As a result of the novel techniques for their synthesis and screening, a number of high affinity thioether monocyclic peptide ligands have been identified in recent years, and for several of these, high resolution x-ray crystal structures have been obtained in complex with a protein target. To the best of our knowledge, all of the co-crystal structures of the monocyclic thioether closed peptide ligands reported to date have involved peptides synthesized in genetically reprogrammed reactions, so as to include N-terminal chloroacetyl groups that cyclize with downstream Cys resides. Co-crystal structures of such peptides bound to transmembrane transporters, enzymes, and other proteins (involved in protein-protein interactions) have been reported, and these are discussed in detail below.
2.2.1. Transporters
In the case of the transmembrane transport proteins, thioether cyclized peptide ligands have been used specifically as co-crystallization ligands in order to facilitate the crystallographic analysis of these relatively intractable proteins. In the first example of this, high affinity macrocyclic peptide ligands were identified to the
Pyrococcus furiosus multidrug and toxic compound extrusion (PfMATE) transporter, a representative member of a diverse family of xenobiotic efflux proteins that confer multidrug resistance to microbial pathogens and neoplastic cells [
4,
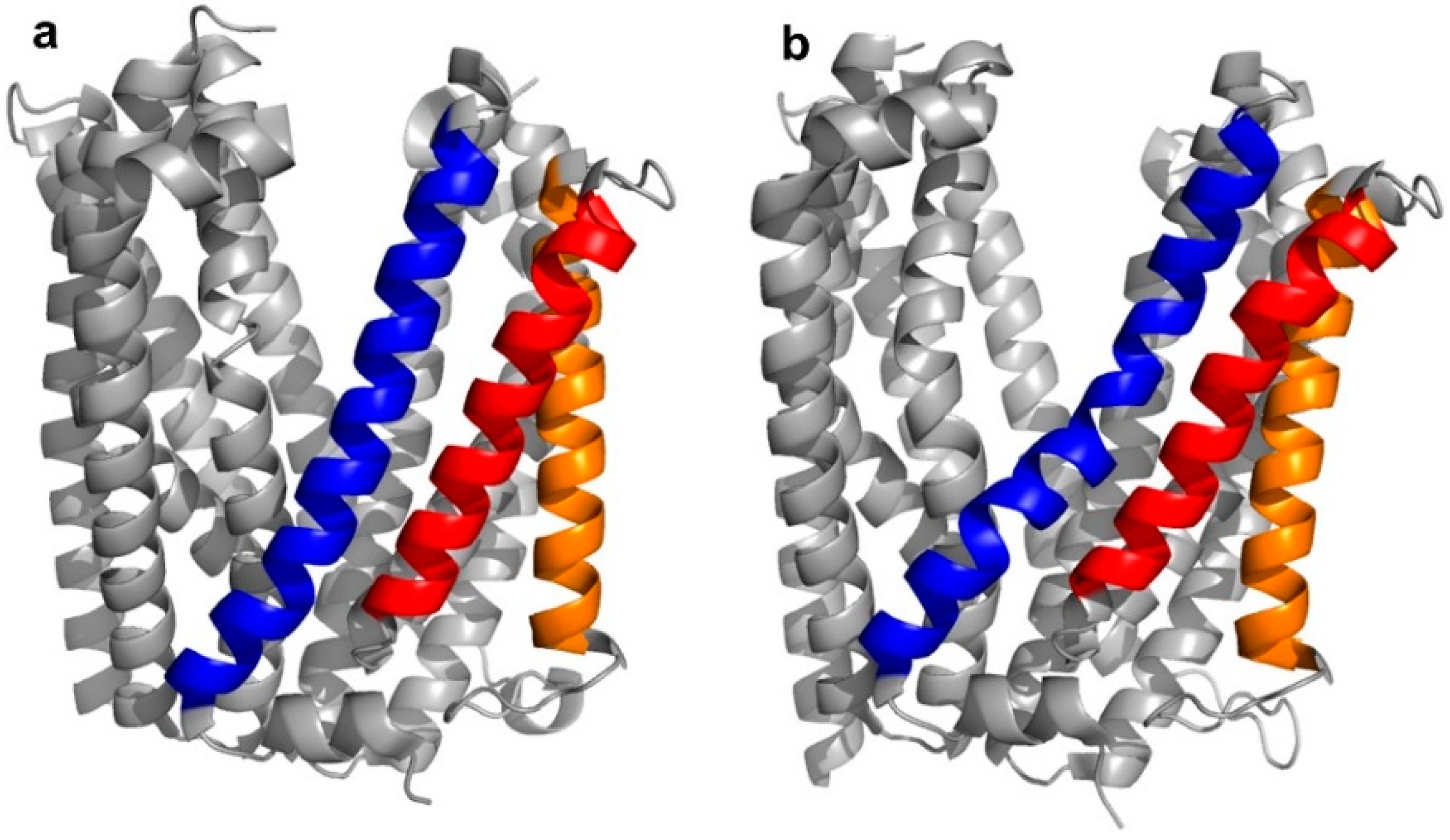
5]. Structurally, the transporter was found to adopt two different “straight” or “bent” conformations, according to the spatial arrangement of its transmembrane (TM) domains, in particular, TM1, TM5, and TM6 (
Figure 3a,b).
