On Zebrafish Disease Models and Matters of the Heart



Abstract
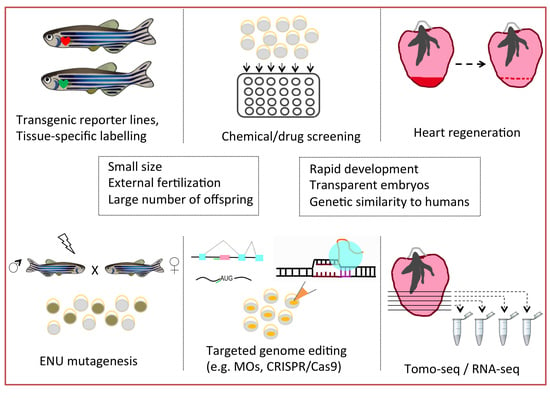
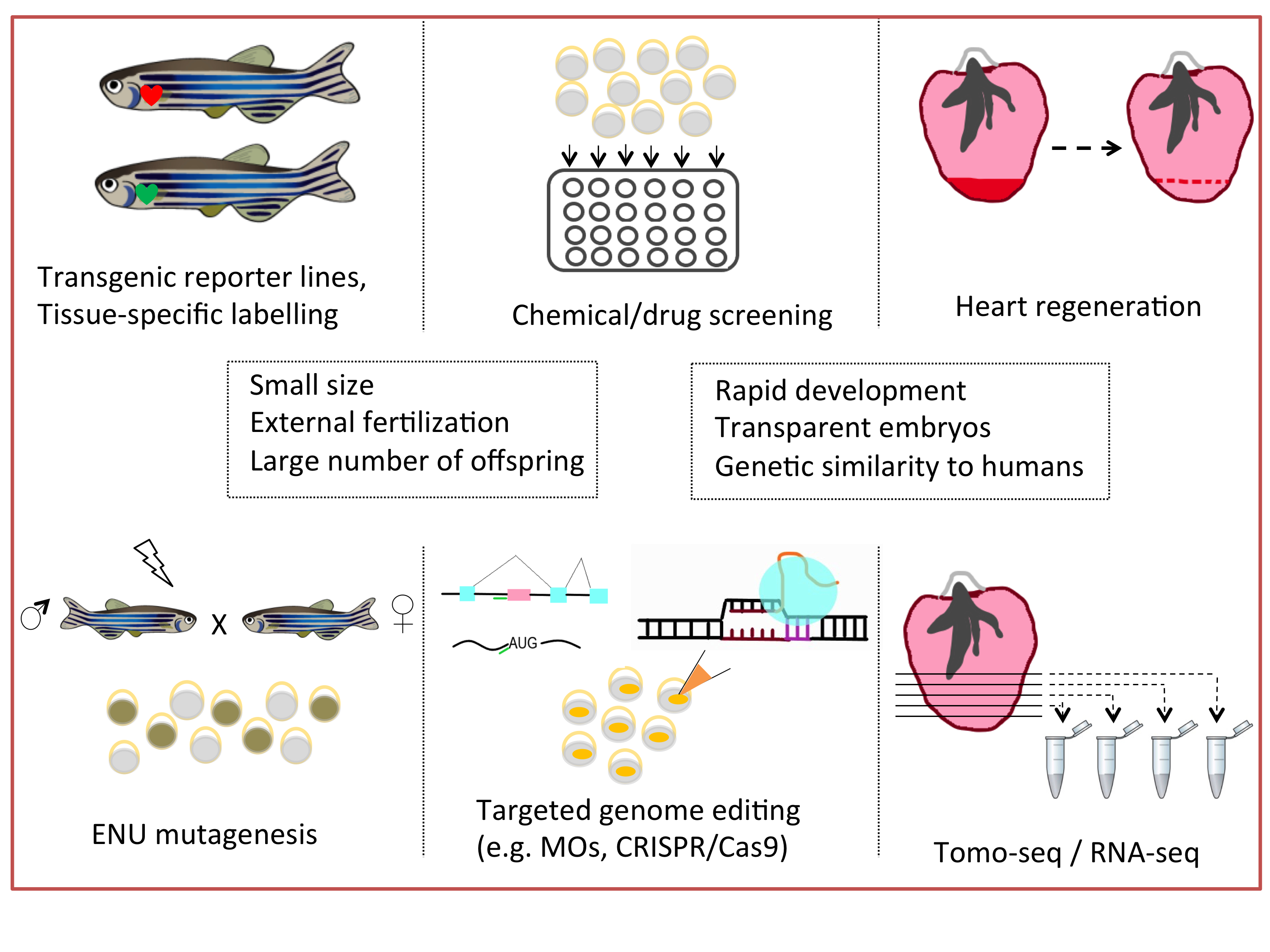
:
1. Introduction
2. Fishing for the Right Animal Model
3. The Pool of Engineering Tools
3.1. Genetic Approaches
3.2. Technical Approaches
4. Investigation of Human Cardiac Diseases under the Light of Zebrafish Research
5. Zebrafish Heart as an Injury Model
6. Future Perspectives
Funding
Conflicts of Interest
References
- World Health Organization. Technical Package for Cardiovascular Disease Management in Primary Health Care. Hearts 2016, 76. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2017 Update: A Report from the American Heart Association; Circulation: Waltham, MA, USA, 2017; Volume 135. [Google Scholar] [CrossRef]
- Veljkovic, N.; Zaric, B.; Djuric, I.; Obradovic, M.; Sudar-Milovanovic, E.; Radak, D.; Isenovic, E.R. Genetic Markers for Coronary Artery Disease. Medicina 2018, 54, 36. [Google Scholar] [CrossRef] [PubMed]
- Mangino, M.; Spector, T. Understanding Coronary Artery Disease Using Twin Studies. Heart 2013, 99, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association Analyses Based on False Discovery Rate Implicate New Loci for Coronary Artery Disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Ahituv, N.; Dunham, M.J.; Kitzman, J.O.; Roth, F.P.; Seelig, G.; Shendure, J.; Fowler, D.M. Variant Interpretation: Functional Assays to the Rescue. Am. J. Hum. Genet. 2017, 101, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.C. Utility and Limitations of Animal Models for the Functional Validation of Human Sequence Variants. Mol. Genet. Genom. Med. 2015, 3, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kim, Y.S.; Kim, J.; Pattison, J.; Kamaid, A.; Miller, Y.I. Modeling Hypercholesterolemia and Vascular Lipid Accumulation in LDL Receptor Mutant Zebrafish. J. Lipid Res. 2018, 59, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.K.; Spitsbergen, J.M. Valvular and Mural Endocardiosis in Aging Zebrafish (Danio Rerio). Vet. Pathol. 2016, 53, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, X.; Chen, Z. Evidence of an Association between Age-Related Functional Modifications and Pathophysiological Changes in Zebrafish Heart. Gerontology 2014, 61, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Stoyek, M.R.; Rog-zielinska, E.A.; Quinn, T.A. Age-Associated Changes in Electrical Function of the Zebra Fish Heart. Prog. Biophys. Mol. Biol. 2018, 138, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Sedmera, D.; Yost, H.J.; Clark, E.B. Structure and Function of the Developing Zebrafish Heart. Anat. Rec. 2000, 260, 148–157. [Google Scholar] [CrossRef]
- Hodgson, P.; Ireland, J.; Grunow, B. Fish, the Better Model in Human Heart Research? Zebrafish Heart Aggregates as a 3D Spontaneously Cardiomyogenic inVitro Model System. Prog. Biophys. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bournele, D.; Beis, D. Zebrafish Models of Cardiovascular Disease. Heart Fail. Rev. 2016, 21, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Bakkers, J. Zebrafish as a Model to Study Cardiac Development and Human Cardiac Disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, R.; Ferrer, T.; Huisken, J.; Spitzer, K.; Stainier, D.Y.R.; Tristani-Firouzi, M.; Chi, N.C. Zebrafish Model for Human Long QT Syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 11316–11321. [Google Scholar] [CrossRef] [PubMed]
