Current Knowledge of IL-6 Cytokine Family Members in Acute and Chronic Kidney Disease


Abstract
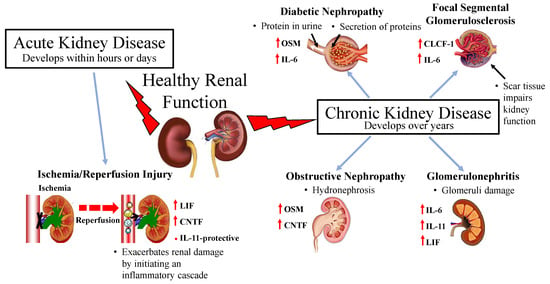
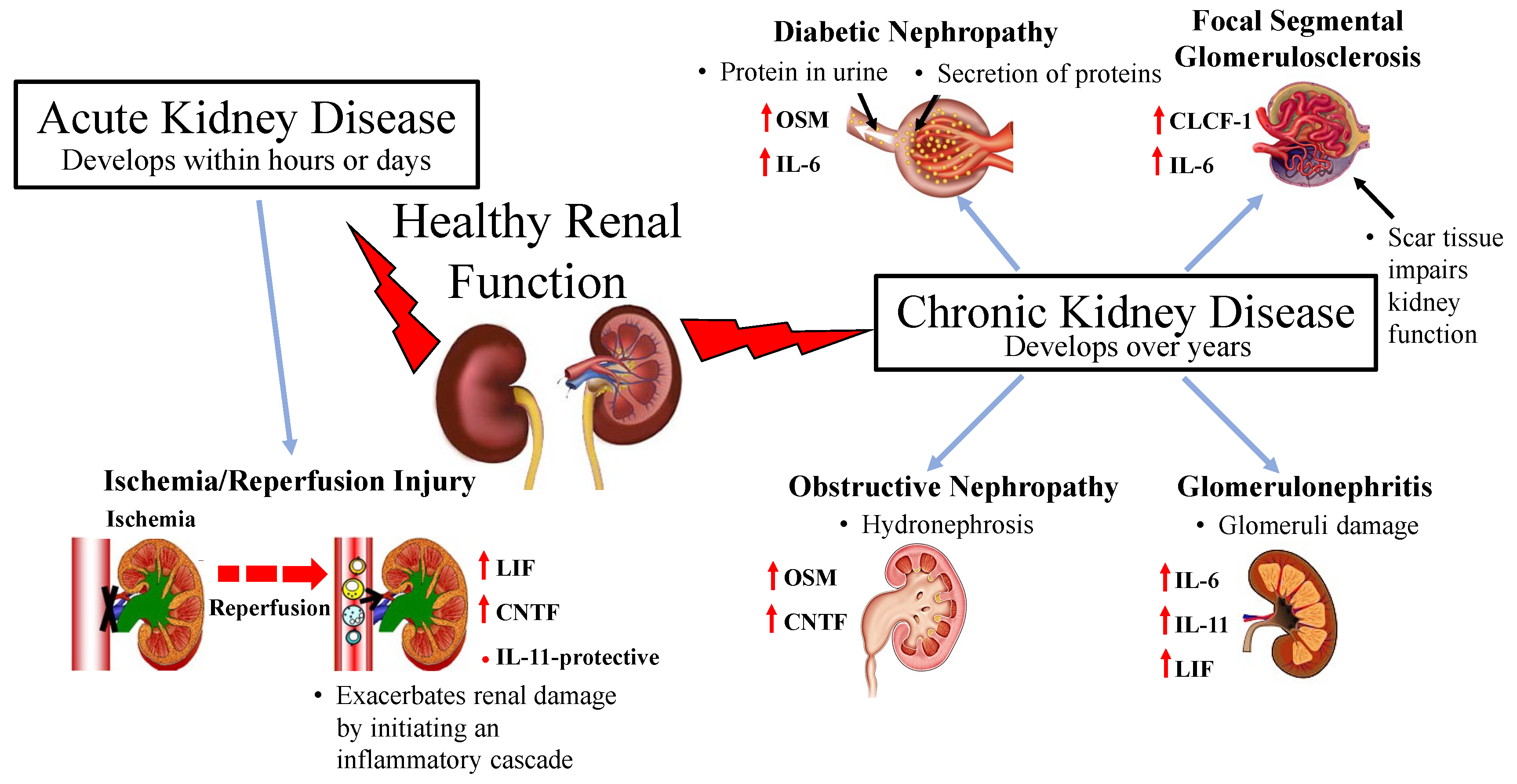
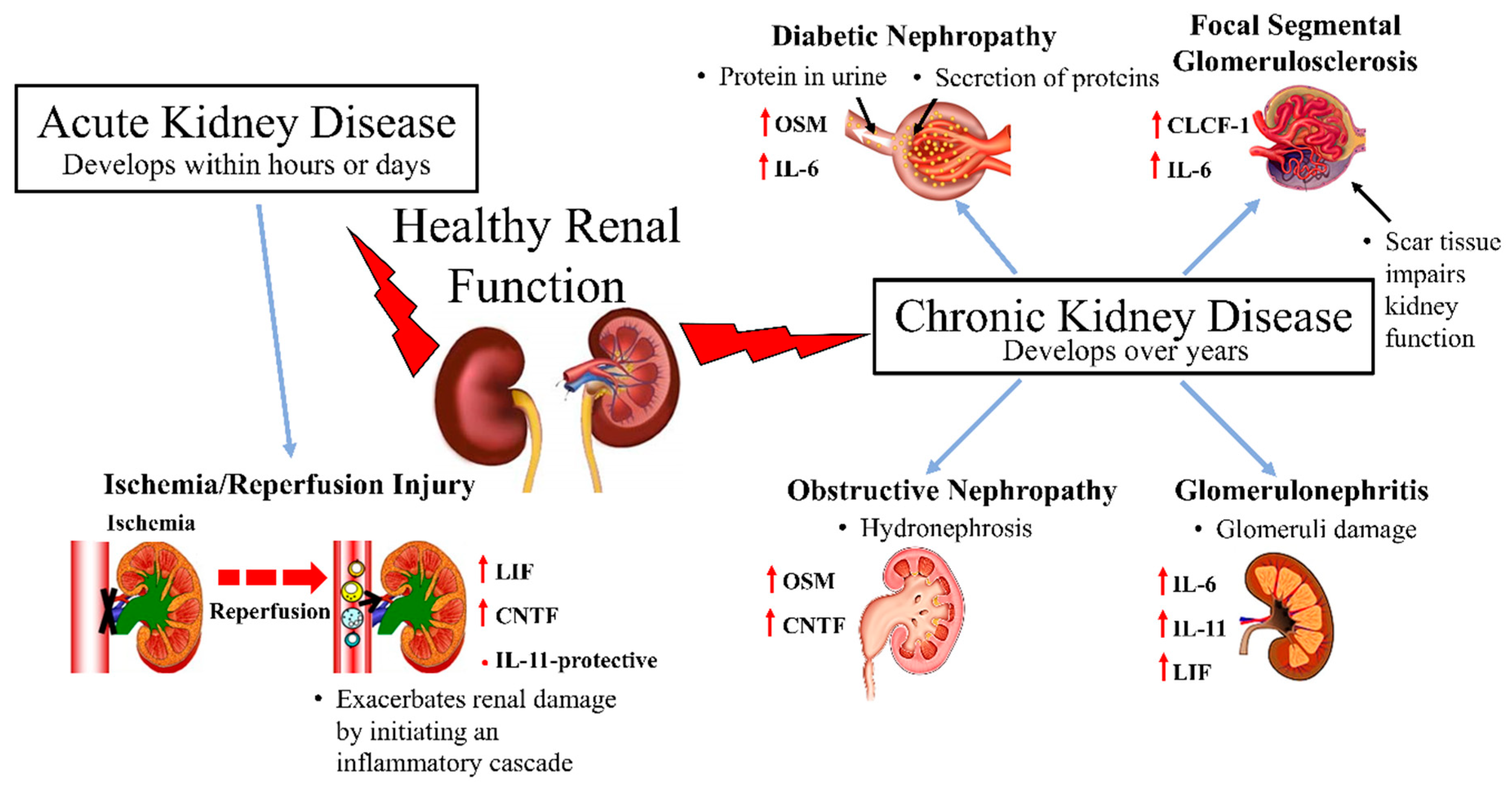
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. IL-6 Cytokine Family Members
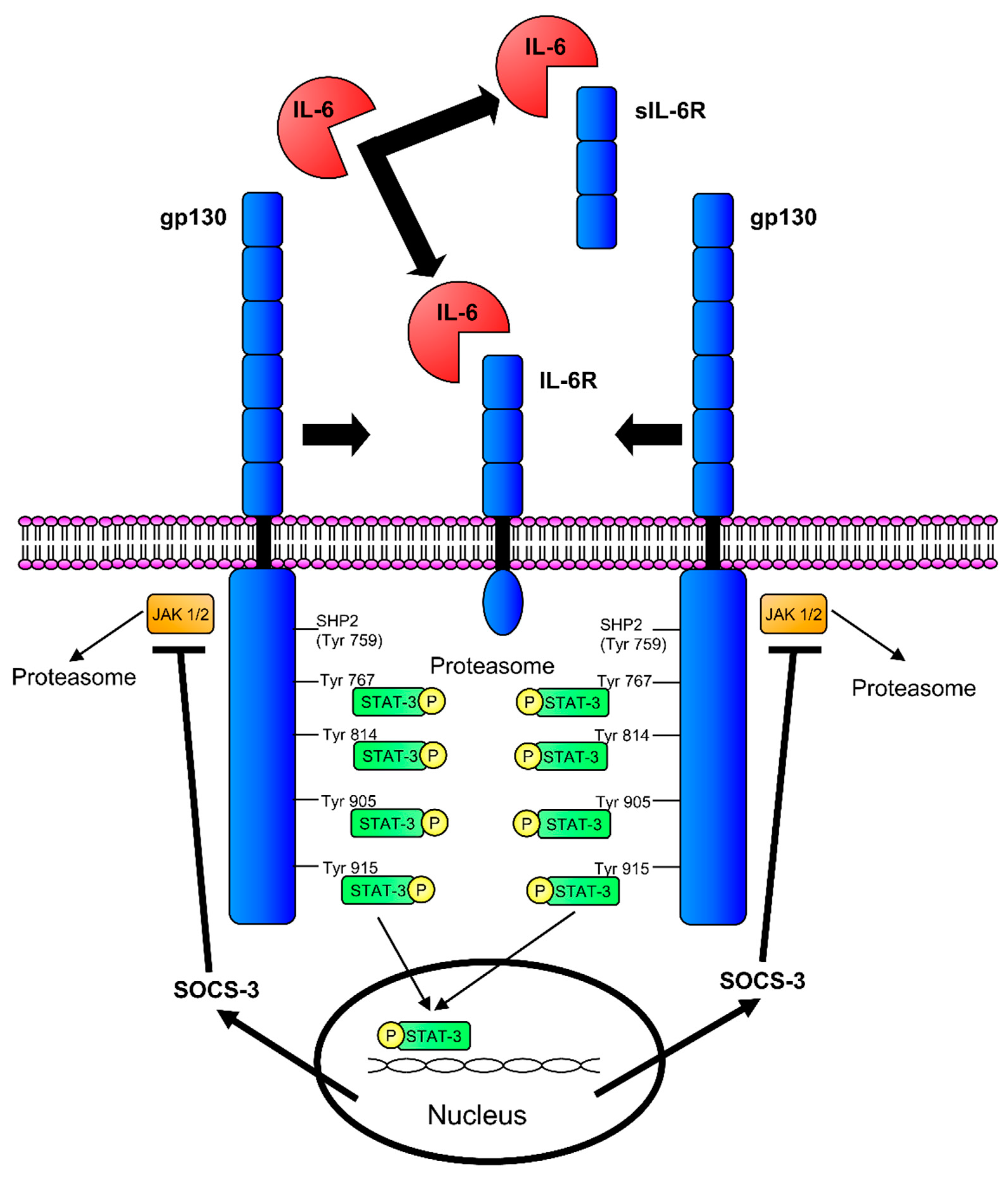
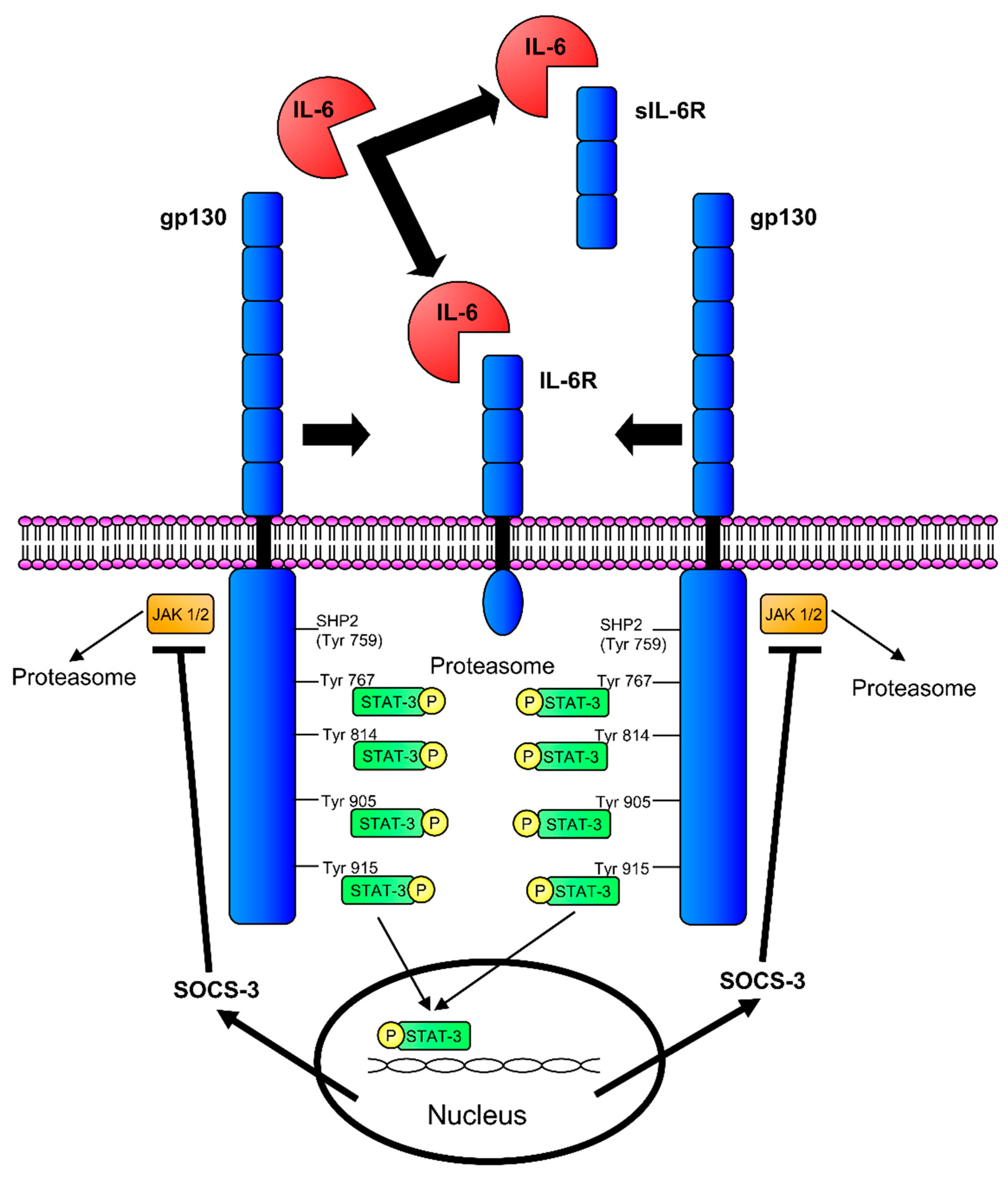
2.1. Interleukin-6
2.2. Interleukin-11
2.3. Leukaemia Inhibitory Factor
2.4. Oncostatin M
2.5. Ciliary Neurotrophic Factor
2.6. Cardiotrophin-1
2.7. Cardiotrophin-Like Cytokine Factor-1
2.8. Granulocyte Colony-Stimulating Factor
3. Implications of IL-6 Cytokine Family Members in Kidney Disease
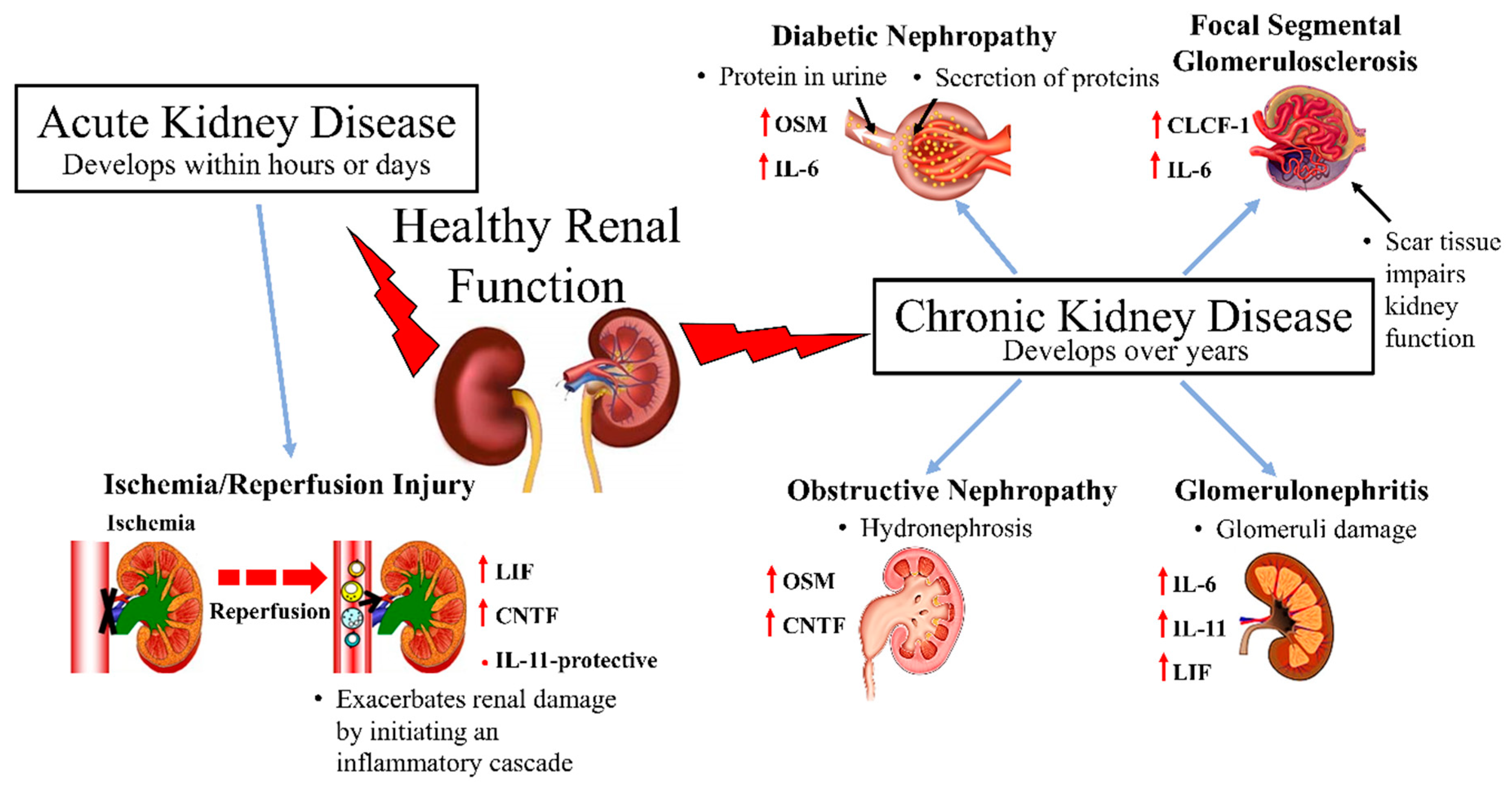
3.1. Acute Kidney Injury
Renal Ischemia and Reperfusion Injury
3.2. Chronic Kidney Disease
3.2.1. Diabetic Nephropathy
3.2.2. Glomerulonephritis
3.2.3. Focal Segmental Glomerulosclerosis
3.2.4. Obstructive Nephropathy
4. Obesity, Hypertension, Diabetes and Kidney Disease
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jones, S.A.; Fraser, D.J.; Fielding, C.A.; Jones, G.W. Interleukin-6 in renal disease and therapy. Nephrol. Dial. Transplant 2015, 30, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Gorshkova, E.A.; Nedospasov, S.A.; Shilov, E.S. Evolutionary plasticity of IL-6 cytokine family. Mol. Biol. (Mosk.) 2016, 50, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Garbers, C.; Hermanns, H.M.; Schaper, F.; Muller-Newen, G.; Grotzinger, J.; Rose-John, S.; Scheller, J. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012, 23, 85–97. [Google Scholar] [CrossRef]
- Hibi, M.; Nakajima, K.; Hirano, T. IL-6 cytokine family and signal transduction: A model of the cytokine system. J. Mol. Med. (Berl.) 1996, 74, 1–12. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Munoz-Canoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef]
- Nechemia-Arbely, Y.; Barkan, D.; Pizov, G.; Shriki, A.; Rose-John, S.; Galun, E.; Axelrod, J.H. IL-6/IL-6R axis plays a critical role in acute kidney injury. J. Am. Soc. Nephrol. 2008, 19, 1106–1115. [Google Scholar] [CrossRef]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.; Schuster, B.; Schutze, S.; Bussmeyer, I.; Ludwig, A.; Hundhausen, C.; Sadowski, T.; Saftig, P.; Hartmann, D.; Kallen, K.J.; et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 2003, 278, 38829–38839. [Google Scholar] [CrossRef]