Four peptide ligands to PfMATE were identified (MaD5, MaD3S, MaD8, and MaL6), and each of these exhibited a strong inhibition of PfMATE extrusion, suggesting their possible use as inhibitors as well as co-crystalization ligands. Three of these (MaD5, MaD3S, and MaD8) included a single
d-Phe residue and exhibited “lariat” structures with relatively small N-terminal macrocyclic regions (5–7 residues) and longer C-terminal tails (9–13 residues). By contrast, in the MaL6 peptide (which did not include any
d- residues), all 17 residues were included in the macrocyclic structure. The co-crystal structures of these peptides bound to PfMATE showed that MaD5 and MaD3S bound within a deep central cleft pocket of a straight conformation-locked PfMATE, with the macrocyclic region of each peptide occupying a substrate recognition site (the N-lobe cavity). The interaction of the macrocyclic domains with PfMATE was mainly mediated through hydrophobic interactions, with the C-terminal tails adopting disordered positions that were not completely defined (
Figure 4a). In contrast, MaL6 and MaD8 were shown to bind to the extracellular opening of the TM domains, with MaD8 binding to a site deep within the channel, and MaL6 binding to the outward face of the transporter (
Figure 4b). The distinct binding mode of each peptide suggested distinct mechanisms of inhibition, with the long tails of the lariat, for example, appearing to restrict the motion of the N- and C-terminal lobes necessary for conformational changes during extrusion. Additionally, the peptides were found to reach the transporter intracellularly after penetrating the bacterial membrane, proceeding to inhibit the transporter’s function through spatial blocking or through restriction of the TM’s dynamic movement. Overall, these studies allowed for the identification of cyclic peptides that both inhibited PfMATE and facilitated its crystallization, providing insight into both the modes of macrocyclic peptide binding and inhibition, and the mechanism of the transporter’s activity.
