N.; Akashi, K. Plasmacytoid dendritic cells activate lymphoid-specific genetic programs irrespective of their cellular origin. Immunity 2004, 21, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Calabro, S.; Liu, D.; Gallman, A.; Nascimento, M.S.; Yu, Z.; Zhang, T.T.; Chen, P.; Zhang, B.; Xu, L.; Gowthaman, U.; et al. Differential Intrasplenic Migration of Dendritic Cell Subsets Tailors Adaptive Immunity. Cell Rep. 2016, 16, 2472–2485. [Google Scholar] [CrossRef] [PubMed]
- Allenspach, E.J.; Lemos, M.P.; Porrett, P.M.; Turka, L.A.; Laufer, T.M. Migratory and lymphoid-resident dendritic cells cooperate to efficiently prime naive CD4 T cells. Immunity 2008, 29, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; von Andrian, U.H. Generation, migration and function of circulating dendritic cells. Curr. Opin. Immunol. 2006, 18, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.; Vollmann, E.H.; von Andrian, U.H. Mechanisms and consequences of dendritic cell migration. Immunity 2008, 29, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Poehlmann, H.; Schefold, J.C.; Zuckermann-Becker, H.; Volk, H.D.; Meisel, C. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: A prospective observational analysis. Crit. Care 2009, 13, R119. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, D.; Louis, S.; Pene, F.; Sirgo, G.; Rousseau, C.; Claessens, Y.E.; Vimeux, L.; Cariou, A.; Mira, J.P.; Hosmalin, A.; et al. Profound and persistent decrease of circulating dendritic cells is associated with ICU-acquired infection in patients with septic shock. Intensive Care Med. 2011, 37, 1438–1446. [Google Scholar] [CrossRef]
- Ding, Y.; Chung, C.S.; Newton, S.; Chen, Y.; Carlton, S.; Albina, J.E.; Ayala, A. Polymicrobial sepsis induces divergent effects on splenic and peritoneal dendritic cell function in mice. Shock 2004, 22, 137–144. [Google Scholar] [CrossRef]
- Benjamim, C.F.; Lundy, S.K.; Lukacs, N.W.; Hogaboam, C.M.; Kunkel, S.L. Reversal of long-term sepsis-induced immunosuppression by dendritic cells. Blood 2005, 105, 3588–3595. [Google Scholar] [CrossRef]
- Scumpia, P.O.; McAuliffe, P.F.; O’Malley, K.A.; Ungaro, R.; Uchida, T.; Matsumoto, T.; Remick, D.G.; Clare-Salzler, M.J.; Moldawer, L.L.; Efron, P.A. CD11c+ dendritic cells are required for survival in murine polymicrobial sepsis. J. Immunol. 2005, 175, 3282–3286. [Google Scholar] [CrossRef]
- Pastille, E.; Didovic, S.; Brauckmann, D.; Rani, M.; Agrawal, H.; Schade, F.U.; Zhang, Y.; Flohe, S.B. Modulation of dendritic cell differentiation in the bone marrow mediates sustained immunosuppression after polymicrobial sepsis. J. Immunol. 2011, 186, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Faivre, V.; Lukaszewicz, A.C.; Alves, A.; Charron, D.; Payen, D.; Haziot, A. Accelerated in vitro differentiation of blood monocytes into dendritic cells in human sepsis. Clin. Exp. Immunol. 2007, 147, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Steinbrink, K.; Graulich, E.; Kubsch, S.; Knop, J.; Enk, A.H. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood 2002, 99, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal ligation and puncture: The gold standard model for polymicrobial sepsis? Trends Microbiol. 2011, 19, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Seemann, S.; Zohles, F.; Lupp, A. Comprehensive comparison of three different animal models for systemic inflammation. J. Biomed. Sci. 2017, 24, 60. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Leelahavanichkul, A.; Yuen, P.S.; Star, R.A. Animal models of sepsis and sepsis-induced kidney injury. J. Clin. Investig. 2009, 119, 2868–2878. [Google Scholar] [CrossRef] [PubMed]
- Tourkova, I.L.; Yurkovetsky, Z.R.; Shurin, M.R.; Shurin, G.V. Mechanisms of dendritic cell-induced T cell proliferation in the primary MLR assay. Immunol. Lett. 2001, 78, 75–82. [Google Scholar] [CrossRef]
- Ben-Sasson, S.Z.; Hu-Li, J.; Quiel, J.; Cauchetaux, S.; Ratner, M.; Shapira, I.; Dinarello, C.A.; Paul, W.E. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 7119–7124. [Google Scholar] [CrossRef]
- Walsh, K.P.; Mills, K.H. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 2013, 34, 521–530. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Chang, K.C.; Swanson, P.E.; Tinsley, K.W.; Hui, J.J.; Klender, P.; Xanthoudakis, S.; Roy, S.; Black, C.; Grimm, E.; et al. Caspase inhibitors improve survival in sepsis: A critical role of the lymphocyte. Nat. Immunol. 2000, 1, 496–501. [Google Scholar] [CrossRef]
- Markwart, R.; Condotta, S.A.; Requardt, R.P.; Borken, F.; Schubert, K.; Weigel, C.; Bauer, M.; Griffith, T.S.; Forster, M.; Brunkhorst, F.M.; et al. Immunosuppression after sepsis: Systemic inflammation and sepsis induce a loss of naive T-cells but no enduring cell-autonomous defects in T-cell function. PLoS ONE 2014, 9, e115094. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Xin Xu, Y.; Ayala, C.A.; Sonefeld, D.E.; Karr, S.M.; Evans, T.A.; Chaudry, I.H. Increased mucosal B-lymphocyte apoptosis during polymicrobial sepsis is a Fas ligand but not an endotoxin-mediated process. Blood 1998, 91, 1362–1372. [Google Scholar] [PubMed]
- Hiramatsu, M.; Hotchkiss, R.S.; Karl, I.E.; Buchman, T.G. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and gut by an endotoxin and TNF-independent pathway. Shock 1997, 7, 247–253. [Google Scholar] [CrossRef] [PubMed]
- de Jong, J.M.; Schuurhuis, D.H.; Ioan-Facsinay, A.; Welling, M.M.; Camps, M.G.; van der Voort, E.I.; Huizinga, T.W.; Ossendorp, F.; Verbeek, J.S.; Toes, R.E. Dendritic cells, but not macrophages or B cells, activate major histocompatibility complex class II-restricted CD4+ T cells upon immune-complex uptake in vivo. Immunology 2006, 119, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.Y.; Clark, E.A. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol. 2009, 21, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Tamoutounour, S.; Guilliams, M.; Montanana Sanchis, F.; Liu, H.; Terhorst, D.; Malosse, C.; Pollet, E.; Ardouin, L.; Luche, H.; Sanchez, C.; et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity 2013, 39, 925–938. [Google Scholar] [CrossRef]
- Poulin, L.F.; Lasseaux, C.; Chamaillard, M. Understanding the Cellular Origin of the Mononuclear Phagocyte System Sheds Light on the Myeloid Postulate of Immune Paralysis in Sepsis. Front. Immunol. 2018, 9, 823. [Google Scholar] [CrossRef]
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef]
- Ganem, M.B.; De Marzi, M.C.; Fernandez-Lynch, M.J.; Jancic, C.; Vermeulen, M.; Geffner, J.; Mariuzza, R.A.; Fernandez, M.M.; Malchiodi, E.L. Uptake and intracellular trafficking of superantigens in dendritic cells. PLoS ONE 2013, 8, e66244. [Google Scholar] [CrossRef]
- Fuentes-Duculan, J.; Suarez-Farinas, M.; Zaba, L.C.; Nograles, K.E.; Pierson, K.C.; Mitsui, H.; Pensabene, C.A.; Kzhyshkowska, J.; Krueger, J.G.; Lowes, M.A. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J. Investig. Dermatol. 2010, 130, 2412–2422. [Google Scholar] [CrossRef]