- Rose-John, S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.R.; Bennett, F.; Calvetti, J.A.; Kelleher, K.; Wood, C.R.; O’Hara, R.M., Jr.; Leary, A.C.; Sibley, B.; Clark, S.C.; Williams, D.A.; et al. Molecular cloning of a cDNA encoding interleukin 11, a stromal cell-derived lymphopoietic and hematopoietic cytokine. Proc. Natl. Acad. Sci. USA 1990, 87, 7512–7516. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Choi, C.; Lee, H.G.; Lim, Y.; Lee, Y.H. Transcriptional regulation of the interleukin-11 gene by oncogenic Ras. Carcinogenesis 2012, 33, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.J. LIF: Lots of interesting functions. Trends Biochem. Sci. 1992, 17, 72–76. [Google Scholar] [CrossRef]
- Nicola, N.A.; Babon, J.J. Leukemia inhibitory factor (LIF). Cytokine Growth Factor Rev. 2015, 26, 533–544. [Google Scholar] [CrossRef]
- Zarling, J.M.; Shoyab, M.; Marquardt, H.; Hanson, M.B.; Lioubin, M.N.; Todaro, G.J. Oncostatin M: A growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9739–9743. [Google Scholar] [CrossRef]
- Hermanns, H.M. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. 2015, 26, 545–558. [Google Scholar] [CrossRef]
- Richards, C.D. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm. 2013, 2013, 512103. [Google Scholar] [CrossRef] [PubMed]
- Negro, A.; Tolosano, E.; Skaper, S.D.; Martini, I.; Callegaro, L.; Silengo, L.; Fiorini, F.; Altruda, F. Cloning and expression of human ciliary neurotrophic factor. Eur. J. Biochem. 1991, 201, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Pasquin, S.; Sharma, M.; Gauchat, J.F. Ciliary neurotrophic factor (CNTF): New facets of an old molecule for treating neurodegenerative and metabolic syndrome pathologies. Cytokine Growth Factor Rev. 2015, 26, 507–515. [Google Scholar] [CrossRef]
- Ettinger, M.P.; Littlejohn, T.W.; Schwartz, S.L.; Weiss, S.R.; McIlwain, H.H.; Heymsfield, S.B.; Bray, G.A.; Roberts, W.G.; Heyman, E.R.; Stambler, N.; et al. Recombinant variant of ciliary neurotrophic factor for weight loss in obese adults: A randomized, dose-ranging study. JAMA 2003, 289, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Yoldi, M.; Moreno-Aliaga, M.J.; Bustos, M. Cardiotrophin-1: A multifaceted cytokine. Cytokine Growth Factor Rev. 2015, 26, 523–532. [Google Scholar] [CrossRef]
- Pennica, D.; King, K.L.; Shaw, K.J.; Luis, E.; Rullamas, J.; Luoh, S.M.; Darbonne, W.C.; Knutzon, D.S.; Yen, R.; Chien, K.R.; et al. Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc. Natl. Acad. Sci. USA 1995, 92, 1142–1146. [Google Scholar] [CrossRef]
- Vlotides, G.; Zitzmann, K.; Stalla, G.K.; Auernhammer, C.J. Novel neurotrophin-1/B cell-stimulating factor-3 (NNT-1/BSF-3)/cardiotrophin-like cytokine (CLC)—A novel gp130 cytokine with pleiotropic functions. Cytokine Growth Factor Rev. 2004, 15, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Kass, D.J. Cytokine-like factor 1 (CLF1): Life after development? Cytokine 2011, 55, 325–329. [Google Scholar] [CrossRef]