- Leong, I.U.S.; Skinner, J.R.; Shelling, A.N.; Love, D.R. Zebrafish as a Model for Long QT Syndrome: The Evidence and the Means of Manipulating Zebrafish Gene Expression. Acta Physiol. 2010, 199, 257–276. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhao, Y.; Gu, J.; Quigley, K.L.; Chi, N.C.; Tai, Y.C.; Hsiai, T.K. Flexible Microelectrode Arrays to Interface Epicardial Electrical Signals with Intracardial Calcium Transients in Zebrafish Hearts. Biomed. Microdevices 2012, 14, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Vornanen, M.; Hassinen, M. Zebrafish Heart as a Model for Human Cardiac Electrophysiology. Channels 2016, 10, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Beis, D.; Kalogirou, S.; Tsigkas, N. Insights into Heart Development and Regeneration. In Introduction to Translational Cardiovascular Research; Springer: Cham, Switzerland, 2015; Chapter 2; pp. 17–30. [Google Scholar] [CrossRef]
- Meyer, A.; Schartl, M. Gene and Genome Duplications in Vertebrates: The One-to-Four (-to-Eight in Fish) Rule and the Evolution of Novel Gene Functions. Curr. Opin. Cell Biol. 1999, 11, 699–704. [Google Scholar] [CrossRef]
- Paone, C.; Rudeck, S.; Etard, C.; Strähle, U.; Rottbauer, W.; Just, S. Loss of Zebrafish Smyd1a Interferes with Myofibrillar Integrity without Triggering the Misfolded Myosin Response. Biochem. Biophys. Res. Commun. 2018, 496, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Sztal, T.E.; McKaige, E.A.; Williams, C.; Ruparelia, A.A.; Bryson-Richardson, R.J. Genetic Compensation Triggered by Actin Mutation Prevents the Muscle Damage Caused by Loss of Actin Protein. PLoS Genet. 2018, 14, e1007212. [Google Scholar] [CrossRef] [PubMed]
- Bouché, N.; Bouchez, D. Arabidopsis Gene Knockout: Phenotypes Wanted. Curr. Opin. Plant Biol. 2001, 4, 111–117. [Google Scholar] [CrossRef]
- Lachowiec, J.; Mason, G.A.; Schultz, K.; Queitsch, C. Redundancy, Feedback, and Robustness in the Arabidopsis Thaliana BZR/BEH Gene Family. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, A.S.; Bornberg-Bauer, E. Robustness by Intrinsically Disordered C-Termini and Translational Readthrough. Nucleic Acids Res. 2018, 46, 10184–10194. [Google Scholar] [CrossRef] [PubMed]
- Keane, O.M.; Toft, C.; Carretero-Paulet, L.; Jones, G.W.; Fares, M.A. Preservation of Genetic and Regulatory Robustness in Ancient Gene Duplicates of Saccharomyces Cerevisiae. Genome Res. 2014, 24, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.N.; Pavlicev, M.; Mitteroecker, P.; Pardo-Manuel de Villena, F.; Spritz, R.A.; Marcucio, R.S.; Hallgrímsson, B. Genetic Structure of Phenotypic Robustness in the Collaborative Cross Mouse Diallel Panel. J. Evol. Biol. 2016, 29, 1737–1751. [Google Scholar] [CrossRef] [PubMed]
- White, J.K.; Gerdin, A.K.; Karp, N.A.; Ryder, E.; Buljan, M.; Bussell, J.N.; Salisbury, J.; Clare, S.; Ingham, N.J.; Podrini, C.; et al. Genome-Wide Generation and Systematic Phenotyping of Knockout Mice Reveals New Roles for Many Genes. Cell 2013, 154, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Kok, F.O.; Shin, M.; Ni, C.W.; Gupta, A.; Grosse, A.S.; vanImpel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse Genetic Screening Reveals Poor Correlation between Morpholino-Induced and Mutant Phenotypes in Zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y.R. Genetic Compensation Induced by Deleterious Mutations but Not Gene Knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Y.; Zhang, D.; Dai, X.; Estelle, M.; Zhao, Y. Auxin Binding Protein 1 (ABP1) Is Not Required for Either Auxin Signaling or Arabidopsis Development. Proc. Natl. Acad. Sci. USA 2015, 112, 2275–2280. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Ganz, K.; Powell, B.E.; Hu, Y.C.; Markoulaki, S.; Cheng, A.W.; Gao, Q.; Kim, J.; Choi, S.W.; Page, D.C.; et al. Tet1 Is Dispensable for Maintaining Pluripotency and Its Loss Is Compatible with Embryonic and Postnatal Development. Cell Stem Cell 2011, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic Compensation: A Phenomenon in Search of Mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.W.; Herzog, W.; Santoro, M.M.; Mitchell, T.S.; Frantsve, J.; Jungblut, B.; Beis, D.; Scott, I.C.; D’Amico, L.A.; Ober, E.A.; et al. A Transgene-Assisted Genetic Screen Identifies Essential Regulators of Vascular Development in Vertebrate Embryos. Dev. Biol. 2007, 307, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Driever, W.; Schier, A.F.; Neuhauss, S.C.F.; Malicki, J.; Stemple, D.L.; Stainier, D.Y.R.; Zwartkruis, F.; Abdelilah, S.; Rangini, Z.; Belak, J.; et al. A Genetic Screen for Mutations Affecting Embryogenesis in Zebrafish. Development 1996, 123, 37–46. [Google Scholar] [PubMed]
- Wang, K.; Huang, Z.; Zhao, L.; Liu, W.; Chen, X.; Meng, P.; Lin, Q.; Chi, Y.; Xu, M.; Ma, N.; et al. Large-Scale Forward Genetic Screening Analysis of Development of Hematopoiesis in Zebrafish. J. Genet. Genom. 2012, 39, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Henke, K.; Daane, J.M.; Hawkins, M.B.; Dooley, C.M.; Busch-Nentwich, E.M.; Stemple, D.L.; Harris, M.P. Genetic Screen for Postembryonic Development in the Zebrafish (Danio Rerio): Dominant Mutations Affecting Adult Form. Genetics 2017, 207, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Zon, L.I. The Art and Design of Genetic Screens: Zebrafish. Nat. Rev. Genet. 2001, 2, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Wu, S.Y.; Baek, J.I.; Choi, S.Y.; Su, Y.; Flynn, C.R.; Gamse, J.T.; Ess, K.C.; Hardiman, G.; Lipschutz, J.H.; et al. A Post-Developmental Genetic Screen for Zebrafish Models of Inherited Liver Disease. PLoS ONE 2015, 10, e0125980. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.; Fouquet, B.; Chen, J.N.; Warren, K.S.; Weinstein, B.M.; Meiler, S.E.; Mohideen, M.A.; Neuhauss, S.C.; Solnica-Krezel, L.; Schier, A.F.; et al. Mutations Affecting the Formation and Function of the Cardiovascular System in the Zebrafish Embryo. Development 1996, 123, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Sehnert, A.J.; Huq, A.; Weinstein, B.M.; Walker, C.; Fishman, M.; Stainier, D.Y.R. Cardiac Troponin T Is Essential in Sarcomere Assembly and Cardiac Contractility. Nat. Genet. 2002, 31, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Beis, D.; Bartman, T.; Jin, S.W.; Scott, I.C.; D’Amico, L.A.; Ober, E.A.; Verkade, H.; Frantsve, J.; Field, H.A.; Wehman, A.; et al. Genetic and Cellular Analyses of Zebrafish Atrioventricular Cushion and Valve Development. Development 2005, 132, 4193–4204. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, K.; Kawakami, K. Targeted Gene Expression by the Gal4-UAS System in Zebrafish. Dev. Growth Differ. 2008, 50, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.M.; Akitake, C.M.; Goll, M.G.; Rhee, J.M.; Gosse, N.; Baier, H.; Halpern, M.E.; Leach, S.D.; Parsons, M.J. Transactivation from Gal4-VP16 Transgenic Insertions for Tissue-Specific Cell Labeling and Ablation in Zebrafish. Dev. Biol. 2007, 304, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Gawdzik, J.C.; Yue, M.S.; Martin, N.R.; Elemans, L.M.H.; Lanham, K.A.; Heideman, W.; Rezendes, R.; Baker, T.R.; Taylor, M.R.; Plavicki, J.S. Sox9b Is Required in Cardiomyocytes for Cardiac Morphogenesis and Function. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Wang, Z.; Hirata, H.; Sehara-Fujisawa, A. Integrin B1 Activity Is Required for Cardiovascular Formation in Zebrafish. Genes Cells 2018, 23, 938–951. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective Targeted Gene “knockdown” in Zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Ekker, S.C.; Larson, J.D. Morphant Technology in Model Developmental Systems. Genesis 2001, 30, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for Morpholino Use in Zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; McCammon, J.M.; Miller, J.C.; Faraji, F.; Ngo, C.; Katibah, G.E.; Amora, R.; Hocking, T.D.; Zhang, L.; Rebar, E.J.; et al. Heritable Targeted Gene Disruption in Zebrafish Using Designed Zinc-Finger Nucleases. Nat. Biotechnol. 2008, 26, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Noyes, M.B.; Zhu, L.J.; Lawson, N.D.; Wolfe, S.A. Targeted Gene Inactivation in Zebrafish Using Engineered Zinc-Finger Nucleases. Nat. Biotechnol. 2008, 26, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Cade, L.; Reyon, D.; Hwang, W.Y.; Tsai, S.Q.; Patel, S.; Khayter, C.; Joung, J.K.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.J. Highly Efficient Generation of Heritable Zebrafish Gene Mutations Using Homo-and Heterodimeric TALENs. Nucleic Acids Res. 2012, 40, 8001–8010. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Carrington, B.; Bishop, K.; Jones, M.P.; Rissone, A.; Candotti, F.; Chandrasekharappa, S.C.; Liu, P. Efficient Methods for Targeted Mutagenesis in Zebrafish Using Zinc-Finger Nucleases: Data from Targeting of Nine Genes Using CompoZr or CoDA ZFNs. PLoS ONE 2013, 8, e0057239. [Google Scholar] [CrossRef] [PubMed]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G.; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.H.; et al. In Vivo Genome Editing Using a High-Efficiency TALEN System. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient Design and Assembly of Custom TALEN and Other TAL Effector-Based Constructs for DNA Targeting. Nucleic Acids Res. 2011, 39, 1–11. [Google Scholar] [CrossRef]
- Varshney, G.K.; Pei, W.; LaFave, M.C.; Idol, J.; Xu, L.; Gallardo, V.; Carrington, B.; Bishop, K.; Jones, M.P.; Li, M.; et al. High-Throughput Gene Targeting and Phenotyping in Zebrafish Using CRISPR/Cas9. Genome Res. 2015, 25, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient Genome Editing in Zebrafish Using a CRISPR-Cas System. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Kettleborough, R.N.W.; Busch-Nentwich, E.M.; Harvey, S.A.; Dooley, C.M.; De Bruijn, E.; Van Eeden, F.; Sealy, I.; White, R.J.; Herd, C.; Nijman, I.J.; et al. A Systematic Genome-Wide Analysis of Zebrafish Protein-Coding Gene Function. Nature 2013, 496, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Tessadori, F.; Roessler, H.I.; Savelberg, S.M.C.; Chocron, S.; Kamel, S.M.; Duran, K.J.; van Haelst, M.M.; van Haaften, G.; Bakkers, J. Effective CRISPR/Cas9-Based Nucleotide Editing in Zebrafish to Model Human Genetic Cardiovascular Disorders. Dis. Model. Mech. 2018, 11, dmm035469. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.; Mali, P.; Moosburner, M.; Church, G.M. Unraveling CRISPR-Cas9 Genome Engineering Parameters via a Library-on-Library Approach. Nat. Methods 2015, 12, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 Variants with Broad PAM Compatibility and High DNA Specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kruse, F.; Junker, J.P.; van Oudenaarden, A.; Bakkers, J. Tomo-Seq: A Method to Obtain Genome-Wide Expression Data with Spatial Resolution; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; Volume 135. [Google Scholar] [CrossRef]
- Junker, J.P.; Noël, E.S.; Guryev, V.; Peterson, K.A.; Shah, G.; Huisken, J.; McMahon, A.P.; Berezikov, E.; Bakkers, J.; Van Oudenaarden, A. Genome-Wide RNA Tomography in the Zebrafish Embryo. Cell 2014, 159, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Kruse, F.; Vasudevarao, M.D.; Junker, J.P.; Zebrowski, D.C.; Fischer, K.; Noël, E.S.; Grün, D.; Berezikov, E.; Engel, F.B.; et al. Spatially Resolved Genome-Wide Transcriptional Profiling Identifies BMP Signaling as Essential Regulator of Zebrafish Cardiomyocyte Regeneration. Dev. Cell 2016, 36, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, S.B.; Bakkers, J. Spatially Resolved RNA-Sequencing of the Embryonic Heart Identifies a Role for Wnt/β-Catenin Signaling in Autonomic Control of Heart Rate. Elife 2018, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Grunow, B.; Mohamet, L.; Shiels, H.A. Generating an in Vitro 3D Cell Culture Model from Zebrafish Larvae for Heart Research. J. Exp. Biol. 2015, 218, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Poss, K.D. Explant Culture of Adult Zebrafish Hearts for Epicardial Regeneration Studies. Nat. Protoc. 2016, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, B.J.; Sala, L.; Tertoolen, L.G.J.; Smith, G.L.; Burton, F.L.; Mummery, C.L. Quantification of Muscle Contraction In Vitro and In Vivo Using MUSCLEMOTION Software: From Stem Cell-Derived Cardiomyocytes to Zebrafish and Human Hearts. Curr. Protoc. Hum. Genet. 2018, 99, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Stouffer, G.A.; Kucharska-newton, A.M.; Qamar, A.; Vaduganathan, M.; Pandey, A.; Mph, D.L.B.; Caughey, M.C. Twenty Year Trends and Sex Differences in Young Adults Hospitalized with Acute Myocardial Infarction: The ARIC Community Surveillance Study. Circulation 2018. [Google Scholar] [CrossRef] [PubMed]
- Sorbets, E.; Steg, P.G.; Young, R.; Danchin, N.; Greenlaw, N.; Ford, I.; Tendera, M.; Ferrari, R.; Merkely, B.; Parkhomenko, A.; et al. β-Blockers, Calcium Antagonists, and Mortality in Stable Coronary Artery Disease: An International Cohort Study. Eur. Heart J. 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pencina, M.J.; Navar, A.M.; Wojdyla, D.; Sanchez, R.J.; Khan, I.; Elassal, J.; D’Agostino, R.B., Sr.; Peterson, E.D.; Sniderman, A.D. Quantifying Importance of Major Risk Factors for Coronary Heart Disease. Circulation 2018. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.E.; Sanghvi, M.M.; Aung, N.; Cooper, J.A.; Paiva, M.; Zemrak, F.; Fung, K.; Lukaschuk, E.; Lee, A.M.; Carapella, V.; et al. The Impact of Cardiovascular Risk Factors on Cardiac Structure and Function: Insights from the UK Biobank Imaging Enhancement Study. PLoS ONE 2017, 12, e0185114. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2014, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schaid, D.J.; Chen, W.; Larson, N.B. From Genome-Wide Associations to Candidate Causal Variants by Statistical Fine-Mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; Webb, T.R.; et al. A Comprehensive 1000 Genomes—Based Genome-Wide Association Meta-Analysis of Coronary Artery Disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.R.; Erdmann, J.; Stirrups, K.E.; Stitziel, N.O.; Masca, N.G.D.; Jansen, H.; Kanoni, S.; Nelson, C.P.; Ferrario, P.G.; Eicher, J.D.; et al. Systematic Evaluation of Pleiotropy Identifies 6 Further Loci Associated With Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69. [Google Scholar] [CrossRef] [PubMed]
- Van Opbergen, C.J.M.; van der Voorn, S.M.; Vos, M.A.; de Boer, T.P.; van Veen, T.A.B. Cardiac Ca2+signalling in Zebrafish: Translation of Findings to Man. Prog. Biophys. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.T.; Hsieh, C.S.; Chang, S.N.; Chuang, E.Y.; Ueng, K.C.; Tsai, C.F.; Lin, T.H.; Wu, C.K.; Lee, J.K.; Lin, L.Y.; et al. Genome-Wide Screening Identifies a KCNIP1 Copy Number Variant as a Genetic Predictor for Atrial Fibrillation. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Hayashi, K.; Fujino, N.; Konno, T.; Tada, H.; Nakanishi, C. Functional Analysis of KCNH2 Gene Mutations of Type 2 Long QT Syndrome in Larval Zebrafish Using Microscopy and Electrocardiography. Heart Vessels 2018. [Google Scholar] [CrossRef] [PubMed]
- Tucker, N.R.; Dolmatova, E.V.; Lin, H.; Cooper, R.R.; Ye, J.; Hucker, W.J.; Jameson, H.S.; Parsons, V.A.; Weng, L.C.; Mills, R.W.; et al. Diminished PRRX1 Expression Is Associated with Increased Risk of Atrial Fibrillation and Shortening of the Cardiac Action Potential. Circ. Cardiovasc. Genet. 2017, 10, e001902. [Google Scholar] [CrossRef] [PubMed]
- Mercer, E.J.; Lin, Y.F.; Cohen-Gould, L.; Evans, T. Hspb7 Is a Cardioprotective Chaperone Facilitating Sarcomeric Proteostasis. Dev. Biol. 2018, 435, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Farr, G.H., 3rd; Imani, K.; Pouv, D.; Maves, L. Functional Testing of a Human PBX3 Variant in Zebrafish Reveals a Potential Modifier Role in Congenital Heart Defects. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Kalogirou, S.; Malissovas, N.; Moro, E.; Argenton, F.; Stainier, D.Y.R.; Beis, D. Intracardiac Flow Dynamics Regulate Atrioventricular Valve Morphogenesis. Cardiovasc. Res. 2014, 104, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Dina, C.; Bouatia-Naji, N.; Tucker, N.; Delling, F.N.; Toomer, K.; Durst, R.; Perrocheau, M.; Fernandez-Friera, L.; Solis, J.; Le Tourneau, T.; et al. Genetic Association Analyses Highlight Biological Pathways Underlying Mitral Valve Prolapse. Nat. Genet. 2015, 47, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Ferese, R.; Bonetti, M.; Consoli, F.; Guida, V.; Sarkozy, A.; Lepri, F.R.; Versacci, P.; Gambardella, S.; Calcagni, G.; Margiotti, K.; et al. Heterozygous Missense Mutations in NFATC1 Are Associated with Atrioventricular Septal Defect. Hum. Mutat. 2018, 39, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Von der Heyde, B.; Emmanouilidou, A.; Klingström, T.; Mazzaferro, E.; Vicenzi, S.; Jumaa, S.; Dethlefsen, O.; Snieder, H.; de Geus, E.; Ingelsson, E.; et al. In Vivo Characterization of Candidate Genes for Heart Rate Variability Identifies Culprits for Sinoatrial Pauses and Arrests. bioRxiv 2018, 1–5. [Google Scholar] [CrossRef]
- Rice, T.; Vogler, G.P.; Perrvb, T.S.; Laskarzewski, P.M. Familial Aggregation of Lipids and Lipoproteins in Families Ascertained through Random and Nonrandom Probands in the Iowa Lipid Research Clinics Family Study. Hum. Hered. 1991, 41, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Damrauer, S.M.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; Duvall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of Blood Lipids among ~300,000 Multiethnic Participants of the Million Veteran Program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, P.; Peloso, G.M.; Zekavat, S.M.; Montasser, M.; Ganna, A.; Chaf, M.; Khera, A.V.; Zhou, W.; Bloom, J.M.; Engreitz, J.M.; et al. Deep-Coverage Whole Genome Sequences and Blood Lipids among 16,324 Individuals. Nat. Commun. 2018, 9, 3391. [Google Scholar] [CrossRef] [PubMed]
- De Ferranti, S.D.; Rodday, A.M.; Mendelson, M.M.; Wong, J.B.; Leslie, L.K.; Sheldrick, R.C. Prevalence of Familial Hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition. Circulation 2016, 133, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.J.; Peloso, G.M.; Yu, H.; Butterworth, A.S.; Wang, X.; Mahajan, A.; Saleheen, D.; Emdin, C.; Alam, D.; Alves, A.C.; et al. Exome-Wide Association Study of Plasma Lipids in >300,000 Individuals. Nat. Genet. 2017, 49, 1758–1766. [Google Scholar] [CrossRef]
- Surakka, I.; Horikoshi, M.; Mägi, R.; Sarin, A.; Mahajan, A.; Lagou, V.; Marullo, L.; Ferreira, T.; Miraglio, B.; Timonen, S.; et al. The Impact of Low-Frequency and Rare Variants on Lipid Levels. Nat. Genet. 2015, 47, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Montasser, M.E.; O’Hare, E.A.; Wang, X.; Howard, A.D.; McFarland, R.; Perry, J.A.; Ryan, K.A.; Rice, K.; Jaquish, C.E.; Shuldiner, A.R.; et al. An APOO Pseudogene on Chromosome 5q Is Associated With Low-Density Lipoprotein Cholesterol Levels. Circulation 2018, 138, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Minchin, J.E.N.; Rawls, J.F. Elucidating the Role of Plexin D1 in Body Fat Distribution and Susceptibility to Metabolic Disease Using a Zebrafish Model System. Adipocyte 2017, 6, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Louw, J.J.; Nunes Bastos, R.; Chen, X.; Verdood, C.; Corveleyn, A.; Jia, Y.; Breckpot, J.; Gewillig, M.; Peeters, H.; Santoro, M.M.; et al. Compound Heterozygous Loss-of-Function Mutations in KIF20A Are Associated with a Novel Lethal Congenital Cardiomyopathy in Two Siblings. PLoS Genet. 2018, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Uva, G.D.; Aharonov, A.; Lauriola, M.; Kain, D.; Yahalom-ronen, Y.; Carvalho, S.; Weisinger, K.; Bassat, E.; Rajchman, D.; Yifa, O.; et al. ERBB2 Triggers Mammalian Heart Regeneration by Promoting Cardiomyocyte Dedifferentiation and Proliferation. Nat. Cell Biol. 2015, 17, 627. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 Regulates Postnatal Cardiomyocyte Cell Cycle Arrest. Nature 2013, 497, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Polizzotti, B.D.; Ganapathy, B.; Walsh, S.; Choudhury, S.; Ammanamanchi, N.; Bennett, D.G.; Remedios, C.G.; Haubner, B.J.; Penninger, J.M.; Kühn, B. Neuregulin Stimulation of Cardiomyocyte Regeneration in Mice and Human Myocardium Reveals a Therapeutic Window. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.; Lechene, C.P.; Lee, R.T. Mammalian Heart Renewal by Pre-Existing Cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Vujic, A.; Lerchenmüller, C.; Wu, T.; Guillermier, C.; Rabolli, C.P.; Gonzalez, E.; Senyo, S.E.; Liu, X.; Steinhauser, M.L.; Lee, R.T.; et al. Exercise Induces New Cardiomyocyte Generation in the Adult Mammalian Heart. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.Y.; Silberstein, L.E.; dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte Proliferation Contributes to Heart Growth in Young Humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.L.; Marín, R.; Didier, J. Immune Responses in Cardiac Repair and Regeneration: A Comparative Point of View. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.E.; Bakovic, M.; Karra, R. Endothelial Contributions to Zebrafish Heart Regeneration. J. Cardiovasc. Dev. Dis. 2018, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.P.; Poss, K.D. The Epicardium as a Hub for Heart Regeneration. Nat. Rev. Cardiol. 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Marín-juez, R.; Marass, M.; Gauvrit, S.; Rossi, A.; Lai, S.; Materna, S.C. Fast Revascularization of the Injured Area Is Essential to Support Zebrafish Heart Regeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 11237–11242. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.L.; Marin-Juez, R.; Moura, P.L.; Kuenne, C.; Lai, J.K.H.; Tsedeke, A.T.; Guenther, S.; Looso, M.; Stainier, D. Reciprocal Analyses in Zebrafish and Medaka Reveal That Harnessing the Immune Response Promotes Cardiac Regeneration. Elife 2017, 6, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Simões, F.C.; Riley, R.P. The Ontogeny, Activation and Function of the Epicardium during Heart Development and Regeneration. Development 2018, 145, dev155994. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-iranzo, H.; Galardi-castilla, M.; Sanz-morejón, A.; González-rosa, J.M. Transient Fibrosis Resolves via Fibroblast Inactivation in the Regenerating Zebrafish Heart. Proc. Natl. Acad. Sci. USA 2018, 115, 4188–4193. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Lindbom, L. Phagocyte Partnership during the Onset and Resolution of Inflammation. Nat. Publ. Gr. 2010, 10, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.P.; Sheng, D.Z.; Sugimoto, K.; Gonzalez-Rajal, A.; Nakagawa, S.; Hesselson, D.; Kikuchi, K. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev. Cell 2017, 43, 659–672.e5. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, W.T.; Lemieux, M.E.; Killen, A.C.; Van Aerle, R.; Yamamoto, Y.; Mommersteeg, M.T.M.; Stockdale, W.T.; Lemieux, M.E.; Killen, A.C.; Zhao, J.; et al. Heart Regeneration in the Mexican Cavefish Report Heart Regeneration in the Mexican Cavefish. Cell Rep. 2018, 25, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Furtado, M.B.; Rosenthal, N. The Interstitium in Cardiac Repair: Role of the Immune—Stromal Cell Interplay. Nat. Rev. Cardiol. 2018, 15, 601–616. [Google Scholar] [CrossRef] [PubMed]
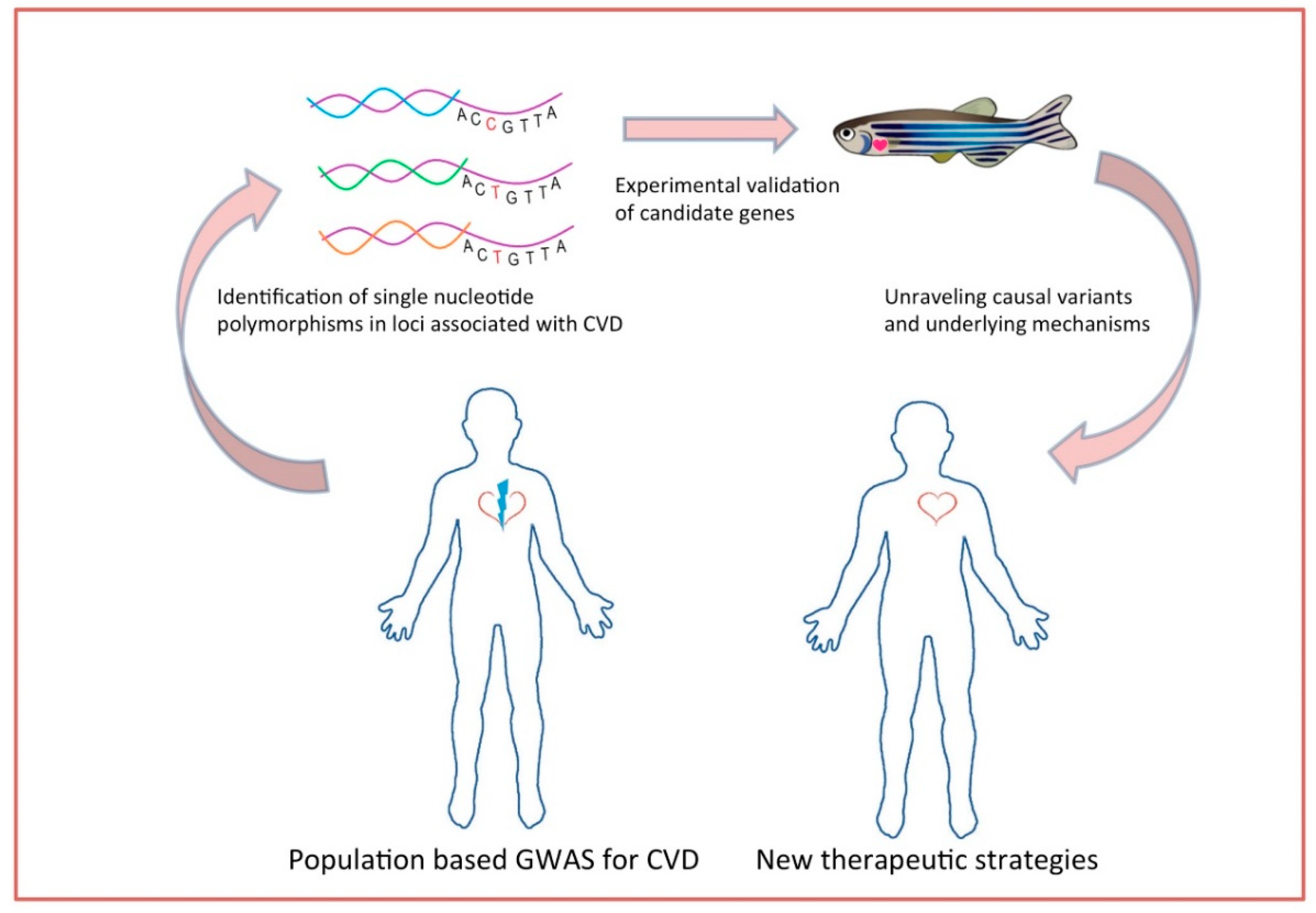

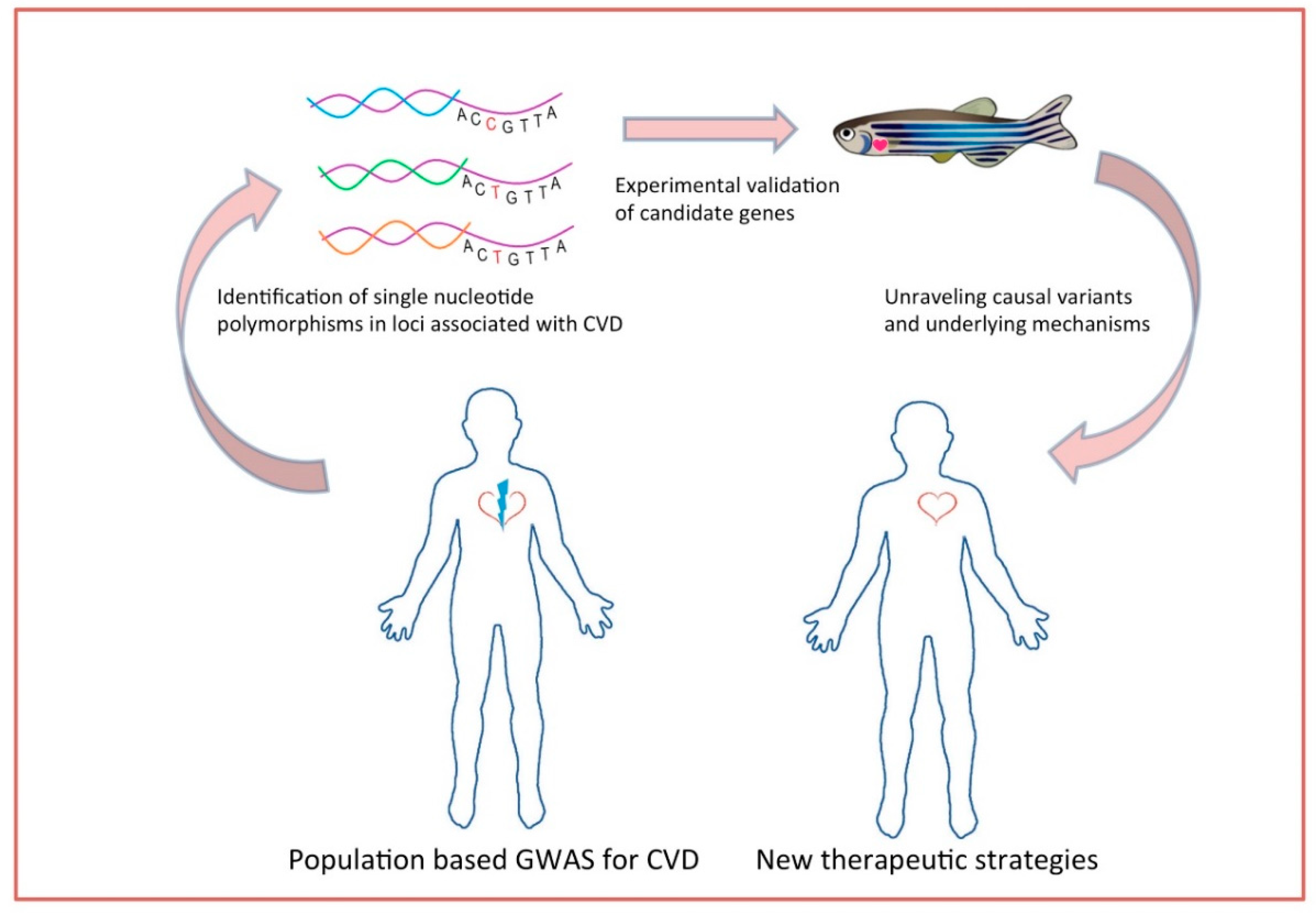
{kind=link}
{kind=link}
| Associated Human Disease | Gene (s) | Zebrafish Genotype | References |
|---|---|---|---|
| Atrial Fibrillation | KCNIP1 | High atrial rate | [80] |
| Long QT Syndrome | KCNH2 | Severe repolarization | [81] |
| Atrial Fibrillation | PRRX1 | Atrial action potential duration | [82] |
| Dilated Cardiomyopathy | HSPB7 | Cardiac fibrosis, cardiomegaly and sarcomeric abnormalities | [83] |
| Congenital Heart Defects | PBX3 | Myocardial morphogenesis defects | [84] |
| Mitral Valve Prolapse | LMCD1, TNS1 | Increased atrioventricular regurgitation, moderate reduction in cardiac looping | [86] |
| Atrioventricular Septal Defect | NFATC1 | Cardiac looping defects and altered atrioventricular canal patterning | [87] |
| Heart Rate Variability | GNG11, SYT10, RGS6, HCN4, NEO1, KIAA1755 | Sinoatrial pauses and arrests, cardiac edema and uncontrolled atrial contractions | [88] |
| Lipid Associated-Cardiomyopathy | APOOP1 | Increased the LDL-C levels, increase in the average number of vascular plaques | [95] |
| Lipid Associated-Cardiomyopathy | PLXND1 | Modulate angiogenesis, reduced capacity to store lipid in visceral adipose tissue | [96] |
| Congenital Cardiomyopathy | KIF20A | Relative tachycardia, red blood cells proximal to the atrium and cardiac edema | [97] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giardoglou, P.; Beis, D. On Zebrafish Disease Models and Matters of the Heart. Biomedicines 2019, 7, 15. https://doi.org/10.3390/biomedicines7010015
Giardoglou P, Beis D. On Zebrafish Disease Models and Matters of the Heart. Biomedicines. 2019; 7(1):15. https://doi.org/10.3390/biomedicines7010015
Chicago/Turabian StyleGiardoglou, Panagiota, and Dimitris Beis. 2019. "On Zebrafish Disease Models and Matters of the Heart" Biomedicines 7, no. 1: 15. https://doi.org/10.3390/biomedicines7010015
APA StyleGiardoglou, P., & Beis, D. (2019). On Zebrafish Disease Models and Matters of the Heart. Biomedicines, 7(1), 15. https://doi.org/10.3390/biomedicines7010015