Monoclonal Antibodies in Multiple Sclerosis: Present and Future


Abstract
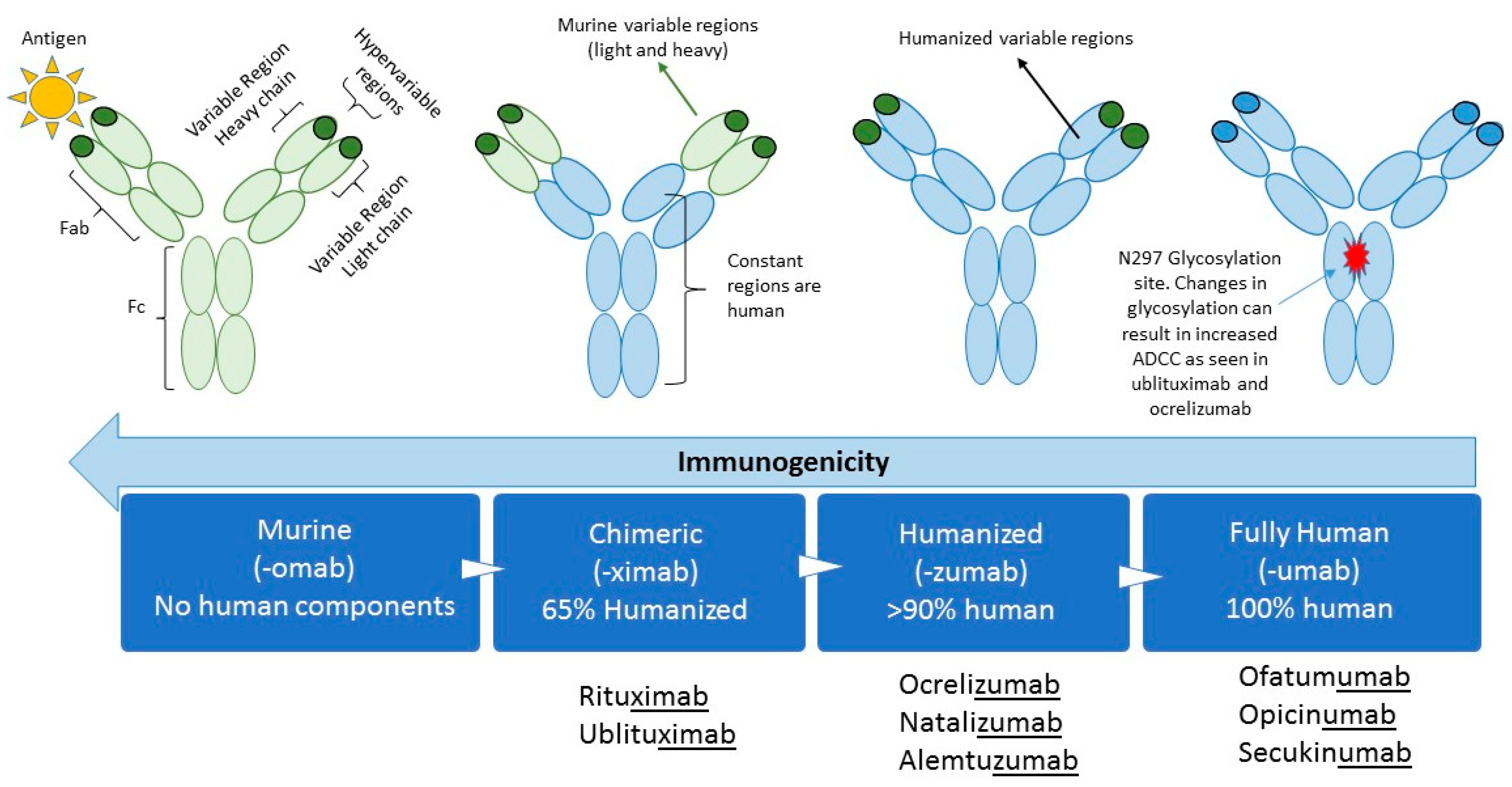
1. Introduction
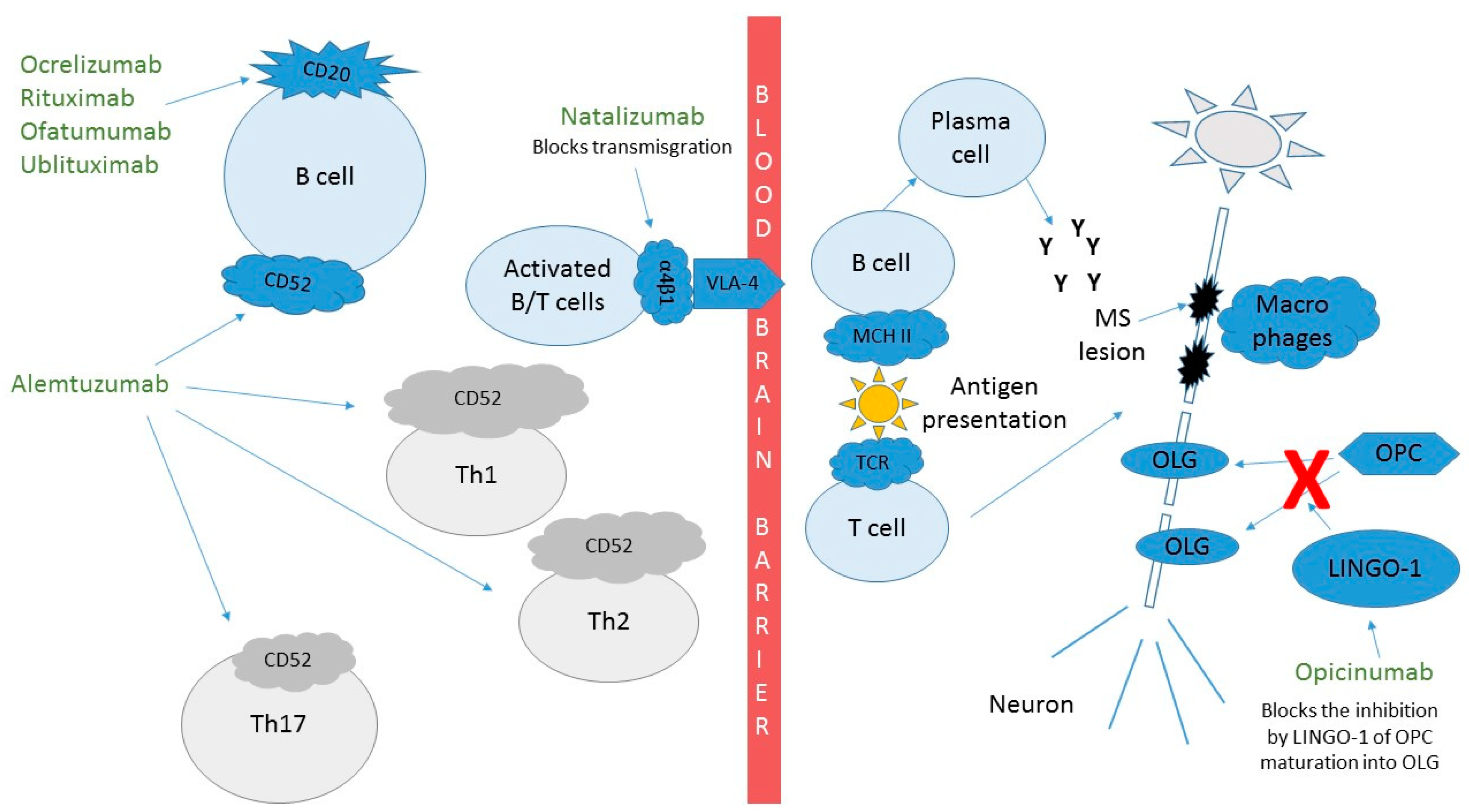
1.1. Natalizumab (Tysabri™)
1.2. Ocrelizumab (Ocrevus™)
1.3. Rituximab (Rituxan™)
1.4. Ofatumumab
1.5. Ublituximab
1.6. Alemtuzumab (Lemtrada™)
1.7. Opicinumab
2. New Horizons and Future Trends for Therapeutic Monoclonal Antibodies
Author Contributions
Funding
Conflicts of Interest
References
- Wootla, B.; Watzlawik, J.O.; Stavropoulos, N.; Wittenberg, N.J.; Dasari, H.; Abdelrahim, M.A.; Henley, J.R.; Oh, S.H.; Warrington, A.E.; Rodriguez, M. Recent Advances in Monoclonal Antibody Therapies for Multiple Sclerosis. Expert Opin. Biol. Ther. 2016, 16, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L.; Carlson, C.M. Monoclonal Antibody Therapy in Multiple Sclerosis. Pract. Neurol. 2018, 28–31. [Google Scholar]
- Smith, S.L. Ten years of Orthoclone OKT3 (muromonab-CD3): A review. J. Transpl. Coord. 1996, 6, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Ober, R.J.; Radu, C.G.; Ghetie, V.; Ward, E.S. Differences in promiscuity for antibody-FcRn interactions across species: Implications for therapeutic antibodies. Int. Immunol. 2001, 13, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharmacol. 2012, 12, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef]
- Lonberg, N. Fully human antibodies from transgenic mouse and phage display platforms. Curr. Opin. Immunol. 2008, 20, 450–459. [Google Scholar] [CrossRef]
- Yaldizli, O.; Putzki, N. Natalizumab in the treatment of multiple sclerosis. Ther. Adv. Neurol. Disord. 2009, 2, 115–128. [Google Scholar] [CrossRef]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.; Frederiksen, J.; Skeen, M.; Jaffe, G.J.; Butzkueven, H.; et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 189–199. [Google Scholar] [CrossRef]
- Giovannoni, G.; Gold, R.; Selmaj, K.; Havrdova, E.; Montalban, X.; Radue, E.W.; Stefoski, D.; McNeill, M.; Amaravadi, L.; Sweetser, M.; et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (selection): A multicentre, randomised, double-blind extension trial. Lancet Neurol. 2014, 13, 472–481. [Google Scholar] [CrossRef]
- Cree, B.; Rosebraugh, M.; Barger, B.; Ziemann, A. A Phase 1, Multiple-Dose Study of Elezanumab (ABT-555) in Patients with Relapsing Forms of Multiple Sclerosis. In Proceedings of the European Committee for Treatment and Research in Multiple Sclerosis Conference, Berlin, Germay, 11 October 2018. [Google Scholar]
- Curtin, F.; Perron, H.; Kromminga, A.; Porchet, H.; Lang, A.B. Preclinical and early clinical development of gnbac1, a humanized igg4 monoclonal antibody targeting endogenous retroviral msrv-env protein. MAbs 2015, 7, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Dörner, T.; Posch, M.; Wagner, F.; Hüser, A.; Fischer, T.; Mooney, L.; Petricoul, O.; Maguire, P.; Pal, P.; Doucet, J.; et al. THU0313 Double-Blind, Randomized Study of VAY736 Single Dose Treatment in Patients with Primary Sjögren’s Syndrome (PSS). Ann. Rheum. Dis. 2016, 75, 300–301. [Google Scholar] [CrossRef]
- Bible, E. Multiple sclerosis: Atacicept increases relapse rates in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 182. [Google Scholar] [CrossRef]
- EMA Recommends Immediate Suspension and Recall of Multiple Sclerosis Medicine Zinbryta. Available online: https://www.ema.europa.eu/en/news/ema-recommends-immediate-suspension-recall-multiple-sclerosis-medicine-zinbryta (accessed on 12 March 2019).