- Demetri, G.D.; Griffin, J.D. Granulocyte colony-stimulating factor and its receptor. Blood 1991, 78, 2791–2808. [Google Scholar]
- Clogston, C.L.; Boone, T.C.; Crandall, B.C.; Mendiaz, E.A.; Lu, H.S. Disulfide structures of human interleukin-6 are similar to those of human granulocyte colony stimulating factor. Arch. Biochem. Biophys. 1989, 272, 144–151. [Google Scholar] [CrossRef]
- Hammacher, A.; Richardson, R.T.; Layton, J.E.; Smith, D.K.; Angus, L.J.; Hilton, D.J.; Nicola, N.A.; Wijdenes, J.; Simpson, R.J. The immunoglobulin-like module of gp130 is required for signaling by interleukin-6, but not by leukemia inhibitory factor. J. Biol. Chem. 1998, 273, 22701–22707. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.C. The role of the granulocyte colony-stimulating factor receptor (G-CSF-R) in disease. Front. Biosci. 2007, 12, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Thomas, B.; Remuzzi, G.; Riella, M.; Nahas, M.E.; Naicker, S.; Dirks, J. Kidney Disease. In Cardiovascular, Respiratory, and Related Disorders, 3rd ed.; Prabhakaran, D., Anand, S., Gaziano, T.A., Mbanya, J.C., Wu, Y., Nugent, R., Eds.; World Bank: Washington, DC, USA, 2017; Chapter 13. [Google Scholar] [CrossRef]
- Bellomo, R.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Palevsky, P. Acute Dialysis Quality Initiative w: Acute renal failure—Definition, outcome measures, animal models, fluid therapy and information technology needs: The Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit. Care 2004, 8, R204–R212. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Cerda, J.; Burdmann, E.A.; Tonelli, M.; Garcia-Garcia, G.; Jha, V.; Susantitaphong, P.; Rocco, M.; Vanholder, R.; Sever, M.S.; et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): A human rights case for nephrology. Lancet 2015, 385, 2616–2643. [Google Scholar] [CrossRef]
- Simmons, E.M.; Himmelfarb, J.; Sezer, M.T.; Chertow, G.M.; Mehta, R.L.; Paganini, E.P.; Soroko, S.; Freedman, S.; Becker, K.; Spratt, D.; et al. Plasma cytokine levels predict mortality in patients with acute renal failure. Kidney Int. 2004, 65, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Yang, C.W.; Lim, S.W.; Han, K.W.; Ahn, H.J.; Park, J.H.; Kim, Y.H.; Kirsh, M.; Cha, J.H.; Park, J.H.; Kim, Y.S.; et al. Upregulation of ciliary neurotrophic factor (CNTF) and CNTF receptor alpha in rat kidney with ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2001, 12, 749–757. [Google Scholar] [PubMed]