- Yu, Y.R.; O’Koren, E.G.; Hotten, D.F.; Kan, M.J.; Kopin, D.; Nelson, E.R.; Que, L.; Gunn, M.D. A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLoS ONE 2016, 11, e0150606. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Chen, Q.; Soncin, I.; Ng, S.L.; Karjalainen, K.; Ruedl, C. A Discrete Subset of Monocyte-Derived Cells among Typical Conventional Type 2 Dendritic Cells Can Efficiently Cross-Present. Cell Rep. 2017, 21, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Lin, K.L.; Yanagita, M.; Charbonneau, C.; Cook, D.N.; Kakiuchi, T.; Gunn, M.D. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat. Immunol. 2009, 10, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Burgents, J.E.; Nakano, K.; Whitehead, G.S.; Cheong, C.; Bortner, C.D.; Cook, D.N. Migratory properties of pulmonary dendritic cells are determined by their developmental lineage. Mucosal Immunol. 2013, 6, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Symons, J.A.; Bundick, R.V.; Suckling, A.J.; Rumsby, M.G. Cerebrospinal fluid interleukin 1 like activity during chronic relapsing experimental allergic encephalomyelitis. Clin. Exp. Immunol. 1987, 68, 648–654. [Google Scholar] [PubMed]
- Jensen, I.J.; Sjaastad, F.V.; Griffith, T.S.; Badovinac, V.P. Sepsis-Induced T Cell Immunoparalysis: The Ins and Outs of Impaired T Cell Immunity. J. Immunol. 2018, 200, 1543–1553. [Google Scholar] [PubMed]
- Eltom, S.; Belvisi, M.G.; Yew-Booth, L.; Dekkak, B.; Maher, S.A.; Dubuis, E.D.; Jones, V.; Fitzgerald, K.A.; Birrell, M.A. TLR4 activation induces IL-1beta release via an IPAF dependent but caspase 1/11/8 independent pathway in the lung. Respir. Res. 2014, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef]
- Eskan, M.A.; Benakanakere, M.R.; Rose, B.G.; Zhang, P.; Zhao, J.; Stathopoulou, P.; Fujioka, D.; Kinane, D.F. Interleukin-1beta modulates proinflammatory cytokine production in human epithelial cells. Infect. Immun. 2008, 76, 2080–2089. [Google Scholar] [CrossRef]
- Deng, M.; Ma, T.; Yan, Z.; Zettel, K.R.; Scott, M.J.; Liao, H.; Frank, A.; Morelli, A.E.; Sodhi, C.P.; Hackam, D.J.; et al. Toll-like Receptor 4 Signaling on Dendritic Cells Suppresses Polymorphonuclear Leukocyte CXCR2 Expression and Trafficking via Interleukin 10 During Intra-abdominal Sepsis. J. Infect. Dis. 2016, 213, 1280–1288. [Google Scholar] [CrossRef]
- Pene, F.; Courtine, E.; Ouaaz, F.; Zuber, B.; Sauneuf, B.; Sirgo, G.; Rousseau, C.; Toubiana, J.; Balloy, V.; Chignard, M.; et al. Toll-like receptors 2 and 4 contribute to sepsis-induced depletion of spleen dendritic cells. Infect. Immun. 2009, 77, 5651–5658. [Google Scholar] [CrossRef] [PubMed]
- Fanning, L.R.; Hegerfeldt, Y.; Tary-Lehmann, M.; Lesniewski, M.; Maciejewski, J.; Weitzel, R.P.; Kozik, M.; Finney, M.; Lazarus, H.M.; Paul, P.; et al. Allogeneic transplantation of multiple umbilical cord blood units in adults: Role of pretransplant-mixed lymphocyte reaction to predict host-vs-graft rejection. Leukemia 2008, 22, 1786–1790. [Google Scholar] [CrossRef][Green Version]
- Hayry, P.; Defendi, V. Mixed lymphocyte cultures produce effector cells: Model in vitro for allograft rejection. Science 1970, 168, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Bradley, B.A.; Edwards, J.M.; Dunn, D.C.; Calne, R.Y. Quantitation of mixed lymphocyte reaction by gene dosage phenomenon. Nat. New Biol. 1972, 240, 54–56. [Google Scholar] [CrossRef]
- Hotta, K.; Oura, T.; Dehnadi, A.; Boskovic, S.; Matsunami, M.; Rosales, I.; Smith, R.N.; Colvin, R.B.; Cosimi, A.B.; Kawai, T. Long-term Nonhuman Primate Renal Allograft Survival Without Ongoing Immunosuppression in Recipients of Delayed Donor Bone Marrow Transplantation. Transplantation 2018, 102, e128–e136. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Shen, X.; Tang, Y.; Fu, Z.; Zheng, Q.; Wang, Q. Astilbin suppresses acute heart allograft rejection by inhibiting maturation and function of dendritic cells in mice. Transpl. Proc. 2010, 42, 3798–3802. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; He, W.; Luo, G.; Wu, J. Mixed lymphocyte reaction induced by multiple alloantigens and the role for IL-10 in proliferation inhibition. Burns Trauma 2014, 2, 24–28. [Google Scholar]