- Weinshenker, B.G.; Bass, B.; Karlik, S.; Ebers, G.C.; Rice, G.P. An open trial of OKT3 in patients with multiple sclerosis. Neurology 1991, 41, 1047–1052. [Google Scholar] [CrossRef]
- Havrdová, E.; Belova, A.; Goloborodko, A.; Tisserant, A.; Wright, A.; Wallstroem, E.; Garren, H.; Maguire, R.P.; Johns, D.R. Activity of secukinumab, an anti-IL-17A antibody, on brain lesions in RRMS: Results from a randomized, proof-of-concept study. J. Neurol. 2016, 263, 1287–1295. [Google Scholar] [CrossRef]
- Silk, M.; Nantz, E. Efficacy and Safety of Tabalumab in Patients with Relapsing-Remitting Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Study (P3.397). Neurology 2018, 90. [Google Scholar]
- Segal, B.M.; Constantinescu, C.S.; Raychaudhuri, A.; Kim, L.; Fidelus-Gort, R.; Kasper, L.H.; Ustekinumab, M.S.I. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: A phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008, 7, 796–804. [Google Scholar] [CrossRef]
- Vatelizumab. Available online: https://multiplesclerosisnewstoday.com/vatelizumab-gbr-500 (accessed on 1 February 2019).
- Wing, M.G.; Moreau, T.; Greenwood, J.; Smith, R.M.; Hale, G.; Isaacs, J.; Waldmann, H.; Lachmann, P.J.; Compston, A. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: Involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J. Clin. Investig. 1996, 98, 2819–2826. [Google Scholar] [CrossRef]
- Klinger, M.; Brandl, C.; Zugmaier, G.; Hijazi, Y.; Bargou, R.C.; Topp, M.S.; Gokbuget, N.; Neumann, S.; Goebeler, M.; Viardot, A.; et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012, 119, 6226–6233. [Google Scholar] [CrossRef]
- Del Boccio, P.; Rossi, C.; di Ioia, M.; Cicalini, I.; Sacchetta, P.; Pieragostino, D. Integration of metabolomics and proteomics in multiple sclerosis: From biomarkers discovery to personalized medicine. Proteom. Clin. Appl. 2016, 10, 470–484. [Google Scholar] [CrossRef]
- McGuigan, C.; Craner, M.; Guadagno, J.; Kapoor, R.; Mazibrada, G.; Molyneux, P.; Nicholas, R.; Palace, J.; Pearson, O.R.; Rog, D.; et al. Stratification and monitoring of natalizumab-associated progressive multifocal leukoencephalopathy risk: Recommendations from an expert group. J. Neurol. Neurosurg. Psychiatry 2016, 87, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Sehr, T.; Proschmann, U.; Thomas, K.; Marggraf, M.; Straube, E.; Reichmann, H.; Chan, A.; Ziemssen, T. New insights into the pharmacokinetics and pharmacodynamics of natalizumab treatment for patients with multiple sclerosis, obtained from clinical and in vitro studies. J. Neuroinflamm. 2016, 13, 164. [Google Scholar] [CrossRef]
- Rudick, R.A.; Stuart, W.H.; Calabresi, P.A.; Confavreux, C.; Galetta, S.L.; Radue, E.W.; Lublin, F.D.; Weinstock-Guttman, B.; Wynn, D.R.; Lynn, F.; et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 911–923. [Google Scholar] [CrossRef]
- Walker, A.; Watson, C.; Alexopoulos, S.T.; Deniz, B.; Arnold, R.; Bates, D. A benefit–risk analysis of natalizumab in the treatment of patients with multiple sclerosis when considering the risk of progressive multifocal leukoencephalopathy. Curr. Med. Res. Opin. 2014, 30, 629–635. [Google Scholar] [CrossRef]
- Thompson, J.P.; Noyes, K.; Dorsey, E.R.; Schwid, S.R.; Holloway, R.G. Quantitative risk-benefit analysis of natalizumab. Neurology 2008, 71, 357–364. [Google Scholar] [CrossRef]
- Bloomgren, G.; Richman, S.; Hotermans, C.; Subramanyam, M.; Goelz, S.; Natarajan, A.; Lee, S.; Plavina, T.; Scanlon, J.V.; Sandrock, A.; et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2012, 366, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.T.; Vartanian, T.K.; Atkins, M.B. Melanoma Complicating Treatment with Natalizumab for Multiple Sclerosis. N. Engl. J. Med. 2008, 358, 647–648. [Google Scholar] [CrossRef] [PubMed]