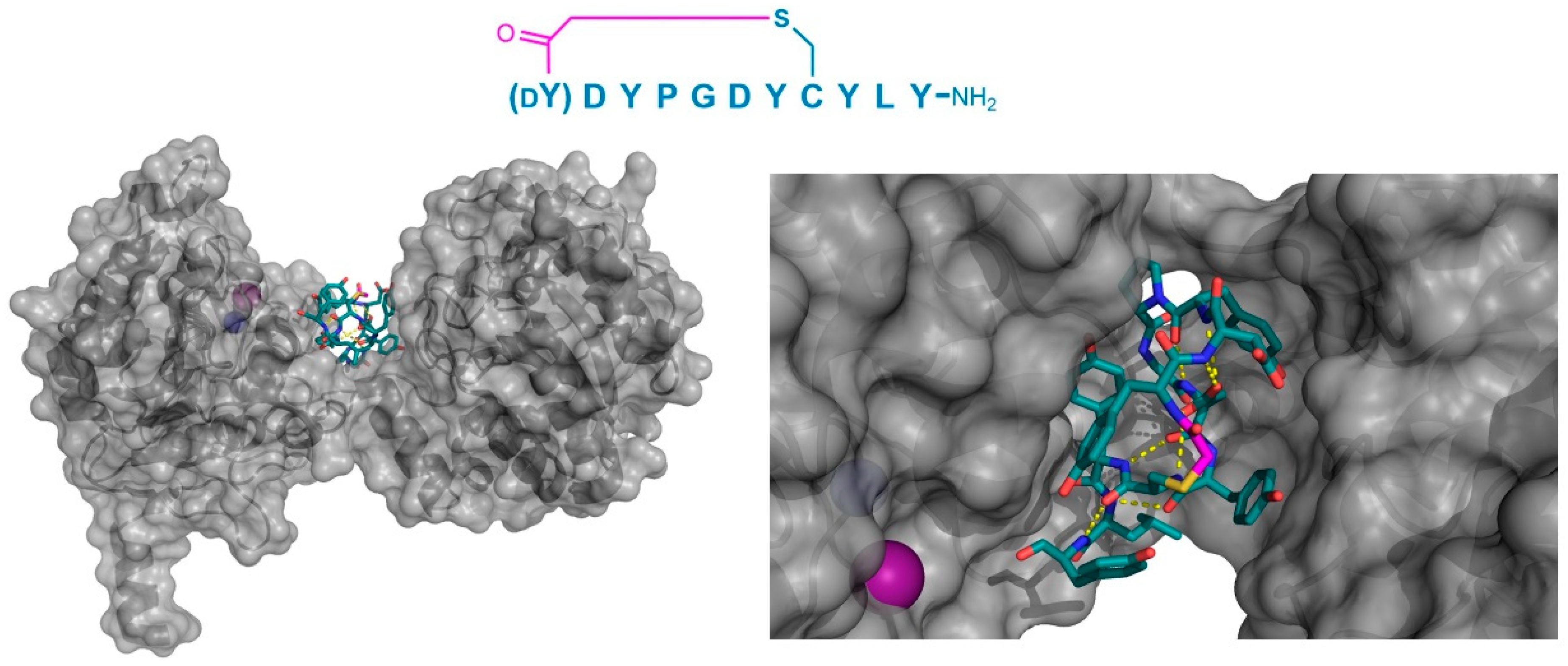
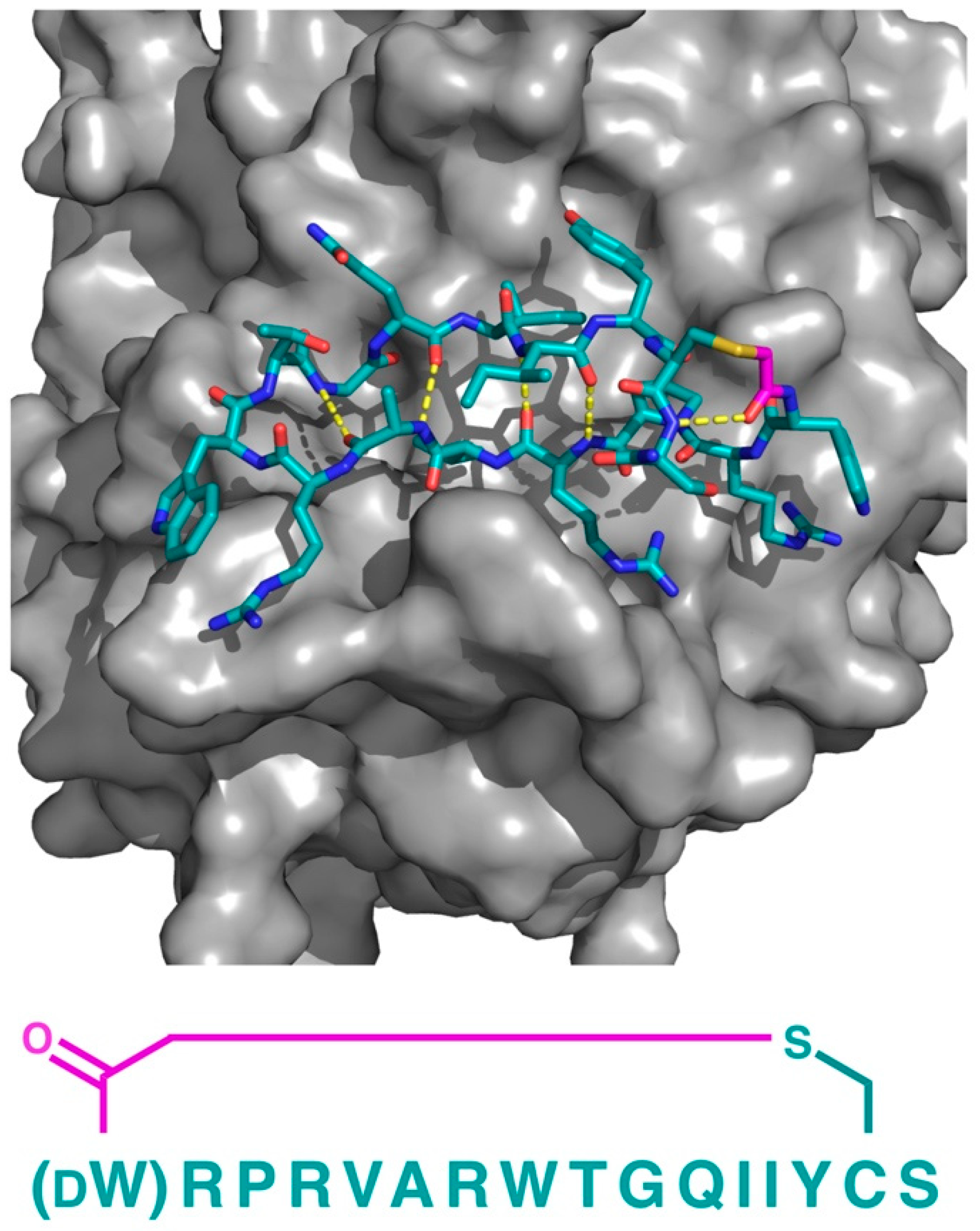
In addition to PfMATE, a different high affinity thioether cyclized peptide ligand was identified against another xenobiotic transporter, the CmABCB1 protein from the red alga Cyanidioschyzon merolae [
6]. In this case, the macrocyclic ligand, aCAP, was an 18 amino acid thioether-cyclized macrocyclic peptide, which, like the PfMATE ligands described above, also functioned as a CmABCB1 inhibitor. The co-crystal structure obtained comprised an inward-open conformation for CmABCB1, with aCAP bound to the extracellular surface (
Figure 5). The peptide was found to interact with a “gate” in the extracellular region of the protein formed by tightly packed TMs, acting as a clamp that restrained their conformation. From the additional mutational and transport studies, a full scheme of the transport mechanism was elucidated. It was found that the extracellular gate in the upper side of the complex is maintained by strong interactions between the TM domains, particularly TM1 and TM6. Upon the binding of the substrate in the cavity of the transporter, it interacts with a Tyr residue in TM5 that promotes the movement of the upper domains in opposite directions, consequently disrupting the interactions of the extracellular gate and generating its opening, while also accelerating the ATPase activity. However, upon the binding of aCAP, the interactions between the TMs in the extracellular gate are reinforced, preventing the opening of the gate and efficiently inhibiting the transport activity.