- Lewis, A.J.; Seymour, C.W.; Rosengart, M.R. Current Murine Models of Sepsis. Surg. Infect. (Larchmt) 2016, 17, 385–393. [Google Scholar] [CrossRef]
- Gautier, E.L.; Huby, T.; Saint-Charles, F.; Ouzilleau, B.; Chapman, M.J.; Lesnik, P. Enhanced dendritic cell survival attenuates lipopolysaccharide-induced immunosuppression and increases resistance to lethal endotoxic shock. J. Immunol. 2008, 180, 6941–6946. [Google Scholar] [CrossRef]
- Li, C.C.; Munitic, I.; Mittelstadt, P.R.; Castro, E.; Ashwell, J.D. Suppression of Dendritic Cell-Derived IL-12 by Endogenous Glucocorticoids Is Protective in LPS-Induced Sepsis. PLoS Biol. 2015, 13, e1002269. [Google Scholar] [CrossRef]
- Hashimoto, D.; Miller, J.; Merad, M. Dendritic cell and macrophage heterogeneity in vivo. Immunity 2011, 35, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Grailer, J.J.; Fattahi, F.; Dick, R.S.; Zetoune, F.S.; Ward, P.A. Cutting edge: Critical role for C5aRs in the development of septic lymphopenia in mice. J. Immunol. 2015, 194, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Deitch, E.A. Gut-origin sepsis: Evolution of a concept. Surgeon 2012, 10, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Krezalek, M.A.; DeFazio, J.; Zaborina, O.; Zaborin, A.; Alverdy, J.C. The Shift of an Intestinal “Microbiome” to a “Pathobiome” Governs the Course and Outcome of Sepsis Following Surgical Injury. Shock 2016, 45, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, K.W.; Grayson, M.H.; Swanson, P.E.; Drewry, A.M.; Chang, K.C.; Karl, I.E.; Hotchkiss, R.S. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J. Immunol. 2003, 171, 909–914. [Google Scholar] [CrossRef]
- Flohe, S.B.; Agrawal, H.; Schmitz, D.; Gertz, M.; Flohe, S.; Schade, F.U. Dendritic cells during polymicrobial sepsis rapidly mature but fail to initiate a protective Th1-type immune response. J. Leukoc. Biol. 2006, 79, 473–481. [Google Scholar] [CrossRef]
- Schindler, D.; Gutierrez, M.G.; Beineke, A.; Rauter, Y.; Rohde, M.; Foster, S.; Goldmann, O.; Medina, E. Dendritic cells are central coordinators of the host immune response to Staphylococcus aureus bloodstream infection. Am. J. Pathol. 2012, 181, 1327–1337. [Google Scholar] [CrossRef]
- Cavassani, K.A.; Carson, W.F.; Moreira, A.P.; Wen, H.; Schaller, M.A.; Ishii, M.; Lindell, D.M.; Dou, Y.; Lukacs, N.W.; Keshamouni, V.G.; et al. The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth. Blood 2010, 115, 4403–4411. [Google Scholar] [CrossRef]
- Kuhlhorn, F.; Rath, M.; Schmoeckel, K.; Cziupka, K.; Nguyen, H.H.; Hildebrandt, P.; Hunig, T.; Sparwasser, T.; Huehn, J.; Potschke, C.; et al. Foxp3+ regulatory T cells are required for recovery from severe sepsis. PLoS ONE 2013, 8, e65109. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 2009, 10, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Yanagita, M.; Gunn, M.D. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 2001, 194, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Cao, W.; Liu, Y.J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B.; Bunin, A.; Ghosh, H.S.; Lewis, K.L.; Sisirak, V. Plasmacytoid dendritic cells: Recent progress and open questions. Annu. Rev. Immunol. 2011, 29, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Tinsley, K.W.; Swanson, P.E.; Grayson, M.H.; Osborne, D.F.; Wagner, T.H.; Cobb, J.P.; Coopersmith, C.; Karl, I.E. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J. Immunol. 2002, 168, 2493–2500. [Google Scholar] [CrossRef] [PubMed]