- Yoshino, J.; Monkawa, T.; Tsuji, M.; Hayashi, M.; Saruta, T. Leukemia inhibitory factor is involved in tubular regeneration after experimental acute renal failure. J. Am. Soc. Nephrol. 2003, 14, 3090–3101. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Park, S.W.; Kim, M.; Ham, A.; Anderson, L.J.; Brown, K.M.; D’Agati, V.D.; Cox, G.N. Interleukin-11 protects against renal ischemia and reperfusion injury. Am. J. Physiol. Renal Physiol. 2012, 303, F1216–F1224. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, M.; Ham, A.; Brown, K.M.; Greene, R.W.; D’Agati, V.D.; Lee, H.T. IL-11 is required for A1 adenosine receptor-mediated protection against ischemic AKI. J. Am. Soc. Nephrol. 2013, 24, 1558–1570. [Google Scholar] [CrossRef]
- Garcia-Cenador, M.B.; Lorenzo-Gomez, M.F.; Herrero-Payo, J.J.; Ruiz, J.; Perez de Obanos, M.P.; Pascual, J.; Lopez-Novoa, J.M.; Garcia-Criado, F.J. Cardiotrophin-1 administration protects from ischemia-reperfusion renal injury and inflammation. Transplantation 2013, 96, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Quiros, Y.; Sanchez-Gonzalez, P.D.; Lopez-Hernandez, F.J.; Morales, A.I.; Lopez-Novoa, J.M. Cardiotrophin-1 administration prevents the renal toxicity of iodinated contrast media in rats. Toxicol. Sci. 2013, 132, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Andres, N.; Rousseau, A.; Akhtar, R.; Calvier, L.; Inigo, C.; Labat, C.; Zhao, X.; Cruickshank, K.; Diez, J.; Zannad, F.; et al. Cardiotrophin 1 is involved in cardiac, vascular, and renal fibrosis and dysfunction. Hypertension 2012, 60, 563–573. [Google Scholar] [CrossRef] [PubMed]
- James, M.T.; Hemmelgarn, B.R.; Tonelli, M. Early recognition and prevention of chronic kidney disease. Lancet 2010, 375, 1296–1309. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Fzeigerlova, E.; Battaglia-Hsu, S.F. IL-6 signaling in diabetic nephropathy: From pathophysiology totherapeutic perspectives. Cytokine Growth Factor Rev. 2017, 37, 57–65. [Google Scholar] [CrossRef]
- Ahmad, J. Management of diabetic nephropathy: Recent progress and future perspective. Diabetes Metab. Syndr. 2015, 9, 343–358. [Google Scholar] [CrossRef]
- Alsaad, K.O.; Herzenberg, A.M. Distinguishing diabetic nephropathy from other causes of glomerulosclerosis: An update. J. Clin. Pathol. 2007, 60, 18–26. [Google Scholar] [CrossRef]
- Giunti, S.; Barit, D.; Cooper, M.E. Mechanisms of diabetic nephropathy: Role of hypertension. Hypertension 2006, 48, 519–526. [Google Scholar] [CrossRef]
- Taslipinar, A.; Yaman, H.; Yilmaz, M.I.; Demirbas, S.; Saglam, M.; Taslipinar, M.Y.; Agilli, M.; Kurt, Y.G.; Sonmez, A.; Azal, O.; et al. The relationship between inflammation, endothelial dysfunction and proteinuria in patients with diabetic nephropathy. Scand. J. Clin. Lab. Invest. 2011, 71, 606–612. [Google Scholar] [CrossRef]