2.2.2. Enzymes
In addition to the use of thioether-cyclized macrocyclic peptides as inhibitors/co-crystallization ligands of membrane transporters, compounds of this class have been identified as inhibitors of specific enzymes [
7,
8,
9,
10], and in several cases, these have been subjected to X-ray crystallography as a basis for the subsequent structure–activity studies. For example, Kawamura et al. initially identified several peptides with strong binding and inhibition properties against the JmjC-domain containing lysine demethylases (JmjC-KDMs), a family of Fe
2+ and 2-oxoglutarate dependent histone modification enzymes [
11]. In particular, the macrocyclic peptide CP2 was found to exhibit the potent inhibition of the KDM4A and KDM4C, and was crystallized bound to KDM4A. This peptide demonstrated a surprising binding mode, localized not to the catalytic 2OG-binding pocket, but to the histone-binding domain, where it formed a two-turn β-sheet stabilized by, and interacting with KDM4A through multiple hydrogen bonds. The Arg6 of CP2 was found to bind to the sub-pocket usually occupied by trimethyl Lys in the histone substrate, mimicking its positive charge, and localized near the protein’s Fe
2+ cofactor (
Figure 6). Based on this co-crystal structure, a number of alterations were made to CP2 (e.g., the N-methylation of specific backbone positions or their substitution with a
d- or fluorinated-amino acids), with the aim of improving the stability and cellular uptake. Most of these modifications were well-tolerated, even when combined, and compounds with an improved activity in cell culture assays were obtained, demonstrating the potential for the structure-based design of macrocyclic peptides.
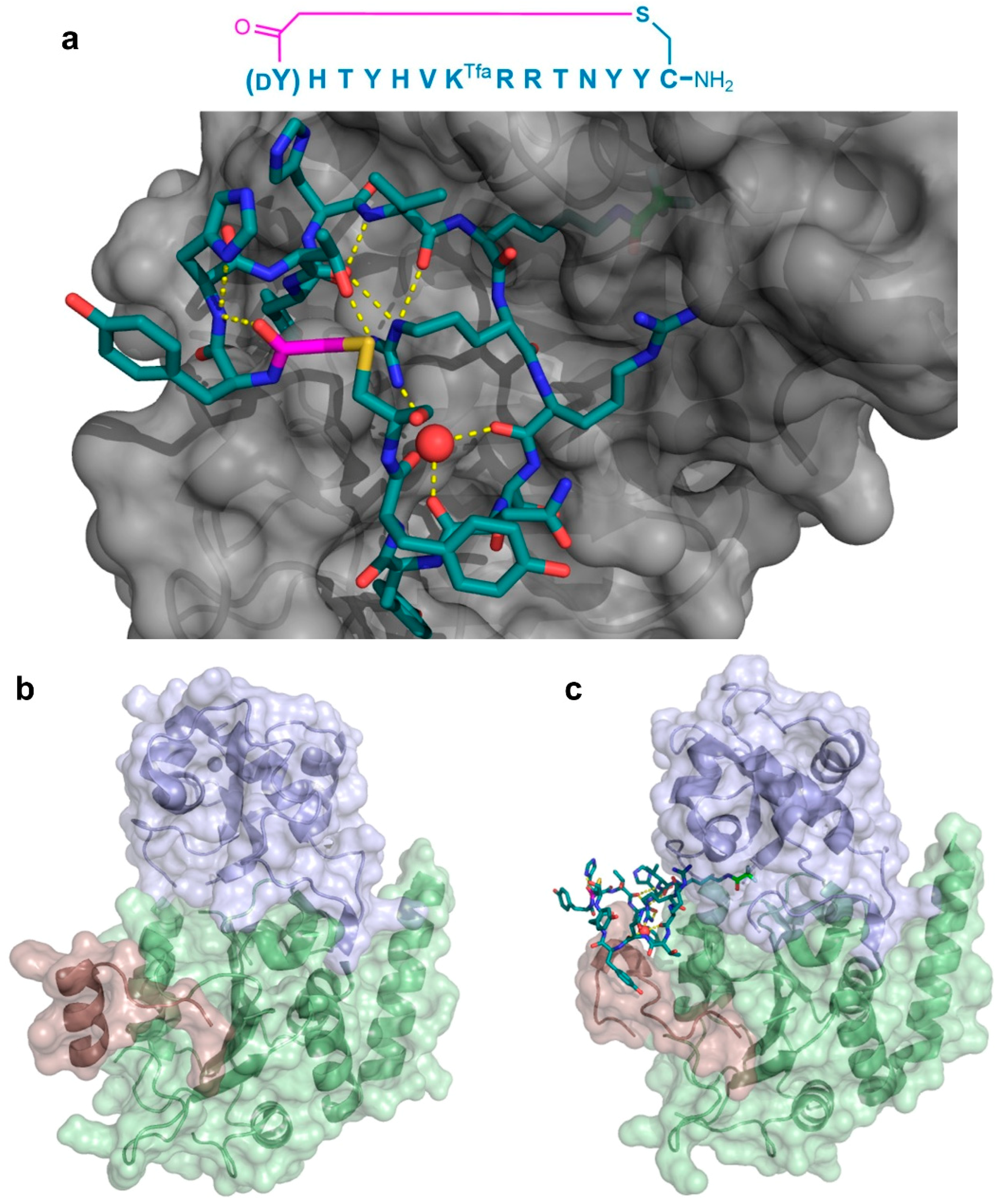
As a second example of the X-ray crystallographic analysis of a thioether-cyclic peptide targeting an enzyme, Yamagata et al. analyzed the structure of the S2iL5 macrocyclic peptide bound to its target, the human NAD
+-dependent deacetylase Sirtuin 2 (SIRT2) [
12,
13]. S2iL5 was originally isolated from a peptide library constructed around a trifluoroacetyl Lys residue (K
Tfa), designed to act as a mechanism-based “warhead” targeted to the SIRT2 active site. The co-crystal structure of S2iL5 and SIRT2 demonstrated that S2iL5 assumed a remarkable structure, in which the cyclic peptide scaffold was stabilized by a central water molecule, effectively coordinated by Arg8, Arg9, and Asn11 (
Figure 7a). This structure allowed for the peptide to bind to a groove of SIRT2, presenting the warhead directly into the active site. Surprisingly, not only did the peptide and protein engage in a β-sheet-like mode of interaction that facilitated the transition of SIRT2 to a more closed state, but the analysis of the unbound SIRT2 showed that the SIRT2-specific insertion region (residues 289–304) went through a drastic structural change from a full α-helix to a loop conformation upon peptide binding (
Figure 7b,c). The subsequent mutational studies on both the peptide and the protein revealed that this region is remarkably flexible and may play an important role in the substrate recognition, with each interaction with the peptide contributing synergistically to the overall kinetics of the conformational change. Furthermore, these studies indicated that this large structural change was facilitated by the macrocyclic skeleton of the peptide, through its structural plasticity and numerous interactions with the target.