- Elsayh, K.I.; Zahran, A.M.; Lotfy Mohamad, I.; Aly, S.S. Dendritic cells in childhood sepsis. J. Crit. Care 2013, 28, 881.e7–881.e13. [Google Scholar] [CrossRef]
- Efron, P.A.; Martins, A.; Minnich, D.; Tinsley, K.; Ungaro, R.; Bahjat, F.R.; Hotchkiss, R.; Clare-Salzler, M.; Moldawer, L.L. Characterization of the systemic loss of dendritic cells in murine lymph nodes during polymicrobial sepsis. J. Immunol. 2004, 173, 3035–3043. [Google Scholar] [CrossRef]
- Hirako, I.C.; Ataide, M.A.; Faustino, L.; Assis, P.A.; Sorensen, E.W.; Ueta, H.; Araujo, N.M.; Menezes, G.B.; Luster, A.D.; Gazzinelli, R.T. Splenic differentiation and emergence of CCR5(+)CXCL9(+)CXCL10(+) monocyte-derived dendritic cells in the brain during cerebral malaria. Nat. Commun. 2016, 7, 13277. [Google Scholar] [CrossRef]
- Cuenca, A.G.; Delano, M.J.; Kelly-Scumpia, K.M.; Moldawer, L.L.; Efron, P.A. Cecal ligation and puncture. Curr. Protoc. Immunol. 2010, 91. [Google Scholar] [CrossRef]
- Ruiz, S.; Vardon-Bounes, F.; Merlet-Dupuy, V.; Conil, J.M.; Buleon, M.; Fourcade, O.; Tack, I.; Minville, V. Sepsis modeling in mice: Ligation length is a major severity factor in cecal ligation and puncture. Intensive Care Med. Exp. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Li, G.; Jing, X.Z.; Li, Y.F.; Ye, Q.Y.; Jia, H.H.; Liu, S.H.; Li, X.J.; Li, H.; Huang, R.; et al. Assessment of clinical sepsis-associated biomarkers in a septic mouse model. J. Int. Med. Re.s 2018, 46, 2410–2422. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Siddiqui, J.; Remick, D.G. Mechanisms of mortality in early and late sepsis. Infect. Immun. 2006, 74, 5227–5235. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Takahashi, I.; Ikeda, J.; Kawahara, K.; Okamoto, T.; Kweon, M.N.; Fukuyama, S.; Groh, V.; Spies, T.; Obata, Y.; et al. Clonal expansion of double-positive intraepithelial lymphocytes by MHC class I-related chain A expressed in mouse small intestinal epithelium. J. Immunol. 2003, 171, 4131–4139. [Google Scholar] [CrossRef]
- Newland, A.; Russ, G.; Krishnan, R. Natural killer cells prime the responsiveness of autologous CD4+ T cells to CTLA4-Ig and interleukin-10 mediated inhibition in an allogeneic dendritic cell-mixed lymphocyte reaction. Immunology 2006, 118, 216–223. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darkwah, S.; Nago, N.; Appiah, M.G.; Myint, P.K.; Kawamoto, E.; Shimaoka, M.; Park, E.J. Differential Roles of Dendritic Cells in Expanding CD4 T Cells in Sepsis. Biomedicines 2019, 7, 52. https://doi.org/10.3390/biomedicines7030052
Darkwah S, Nago N, Appiah MG, Myint PK, Kawamoto E, Shimaoka M, Park EJ. Differential Roles of Dendritic Cells in Expanding CD4 T Cells in Sepsis. Biomedicines. 2019; 7(3):52. https://doi.org/10.3390/biomedicines7030052
Chicago/Turabian StyleDarkwah, Samuel, Nodoka Nago, Michael G. Appiah, Phyoe Kyawe Myint, Eiji Kawamoto, Motomu Shimaoka, and Eun Jeong Park. 2019. "Differential Roles of Dendritic Cells in Expanding CD4 T Cells in Sepsis" Biomedicines 7, no. 3: 52. https://doi.org/10.3390/biomedicines7030052
APA StyleDarkwah, S., Nago, N., Appiah, M. G., Myint, P. K., Kawamoto, E., Shimaoka, M., & Park, E. J. (2019). Differential Roles of Dendritic Cells in Expanding CD4 T Cells in Sepsis. Biomedicines, 7(3), 52. https://doi.org/10.3390/biomedicines7030052