- Lei, C.T.; Su, H.; Ye, C.; Tang, H.; Gao, P.; Wan, C.; He, F.F.; Wang, Y.M.; Zhang, C. The classic signalling and trans-signalling of interleukin-6 are both injurious in podocyte under high glucose exposure. J. Cell. Mol. Med. 2018, 22, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xing, L.; Wang, L.; Yao, F.; Liu, S.; Hao, J.; Liu, W.; Duan, H. Therapeutic effects of suppressors of cytokine signaling in diabetic nephropathy. J. Histochem. Cytochem. 2014, 62, 119–128. [Google Scholar] [CrossRef]
- Sarkozi, R.; Flucher, K.; Haller, V.M.; Pirklbauer, M.; Mayer, G.; Schramek, H. Oncostatin M inhibits TGF-beta1-induced CTGF expression via STAT3 in human proximal tubular cells. Biochem. Biophys. Res. Commun. 2012, 424, 801–806. [Google Scholar] [CrossRef]
- Brosius, F.C.; Tuttle, K.R.; Kretzler, M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia 2016, 59, 1624–1627. [Google Scholar] [CrossRef]
- Recio, C.; Lazaro, I.; Oguiza, A.; Lopez-Sanz, L.; Bernal, S.; Blanco, J.; Egido, J.; Gomez-Guerrero, C. Suppressor of Cytokine Signaling-1 Peptidomimetic Limits Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 575–585. [Google Scholar] [CrossRef] [PubMed]
- So, B.I.; Song, Y.S.; Fang, C.H.; Park, J.Y.; Lee, Y.; Shin, J.H.; Kim, H.; Kim, K.S. G-CSF prevents progression of diabetic nephropathy in rat. PLoS ONE 2013, 8, e77048. [Google Scholar] [CrossRef]
- Suematsu, S.; Matsuda, T.; Aozasa, K.; Akira, S.; Nakano, N.; Ohno, S.; Miyazaki, J.; Yamamura, K.; Hirano, T.; Kishimoto, T. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA 1989, 86, 7547–7551. [Google Scholar] [CrossRef]
- Horii, Y.; Muraguchi, A.; Iwano, M.; Matsuda, T.; Hirayama, T.; Yamada, H.; Fujii, Y.; Dohi, K.; Ishikawa, H.; Ohmoto, Y.; et al. Involvement of IL-6 in mesangial proliferative glomerulonephritis. J. Immunol. 1989, 143, 3949–3955. [Google Scholar]
- Liu, D.; Zhang, N.; Zhang, J.; Zhao, H.; Wang, X. miR-410 suppresses the expression of interleukin-6 as well as renal fibrosis in the pathogenesis of lupus nephritis. Clin. Exp. Pharmacol. Physiol. 2016, 43, 616–625. [Google Scholar] [CrossRef]
- Chien, J.W.; Chen, W.L.; Tsui, Y.G.; Lee, M.C.; Lin, A.Y.; Lin, C.Y. Daily urinary interleukin-11 excretion correlated with proteinuria in IgA nephropathy and lupus nephritis. Pediatr. Nephrol. 2006, 21, 490–496. [Google Scholar] [CrossRef]
- Berghea, E.C.; Balgradean, M.; Popa, I.L. Correlation Between Idiopathic Nephrotic Syndrome and Atopy in Children—Short Review. Maedica 2017, 12, 55–58. [Google Scholar] [PubMed]
- Shen, M.M.; Skoda, R.C.; Cardiff, R.D.; Campos-Torres, J.; Leder, P.; Ornitz, D.M. Expression of LIF in transgenic mice results in altered thymic epithelium and apparent interconversion of thymic and lymph node morphologies. EMBO J. 1994, 13, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.S.; Taupin, J.L.; Potier, M.; Deminiere, C.; Potaux, L.; Gualde, N.; Moreau, J.F. Renal synthesis of leukaemia inhibitory factor (LIF), under normal and inflammatory conditions. Cytokine 2000, 12, 265–271. [Google Scholar] [CrossRef]