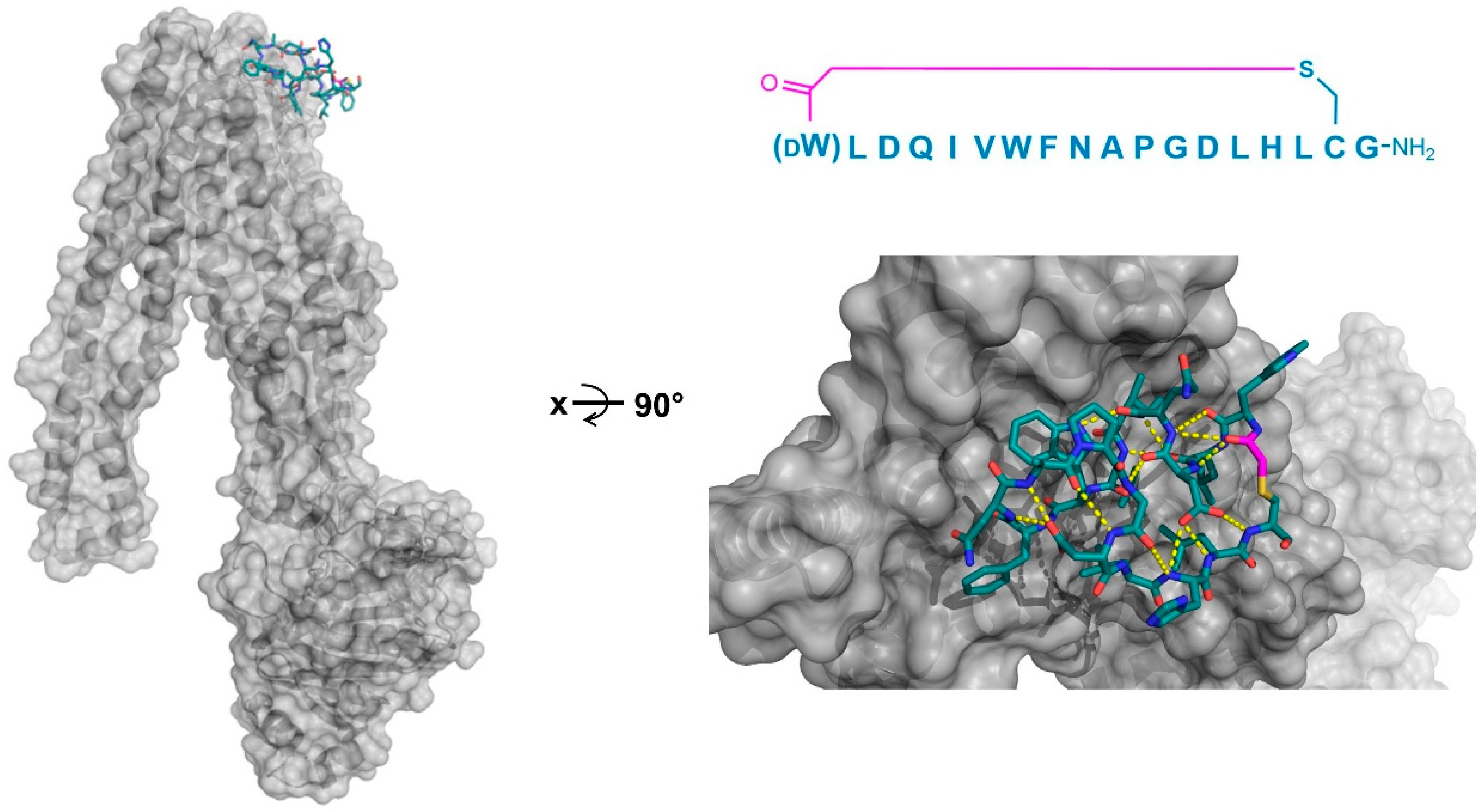
In a third example, the co-crystal structure of a thioether-cyclized macrocycle inhibitor (Ce-2d) of the
Caenorhabditis elegans co-factor independent phosphoglycerate mutase enzyme (iPGM) was determined [
14]. Similar to the MaD5, MaD3S, and MaD8 peptides described above, Ce-2d exhibited a “lariat” structure with a
d-Tyr initiated, thioether-closed macrocycle of eight residues, followed by a linear “tail” of three amino acids. The crystal structure of Ce-2d with the
C. elegans iPGM demonstrated that the macrocycle of Ce-2d was bound between the phosphatase and transferase domains of iPGM in a dominantly polar cavity, leaving the four amino acid tail free to form a small α-helical structure in contact with the solvent, with Tyr11 in close proximity to the Zn
2+ and Mn
2+ ion cofactors (
Figure 8). The superposition of this structure with the 2-phosphoglycerate substrate showed that Ce-2d did not interact directly with either the substrate or the active site, evidencing an allosteric effect. Studies of the truncated variants of Ce-2d demonstrated that the terminal Tyr 11 was critical for activity, probably because of the hydrogen bonding of the carboxamide of this residue with iPGM Glu87. These studies also identified a more potent analogue of Ce-2d, Ce-2a, which exhibited the sub-nanomolar inhibition of iPGM, but for which the co-crystal structure could not be determined. Ce-2a includes a longer (seven residue) “tail” than Ce-2d and a terminal Cys residue. The modeling of this analog suggested an interaction of this terminal Cys with the Zn
2+ co-factor of iPGM, explaining the considerably stronger inhibitory activity of Ce-2a compared with Ce-2d. Overall, these thioether-closed macrocycles were found to induce allosteric inhibition, stabilizing a locked-open structure of iPGM, and, in the case of Ce-2a, sequestering its Zn
2+ co-factor.
