Evaluation of Chemical Strategies for Improving the Stability and Oral Toxicity of Insecticidal Peptides





Abstract
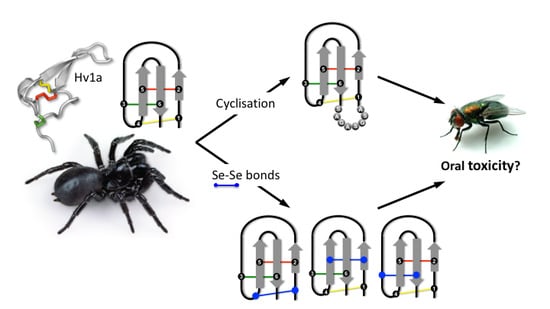
1. Introduction
2. Results
2.1. Synthesis of Diselenide and Cyclic Analogs of Hv1a
2.2. Synthesis of Cyclic Hv1a
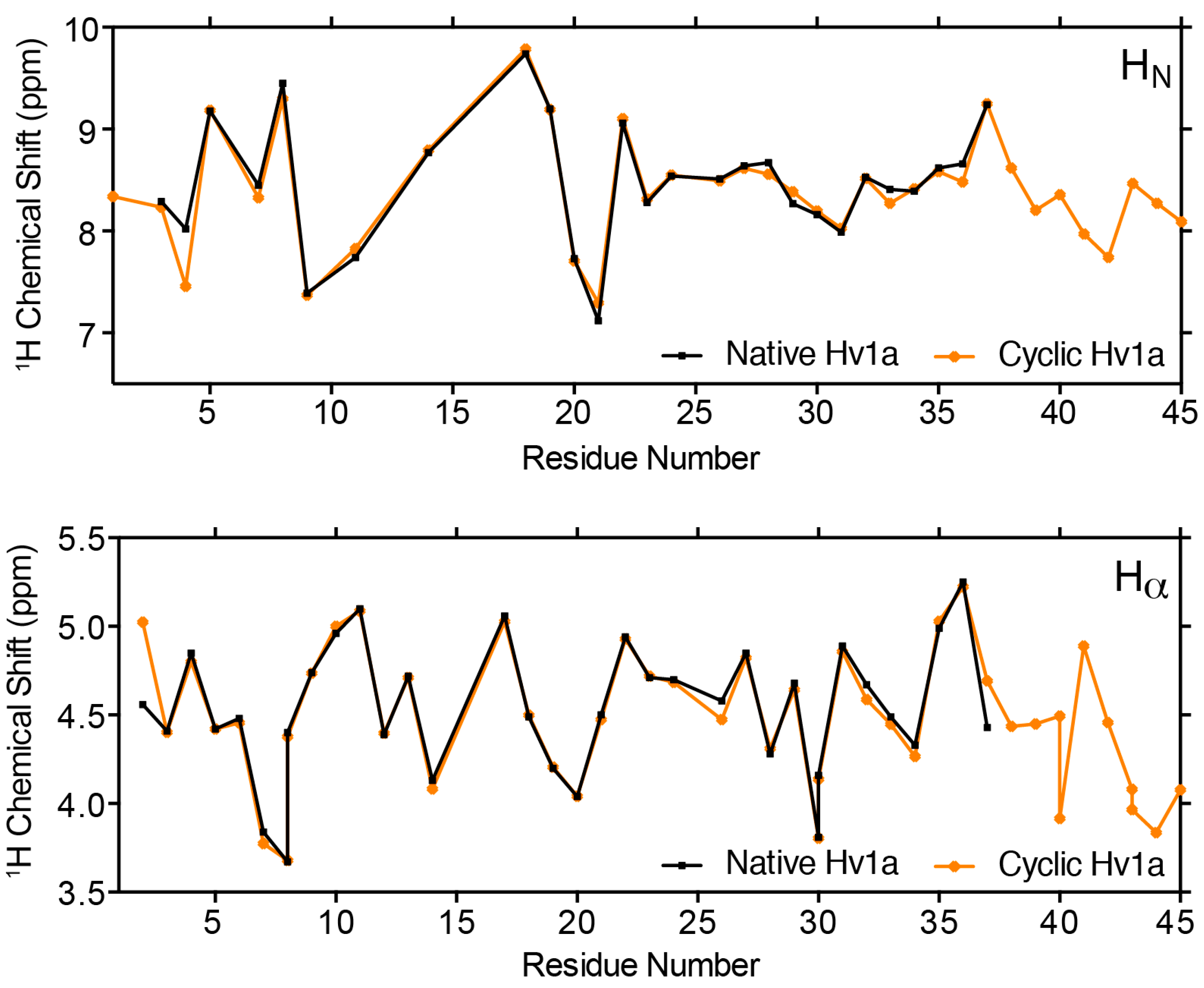
2.3. Structural Comparison of Native and Cyclic Hv1a
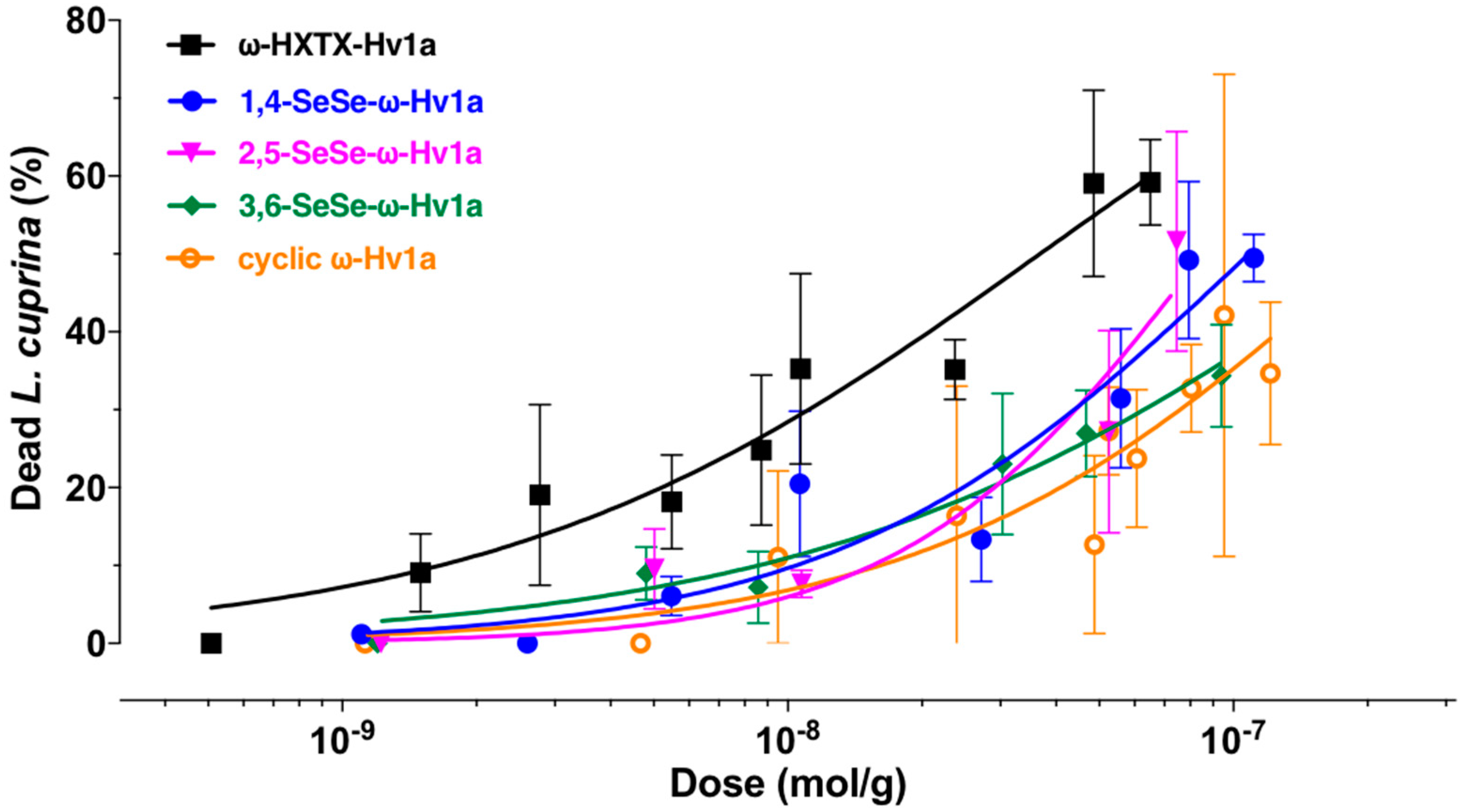
2.4. Blowfly Toxicity Assays
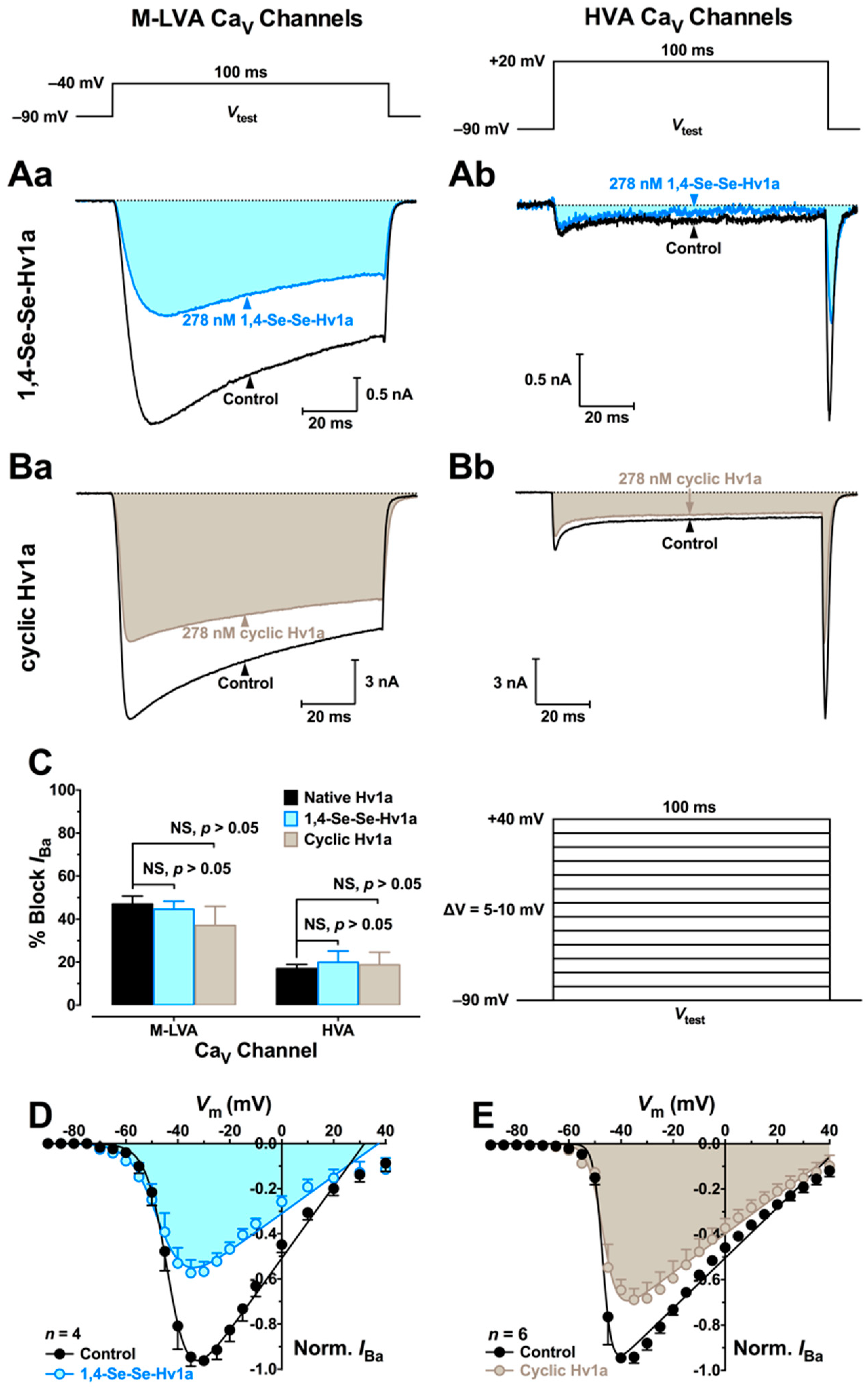
2.5. Electrophysiology
2.6. Midgut Permeation Assay
3. Discussion
4. Experimental Section
4.1. Peptide Synthesis
4.1.1. Chemicals
4.1.2. Synthesis of Native Hv1a and Diselenide Analogs
4.1.3. Synthesis of Cyclic Hv1a
4.2. Suceptibility to Scrambling of Hv1a Diselenide Analogs
4.3. NMR Spectroscopy and Resonance Assignments of Cyclic Hv1a
4.4. Blowfly Toxicity Assay
4.5. Electrophysiology
4.6. Midgut Permeation Assay
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: Mining the complexity of spider venoms via a combined cDNA and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; King, G.F. Spider toxins and their potential for insect control. In Insect Pharmacology; Gilbert, L.I., Gill, S.S., Eds.; Elsevier: Oxford, UK, 2010; pp. 119–123. [Google Scholar]
- King, G.F.; Hardy, M.C. Spider-venom peptides: Structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 2013, 58, 475–496. [Google Scholar] [CrossRef] [PubMed]
- Windley, M.J.; Herzig, V.; Dziemborowicz, S.A.; Hardy, M.C.; King, G.F.; Nicholson, G.M. