Pyrrole-Mediated Peptide Cyclization Identified through Genetically Reprogrammed Peptide Synthesis


Abstract
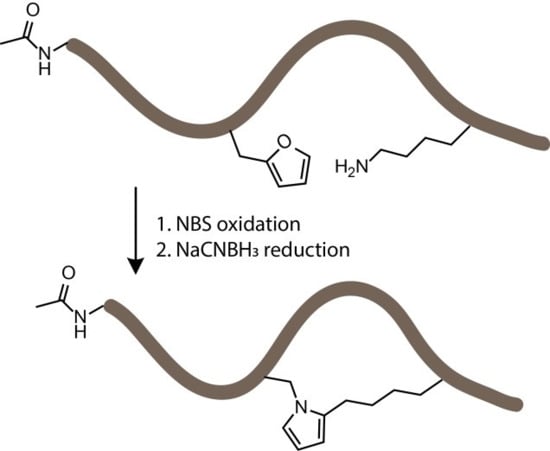
1. Introduction
2. Experimental Section
2.1. Synthesis of Cyanomethyl Ester (CME)-Activated Amino Acid Substrates
2.2. Flexible In Vitro Translation
2.3. Solid-Phase Peptide Synthesis
3. Results and Discussion
3.1. FIT-Based Synthesis of Furan-Modified Peptides
3.2. NBS Oxidation of Furan-Containing Peptides Obtained through FIT
3.3. Peptide Scale up by SPPS and Subsequent NBS Oxidation
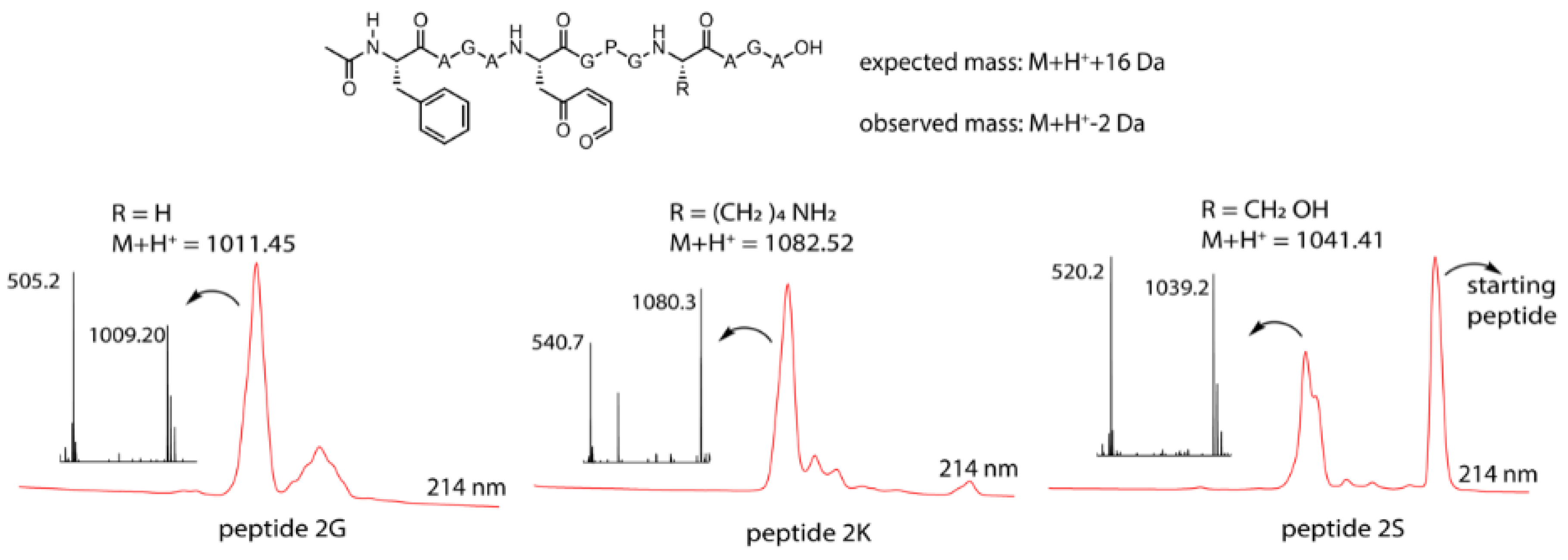
3.4. Identification of a Pyrrole Moiety as Cyclisation Motif
3.5. Extending the Scope of Pyrrole-Mediated Cyclisation: Varying the Positioning and Conformation within the Template Peptide
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C. Constrained peptides’ time to shine? Nat. Rev. Drug Discov. 2018, 17, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Torres, O.; Yüksel, D.; Bernardina, M.; Kumar, K.; Bong, D. Peptide tertiary structure nucleation by side-chain crosslinking with metal complexation and double “click” cycloaddition. ChemBioChem 2008, 9, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Kumita, J.R.; Flint, D.G.; Woolley, G.A.; Smart, O.S. Achieving photo-control of protein conformation and activity: Producing a photo-controlled leucine zipper. Faraday Discuss. 2002, 122, 89–103. [Google Scholar] [CrossRef]
- Lautrette, G.; Touti, F.; Lee, H.G.; Dai, P.; Pentelute, B.L. Nitrogen arylation for macrocyclization of unprotected peptides. J. Am. Chem. Soc. 2016, 138, 8340–8343. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Meinhardt, N.; Wu, Y.; Kulkarni, S.; Hu, X.; Low, K.E.; Davies, P.L.; Degrado, W.F.; Greenbaum, D.C. Development of α-helical calpain probes by mimicking a natural protein-protein interaction. J. Am. Chem. Soc. 2012, 134, 17704–17713. [Google Scholar] [CrossRef] [PubMed]
- Fairlie, D.P.; Dantas de Araujo, A. Review stapling peptides using cysteine crosslinking. Pept. Sci. 2016, 106, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Chorev, M.; Roubini, E.; McKee, R.L.; Gibbons, S.W.; Goldman, M.E.; Caulfield, M.P.; Rosenblatt, M. Cyclic parathyroid hormone related protein antagonists: Lysine 13 to aspartic acid 17 [I to (I + 4)] side chain to side chain lactamization. Biochemistry 1991, 30, 5968–5974. [Google Scholar] [CrossRef] [PubMed]
- Van Lysebetten, D.; Felissati, S.; Antonatou, E.; Carrette, L.L.G.; Espeel, P.; Focquet, E.; Du Prez, F.E.; Madder, A. A thiolactone strategy for straightforward synthesis of disulfide-linked side-chain-to-tail cyclic peptides featuring an N-terminal modification handle. ChemBioChem 2018, 19, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Walsh, C.T.; Burkart, M.D. Biomimetic synthesis and optimization of cyclic peptide antibiotics. Nature 2002, 418, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Grossmann, T.N.; Verdine, G.L. Synthesis of all-hydrocarbon stapled α-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011, 6, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.A.; Coleska, A.; Ran, X.; Yi, H.; Yang, C.Y.; Wang, S. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell cll/lymphoma 9 (BCL9) protein-protein interaction. J. Med. Chem. 2012, 55, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Madden, M.M.; Muppidi, A.; Li, Z.; Li, X.; Chen, J.; Lin, Q. Synthesis of cell-permeable stapled peptide dual inhibitors of the p53-Mdm2/Mdmx interactions via photoinduced cycloaddition. Bioorg. Med. Chem. Lett. 2011, 21, 1472–1475. [Google Scholar] [CrossRef] [PubMed]
- Cistrone, P.A.; Silvestri, A.P.; Hintzen, J.C.J.; Dawson, P.E. Rigid peptide macrocycles from on-resin glaser stapling. ChemBioChem 2018, 19, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Haney, C.M.; Loch, M.T.; Horne, W.S. Promoting peptide α-helix formation with dynamic covalent oxime side-chain cross-links. Chem. Commun. 2011, 47, 10915–10917. [Google Scholar] [CrossRef] [PubMed]
- Kamens, A.J.; Mientkiewicz, K.M.; Eisert, R.J.; Walz, J.A.; Mace, C.R.; Kritzer, J.A. Thioether-stapled macrocyclic inhibitors of the EH domain of EHD1. Bioorg. Med. Chem. 2018, 26, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Malins, L.R.; Degruyter, J.N.; Robbins, K.J.; Scola, P.M.; Eastgate, M.D.; Ghadiri, M.R.; Baran, P.S. Peptide macrocyclization inspired by non-ribosomal imine natural products. J. Am. Chem. Soc. 2017, 139, 5233–5241. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Katoh, T.; Suga, H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011, 6, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, Y.; Ashigai, H.; Goto, Y.; Murakami, H.; Suga, H. Ribosomal synthesis of cyclic peptides with a fluorogenic oxidative coupling reaction. ChemBioChem 2009, 10, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ohta, A.; Sako, Y.; Yamagishi, Y.; Murakami, H.; Suga, H. Reprogramming the translation initiation for the synthesis of physiologically stable cyclic peptides. ACS Chem. Biol. 2008, 3, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Sako, Y.; Goto, Y.; Murakami, H.; Suga, H. Ribosomal synthesis of peptidase-resistant peptides closed by a nonreducible inter-side-chain bond. ACS Chem. Biol. 2008, 3, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Sako, Y.; Morimoto, J.; Murakami, H.; Suga, H. Ribosomal synthesis of bicyclic peptides via two orthogonal inter-side-chain reactions. J. Am. Chem. Soc. 2008, 130, 7232–7234. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Iwasaki, K.; Torikai, K.; Murakami, H.; Suga, H. Ribosomal synthesis of dehydrobutyrine- and methyllanthionine-containing peptides. Chem. Commun. 2009, 3419–3421. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Ohta, A.; Ohuchi, M.; Ashigai, H.; Murakami, H.; Suga, H. Diverse backbone-cyclized peptides via codon reprogramming. Nat. Chem. Biol. 2009, 5, 888–890. [Google Scholar] [CrossRef] [PubMed]
- Deceuninck, A.; Madder, A. From DNA cross-linking to peptide labeling: On the versatility of the furan-oxidation-conjugation strategy. Chem. Commun. 2009, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Antonatou, E.; Hoogewijs, K.; Kalaitzakis, D.; Baudot, A.; Vassilikogiannakis, G.; Madder, A. Singlet oxygen-induced furan oxidation for site-specific and chemoselective peptide ligation. Chem. A Eur. J. 2016, 22, 8457–8461. [Google Scholar] [CrossRef] [PubMed]
- Vannecke, W.; Ampe, C.; Van Troys, M.; Beltramo, M.; Madder, A. Cross-linking furan-modified kisspeptin-10 to the KISS receptor. ACS Chem. Biol. 2017, 12, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ashigai, H.; Sako, Y.; Murakami, H.; Suga, H. Translation initiation by using various N-acylaminoacyl tRNAs. Nucleic Acids Symp. Ser. 2006, 50, 293–294. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Peptide Name and Sequence | Calculated Exact Mass (Da) | M + H+ (Da) | Experimental M + Na+ (Da) | M + K+ (Da) |
|---|---|---|---|---|---|
| 1C | AcFAGAFuaGPGCAGA | 1056.43 | / | 1079.58 | 1095.55 |
| 1H | AcFAGAFuaGPGHAGA | 1090.48 | 1091.66 | 1113.67 | 1129.68 |
| 1K | AcFAGAFuaGPGKAGA | 1081.52 | 1082.69 | 1104.69 | 1120.66 |
| 1R | AcFAGAFuaGPGRAGA | 1109.53 | 1110.75 | 1132.71 | 1148.70 |
| 1S | AcFAGAFuaGPGSAGA | 1040.46 | / | 1063.65 | 1079.63 |
| 1Y | AcFAGAFuaGPGYAGA | 1116.49 | / | 1139.70 | 1155.69 |

| Peptides 1 Peptides 2 | M + H+ | M + Na+ | M + K+ | Other |
| 1C 2C | / | / | / | 1214.97 (K+ + cysteinylation) 1230.97 (K+ + cysteinylation + 16) |
| 1H 2H | / | / | 1130.18 1146.21 | / |
| 1K 2K | 1082.85 | 1105.13 | 1121.06 1137.09 | 1062.11 (M + H − 20 Da) |
| 1R 2R | 1111.13 1127.17 | / | 1149.17 | / |
| 1S 2S | / | 1064.06 | 1080.00 1096.00 | 1021.43 (M + H − 20 Da) |
| 1Y 2Y | / | / | / | 1313.91 (dibromination) 1329.87 (dibromination + 16) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decoene, K.W.; Vannecke, W.; Passioura, T.; Suga, H.; Madder, A. Pyrrole-Mediated Peptide Cyclization Identified through Genetically Reprogrammed Peptide Synthesis. Biomedicines 2018, 6, 99. https://doi.org/10.3390/biomedicines6040099
Decoene KW, Vannecke W, Passioura T, Suga H, Madder A. Pyrrole-Mediated Peptide Cyclization Identified through Genetically Reprogrammed Peptide Synthesis. Biomedicines. 2018; 6(4):99. https://doi.org/10.3390/biomedicines6040099
Chicago/Turabian StyleDecoene, Klaas W., Willem Vannecke, Toby Passioura, Hiroaki Suga, and Annemieke Madder. 2018. "Pyrrole-Mediated Peptide Cyclization Identified through Genetically Reprogrammed Peptide Synthesis" Biomedicines 6, no. 4: 99. https://doi.org/10.3390/biomedicines6040099
APA StyleDecoene, K. W., Vannecke, W., Passioura, T., Suga, H., & Madder, A. (2018). Pyrrole-Mediated Peptide Cyclization Identified through Genetically Reprogrammed Peptide Synthesis. Biomedicines, 6(4), 99. https://doi.org/10.3390/biomedicines6040099