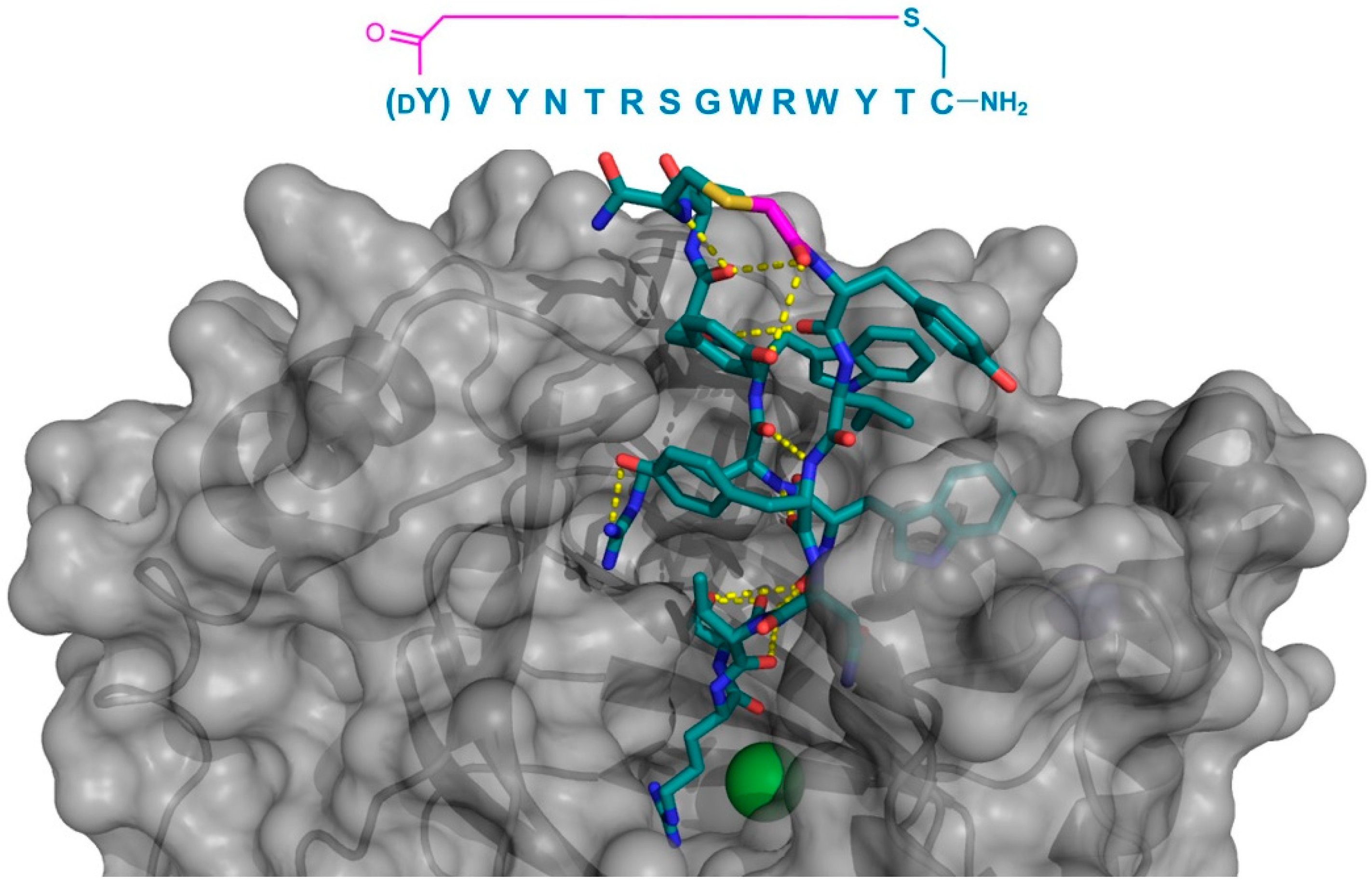
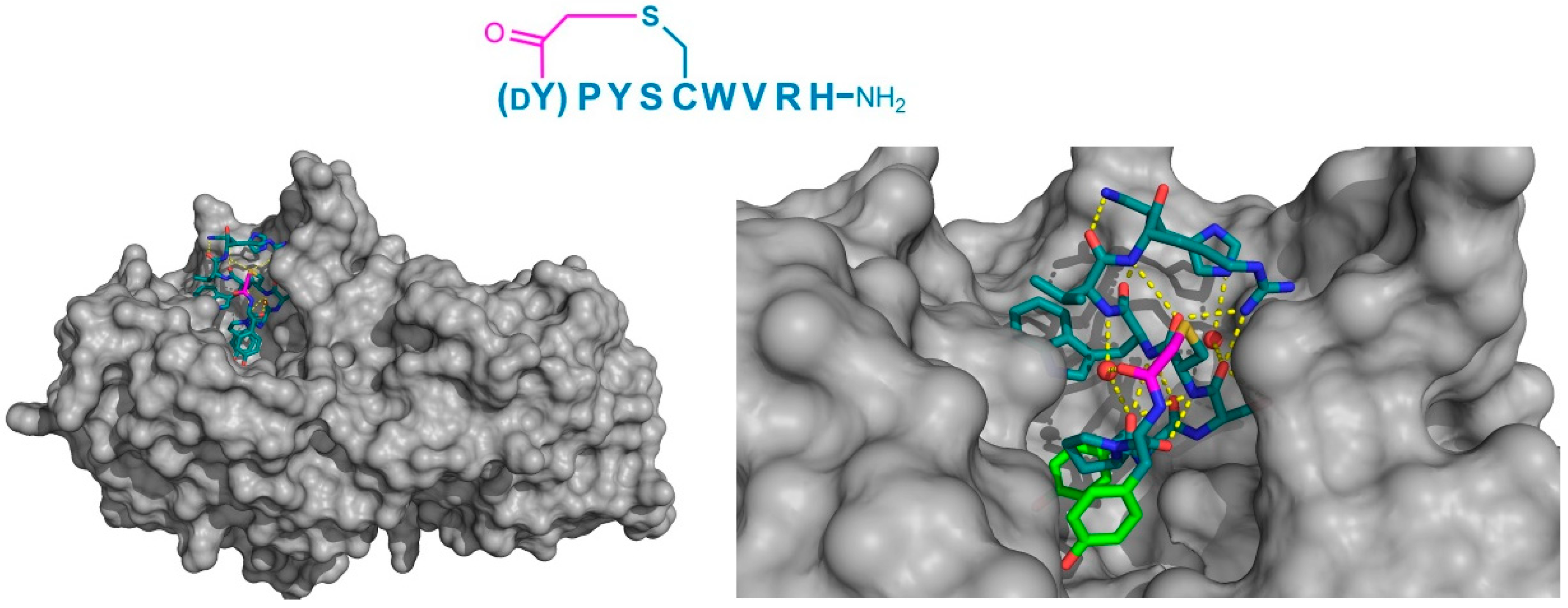
Finally, the thioether cyclized peptide ligands have also been co-crystalized bound to the human pancreatic amylase (HPA), an enzyme involved in starch digestion and implicated as playing a role in type-2 diabetes [
15]. These peptides exhibited conserved RFGYAY and (
dY)PYSCWXRH motifs, and a lariat architecture, and were potent competitive inhibitors of HPA, with inhibition constants in the single digit nanomolar range. The co-crystal structure of one of these peptides, a nonapeptide termed piHA-Dm, showed that it occupied the catalytic site of the protein, consistent with its competitive mechanism. The tail region of the lariat assumed a highly ordered α-helical structure, while the five-membered cycle was localized and tightly compacted within the catalytic site and bound directly (or through water molecules) to several key catalytic amino acids in the protein (
Figure 9). Furthermore, even the initiating
d-Tyr was shown to form important interactions with the protein, through both its side chain and amide moieties. The
d-stereochemistry of the initiating Tyr was found to allow a parallel arrangement with Tyr3, forming a tripeptide motif with Pro2 that made crucial interactions with the protein that were strikingly similar to the known inhibitors of HPA. On the basis of these findings, the authors hypothesized that the (
dY)PY and YAY motifs may be responsible for the inhibitory activity observed in all of the peptides identified, and that similar two-phenolic moiety motifs may have the potential for the development of inhibitors targeting amylases more generally.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