- Bezabeh, S.; Flowers, C.M.; Kortepeter, C.; Avigan, M. Clinically significant liver injury in patients treated with natalizumab. Aliment. Pharmacol. Ther. 2010, 31, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Lalive, P.H.; Chofflon, M.; Simon, H.-U.; Chizzolini, C.; Ribi, C. Hypereosinophilia in patients with multiple sclerosis treated with natalizumab. Neurology 2011, 77, 1561–1564. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.S.; Blinkenberg, M. The potential role for ocrelizumab in the treatment of multiple sclerosis: Current evidence and future prospects. Ther. Adv. Neurol. Disord. 2016, 9, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Pasic, I.; Lipton, J.H. Current approach to the treatment of chronic myeloid leukaemia. Leuk. Res. 2017, 55, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A. B-Cell Depletion—A Frontier in Monoclonal Antibodies for Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Diem, L.; Nedeltchev, K.; Kahles, T.; Achtnichts, L.; Findling, O. Long-term evaluation of NEDA-3 status in relapsing-remitting multiple sclerosis patients after switching from natalizumab to fingolimod. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Guo, R.; Zhang, F.; Zhang, C.; Dong, S.; Zhou, H. Rituximab for relapsing-remitting multiple sclerosis. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Di Gaetano, N.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement Activation Determines the Therapeutic Activity of Rituximab In Vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef]
- Bar-Or, A.; Calabresi, P.A.; Arnold, D.; Markowitz, C.; Shafer, S.; Kasper, L.H.; Waubant, E.; Gazda, S.; Fox, R.J.; Panzara, M.; et al. Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Ann. Neurol. 2008, 63, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.H.; Stark, J.L.; Lauber, J.; Ramsbottom, M.J.; Lyons, J.A. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 2006, 180, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef]
- Memon, A.B.; Javed, A.; Caon, C.; Srivastawa, S.; Bao, F.; Bernitsas, E.; Chorostecki, J.; Tselis, A.; Seraji-Bozorgzad, N.; Khan, O. Long-term safety of rituximab induced peripheral B-cell depletion in autoimmune neurological diseases. PLoS ONE 2018, 13, e0190425. [Google Scholar] [CrossRef] [PubMed]
- Dunn, N.; Juto, A.; Ryner, M.; Manouchehrinia, A.; Piccoli, L.; Fink, K.; Piehl, F.; Fogdell-Hahn, A. Rituximab in multiple sclerosis: Frequency and clinical relevance of anti-drug antibodies. Mult. Scler. J. 2018, 24, 1224–1233. [Google Scholar] [CrossRef]
- Bar-Or, A.; Grove, R.A.; Austin, D.J.; Tolson, J.M.; VanMeter, S.A.; Lewis, E.W.; Derosier, F.J.; Lopez, M.C.; Kavanagh, S.T.; Miller, A.E.; et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: The MIRROR study. Neurology 2018, 90, e1805–e1814. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.S.; Lisby, S.; Grove, R.; Derosier, F.; Shackelford, S.; Havrdova, E.; Drulovic, J.; Filippi, M. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: A phase 2 study. Neurology 2014, 82, 573–581. [Google Scholar] [CrossRef]
- Kurrasch, R.; Brown, J.C.; Chu, M.; Craigen, J.; Overend, P.; Patel, B.; Wolfe, S.; Chang, D.J. Subcutaneously administered ofatumumab in rheumatoid arthritis: A phase I/II study of safety, tolerability, pharmacokinetics, and pharmacodynamics. J. Rheumatol. 2013, 40, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Inglese, M.; Petracca, M.; Cocozza, S.; Wray, S.; Racke, M.; Shubin, R.; Twyman, C.; Eubanks, J.L.; Mok, K.; Weiss, M.; Fox, E. Final MRI results at 6 months from a Phase 2 Multicenter Study of Ublituximab, A Novel Glycoengineered Anti-CD20 Monoclonal Antibody, In Patients With Relapsing Forms of Multiple Sclerosis (RMS), Demonstrates Complete Elimination of Gd-Enhancing Lesions. In Proceedings of the AAN Enterprises, Los Angeles, CA, USA, 21–27 April 2018. [Google Scholar]
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Coles, A.J.; Compston, A. Product licences for alemtuzumab and multiple sclerosis. Lancet 2014, 383, 867–868. [Google Scholar] [CrossRef]
- FDA. FDA Warns about Rare but Serious Risks of Stroke and Blood Vessel Wall Tears with Multiple Sclerosis Drug Lemtrada (Alemtuzumab). Available online: https://www.fda.gov/Drugs/DrugSafety/ucm624247.htm (accessed on 3 July 2019).
- Decallonne, B.; Bartholome, E.; Delvaux, V.; D’Haeseleer, M.; El Sankari, S.; Seeldrayers, P.; Van Wijmeersch, B.; Daumerie, C. Thyroid disorders in alemtuzumab-treated multiple sclerosis patients: A Belgian consensus on diagnosis and management. Acta Neurol. Belg. 2018, 118, 153–159. [Google Scholar] [CrossRef] [PubMed]
- LaCasce, A.S.; Castells, M.C.; Burstein, H.J.; Meyerhardt, J.A. Infusion-Related Reactions to Therapeutic Monoclonal Antibodies Used for Cancer Therapy. Available online: https://www.uptodate.com/contents/infusion-related-reactions-to-therapeutic-monoclonal-antibodies-used-for-cancer-therapy (accessed on 6 February 2019).
- Daniels, G.H.; Vladic, A.; Brinar, V.; Zavalishin, I.; Valente, W.; Oyuela, P.; Palmer, J.; Margolin, D.H.; Hollenstein, J. Alemtuzumab-related thyroid dysfunction in a phase 2 trial of patients with relapsing-remitting multiple sclerosis. J. Clin. Endocrinol. Metab. 2014, 99, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Van Den Neste, E.; Cazin, B.; Janssens, A.; Gonzalez-Barca, E.; Terol, M.J.; Levy, V.; Perez de Oteyza, J.; Zachee, P.; Saunders, A.; de Frias, M.; et al. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (cll): A multicenter phase i/ii study. Cancer Chemother. Pharmacol. 2013, 71, 581–591. [Google Scholar] [CrossRef]
- McCroskery, P.; Selmaj, K.; Fernandez, O.; Grimaldi, L.M.E.; Silber, E.; Pardo, G.; Freedman, S.M.; Zhang, Y.; Xu, L.; Cadavid, D.; et al. Safety and Tolerability of Opicinumab in Relapsing Multiple Sclerosis: The Phase 2b SYNERGY Trial (P5.369). Neurology 2017, 88. [Google Scholar]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.J.; Leocani, L.; Freedman, M.S.; Plant, G.T.; Preiningerova, J.L.; et al. Predictors of response to opicinumab in acute optic neuritis. Ann. Clin. Transl. Neurol. 2018, 5, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Mellion, M.; Edwards, K.R.; Hupperts, R.; Drulović, J.; Montalban, X.; Hartung, H.-P.; Brochet, B.; Calabresi, P.A.; Rudick, R.; Ibrahim, A.; et al. Efficacy Results from the Phase 2b SYNERGY Study: Treatment of Disabling Multiple Sclerosis with the Anti-LINGO-1 Monoclonal Antibody Opicinumab (S33.004). Neurology 2017, 88, S33.004. [Google Scholar]



{kind=link}
{kind=link}
| Therapeutic Monoclonal Antibodies | |||||
|---|---|---|---|---|---|
| MAB | Composition | Target | Mechanism of Action | Administration | FDA Approval Date for MS |
| Alemtuzumab | Humanized MAB IgGk | CD52 | ADCC | Intravenous | November 2014 |
| Elezanumab [11] | Fully human MAB | RGMa | Binds and neutralizes RGMa which modulates T cell responses and dendritic cells in CNS lesions | Intravenous | N/A |
| GNbAC1 [12] | Humanized IgG4 MAB | Envelope protein of HERV-W MSRV | Targets the envelope protein of HERV-W MSRV, which may play a critical role in multiple sclerosis | Intravenous | N/A |
| Natalizumab | Humanized monoclonal IgG1 | Cell adhesion molecule α4-integrin | Preventing lymphocyte transport across the blood brain barrier | Intravenous | November 2004 and reapproved on June 2006 |
| Ocrelizumab | Humanized IgG1 | Phosphorylated glycoprotein CD20 on B lymphocytes | ADCC > CDC | Intravenous | March 2017 |
| Ofatumumab | Fully humanized IgG1 | CD20 | CDC > ADCC | Subcutaneous | N/A |