- Liu, Q.; Du, Y.; Li, K.; Zhang, W.; Feng, X.; Hao, J.; Li, H.; Liu, S. Anti-OSM Antibody Inhibits Tubulointerstitial Lesion in a Murine Model of Lupus Nephritis. Mediat. Inflamm. 2017, 2017, 3038514. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, W.; Yu, H.; Zhang, Y.; Dai, Y.; Ning, C.; Tao, L.; Sun, H.; Kellems, R.E.; Blackburn, M.R.; et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension 2012, 59, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; van Vollenhoven, R.F.; Aranow, C.; Wagner, C.; Gordon, R.; Zhuang, Y.; Belkowski, S.; Hsu, B. A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Treatment With Sirukumab (CNTO 136) in Patients With Active Lupus Nephritis. Arthritis Rheumatol. 2016, 68, 2174–2183. [Google Scholar] [CrossRef] [PubMed]
- Kiberd, B.A. Interleukin-6 receptor blockage ameliorates murine lupus nephritis. J. Am. Soc. Nephrol. 1993, 4, 58–61. [Google Scholar]
- Tsantikos, E.; Maxwell, M.J.; Putoczki, T.; Ernst, M.; Rose-John, S.; Tarlinton, D.M.; Hibbs, M.L. Interleukin-6 trans-signaling exacerbates inflammation and renal pathology in lupus-prone mice. Arthritis Rheumatism 2013, 65, 2691–2702. [Google Scholar] [CrossRef]
- Lai, P.C.; Cook, H.T.; Smith, J.; Keith, J.C., Jr.; Pusey, C.D.; Tam, F.W. Interleukin-11 attenuates nephrotoxic nephritis in Wistar Kyoto rats. J. Am. Soc. Nephrol. 2001, 12, 2310–2320. [Google Scholar] [PubMed]
- Lai, P.C.; Smith, J.; Bhangal, G.; Chaudhry, K.A.; Chaudhry, A.N.; Keith, J.C., Jr.; Tam, F.W.; Pusey, C.D.; Cook, H.T. Interleukin-11 reduces renal injury and glomerular NF-kappa B activity in murine experimental glomerulonephritis. Nephron Exp. Nephrol. 2005, 101, e146–e154. [Google Scholar] [CrossRef] [PubMed]
- Stangou, M.; Bhangal, G.; Lai, P.C.; Smith, J.; Keith, J.C., Jr.; Boyle, J.J.; Pusey, C.D.; Cook, T.; Tam, F.W. Effect of IL-11 on glomerular expression of TGF-beta and extracellular matrix in nephrotoxic nephritis in Wistar Kyoto rats. J. Nephrol. 2011, 24, 106–111. [Google Scholar] [CrossRef]
- Zavala, F.; Masson, A.; Hadaya, K.; Ezine, S.; Schneider, E.; Babin, O.; Bach, J.F. Granulocyte-colony stimulating factor treatment of lupus autoimmune disease in MRL-lpr/lpr mice. J. Immunol. 1999, 163, 5125–5132. [Google Scholar]
- Yan, J.J.; Jambaldorj, E.; Lee, J.G.; Jang, J.Y.; Shim, J.M.; Han, M.; Koo, T.Y.; Ahn, C.; Yang, J. Granulocyte colony-stimulating factor treatment ameliorates lupus nephritis through the expansion of regulatory T cells. BMC Nephrol. 2016, 17, 175. [Google Scholar] [CrossRef] [PubMed]