| Opicinumab | Humanized MAB | Targets LINGO-1 | Allows OPCs to differentiate into mature OLG for remyelination | Intravenous | N/A |
| Ublituximab | Chimeric IgG1 MAB | CD20 | CDC and ADCC | Intravenous | N/A |
| Rituximab | Chimeric (murine/human) MAB | CD20 | CDC and ADCC | Intravenous | N/A |
| VAY736 [13] | Defucosylated, human IgG1 MAB | Targets the receptor for BAFF-R | ADCC and blockade of BAFF:BAFF-R signaling that drives B cell differentiation, proliferation and survival | Intravenous | N/A |
| MAB | Composition | Target/Mechanism | Withdrawn |
|---|---|---|---|
| Atacicept [14] | Fully humanized recombinant fusion protein containing the extracellular ligand-binding portion of the human TACI receptor | Binds to the cytokines BLyS and APRIL, involved in B-cell differentiation, maturation, and survival. | Increases relapse rates in multiple sclerosis reflected on an increase in annualized relapse rates. |
| Daclizumab [15] | Humanized IgG1 MAB | CD25, which is attached to the Tac epitope on the alpha chain of CD25 (IL-2 receptor) on activated lymphocytes | Post-marketing vigilance helped to detect secondary autoimmune events, including inflammatory encephalitis in 12 patients worldwide leading to at least 3 deaths where an interaction with the drug could not be ruled out. |
| Muromonab [16] | Chimeric MAB, first MAB to ever be approved | Inhibition of CD3 receptor | High toxicity made it unlikely to be a preferred treatment for MS. |
| Secukinumab [17] | Humanized IgG1kappa MAB | IL-17 receptor, inhibits proinflammatory IL-17A | Discontinued due to the development of a fully-human anti IL-17 MAB with better potential |
| Tabalumab [18] | Selective and fully human IgG4 MAB | Neutralization of membrane-bound and soluble B-cell activating factor (BAFF) | Results from phase 2 clinical trials in patients with RMS, showed no evidence of reduction Gd-enhancing lesions versus placebo, further analysis were discontinued. |
| Ustekinumab [19] | Fully humanized IgG1 MAB | Targets subunit P40 on cytokines IL-12 and IL-23 preventing them from differentiating and activating Th1 cells | Discontinued after phase 2 trials for low/lack of efficacy. |
| Vatelizumab [20] | Fully humanized MAB that targets VLA-2, a collagen binding integrin expressed on activated lymphocytes | Preventing the crossing of inflammatory cells into the brain, reducing inflammation and tested on RMS | Primary efficacy endpoint was not met after phase 2a and 2b studies halting further development for MS. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voge, N.V.; Alvarez, E. Monoclonal Antibodies in Multiple Sclerosis: Present and Future. Biomedicines 2019, 7, 20. https://doi.org/10.3390/biomedicines7010020
Voge NV, Alvarez E. Monoclonal Antibodies in Multiple Sclerosis: Present and Future. Biomedicines. 2019; 7(1):20. https://doi.org/10.3390/biomedicines7010020
Chicago/Turabian StyleVoge, Natalia V., and Enrique Alvarez. 2019. "Monoclonal Antibodies in Multiple Sclerosis: Present and Future" Biomedicines 7, no. 1: 20. https://doi.org/10.3390/biomedicines7010020
APA StyleVoge, N. V., & Alvarez, E. (2019). Monoclonal Antibodies in Multiple Sclerosis: Present and Future. Biomedicines, 7(1), 20. https://doi.org/10.3390/biomedicines7010020