- Vasiliu, I.M.; Petri, M.A.; Baer, A.N. Therapy with granulocyte colony-stimulating factor in systemic lupus erythematosus may be associated with severe flares. J. Rheumatol. 2006, 33, 1878–1880. [Google Scholar]
- McCarthy, E.T.; Sharma, M.; Savin, V.J. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2010, 5, 2115–2121. [Google Scholar] [CrossRef]
- Savin, V.J.; Sharma, M.; Zhou, J.; Gennochi, D.; Fields, T.; Sharma, R.; McCarthy, E.T.; Srivastava, T.; Domen, J.; Tormo, A.; et al. Renal and Hematological Effects of CLCF-1, a B-Cell-Stimulating Cytokine of the IL-6 Family. J. Immunol. Res. 2015, 2015, 714964. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Zhou, J.; Gauchat, J.F.; Sharma, R.; McCarthy, E.T.; Srivastava, T.; Savin, V.J. Janus kinase 2/signal transducer and activator of transcription 3 inhibitors attenuate the effect of cardiotrophin-like cytokine factor 1 and human focal segmental glomerulosclerosis serum on glomerular filtration barrier. Transl. Res. 2015, 166, 384–398. [Google Scholar] [CrossRef]
- Savin, V.J.; Sharma, M.; Zhou, J.; Genochi, D.; Sharma, R.; Srivastava, T.; Ilahe, A.; Budhiraja, P.; Gupta, A.; McCarthy, E.T. Multiple Targets for Novel Therapy of FSGS Associated with Circulating Permeability Factor. Biomed. Res. Int. 2017, 2017, 6232616. [Google Scholar] [CrossRef] [PubMed]
- Isobe, S.; Ohashi, N.; Katahashi, N.; Ishigaki, S.; Tsuji, N.; Tsuji, T.; Kato, A.; Fujigaki, Y.; Shimizu, A.; Yasuda, H. Focal segmental glomerulosclerosis associated with cutaneous and systemic plasmacytosis. CEN Case Rep. 2017, 6, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Elbjeirami, W.M.; Truong, L.D.; Tawil, A.; Wang, W.; Dawson, S.; Lan, H.Y.; Zhang, P.; Garcia, G.E.; Wayne Smith, C. Early differential expression of oncostatin M in obstructive nephropathy. J. Interferon Cytok. Res. 2010, 30, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Choi, J.Y.; Cha, J.H. Expression of ciliary neurotrophic factor and its receptor in experimental obstructive nephropathy. Anat. Cell. Biol. 2011, 44, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Decleves, A.E.; Sharma, K. Obesity and kidney disease: Differential effects of obesity on adipose tissue and kidney inflammation and fibrosis. Curr Opin Nephrol Hypertens 2015, 24, 28–36. [Google Scholar] [CrossRef] [PubMed]
- The GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, R.; Suhail, F.; Lerma, E.V. Hypertension and chronic kidney disease. Dis. Mon. 2015, 61, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Brands, M.W.; Henegar, J.R. Mechanisms of hypertension and kidney disease in obesity. Ann. N. Y. Acad. Sci. 1999, 892, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.E.; Forbes, J.M.; Matthews, V.B. Obesity-induced renal impairment is exacerbated in interleukin-6-knockout mice. Nephrology (Carlton) 2012, 17, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Malone, J.I.; Hansen, B.C. Does obesity cause type 2 diabetes mellitus (T2DM)? Or is it the opposite? Pediatr. Diabetes 2018, 20, 5–9. [Google Scholar] [CrossRef]
- Toth-Manikowski, S.; Atta, M.G. Diabetic Kidney Disease: Pathophysiology and Therapeutic Targets. J. Diabetes Res. 2015, 2015, 697010. [Google Scholar] [CrossRef]
- DeMarco, V.G.; Aroor, A.R.; Sowers, J.R. The pathophysiology of hypertension in patients with obesity. Nat. Rev. Endocrinol. 2014, 10, 364–376. [Google Scholar] [CrossRef]
- Schlaich, M.P.; Socratous, F.; Hennebry, S.; Eikelis, N.; Lambert, E.A.; Straznicky, N.; Esler, M.D.; Lambert, G.W. Sympathetic activation in chronic renal failure. J. Am. Soc. Nephrol. 2009, 20, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.; University of Western Australia, Perth, Western Australia, Australia; Matthews, V.B.; University of Western Australia, Perth, Western Australia, Australia. Personal communication, 2018.
- Matthews, V.B.; Elliot, R.H.; Rudnicka, C.; Hricova, J.; Herat, L.; Schlaich, M.P. Role of the sympathetic nervous system in regulation of the sodium glucose cotransporter 2. J. Hypertens. 2017, 35, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Cervantes, M.I.; Galicia, O.G.; Moreno-Jaime, B.; Zapata-Morales, J.R.; Montoya-Contreras, A.; Bautista-Perez, R.; Martinez-Morales, F. Autocrine modulation of glucose transporter SGLT2 by IL-6 and TNF-alpha in LLC-PK(1) cells. J. Physiol. Biochem. 2012, 68, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magno, A.L.; Herat, L.Y.; Carnagarin, R.; Schlaich, M.P.; Matthews, V.B. Current Knowledge of IL-6 Cytokine Family Members in Acute and Chronic Kidney Disease. Biomedicines 2019, 7, 19. https://doi.org/10.3390/biomedicines7010019
Magno AL, Herat LY, Carnagarin R, Schlaich MP, Matthews VB. Current Knowledge of IL-6 Cytokine Family Members in Acute and Chronic Kidney Disease. Biomedicines. 2019; 7(1):19. https://doi.org/10.3390/biomedicines7010019
Chicago/Turabian StyleMagno, Aaron L., Lakshini Y. Herat, Revathy Carnagarin, Markus P. Schlaich, and Vance B. Matthews. 2019. "Current Knowledge of IL-6 Cytokine Family Members in Acute and Chronic Kidney Disease" Biomedicines 7, no. 1: 19. https://doi.org/10.3390/biomedicines7010019
APA StyleMagno, A. L., Herat, L. Y., Carnagarin, R., Schlaich, M. P., & Matthews, V. B. (2019). Current Knowledge of IL-6 Cytokine Family Members in Acute and Chronic Kidney Disease. Biomedicines, 7(1), 19. https://doi.org/10.3390/biomedicines7010019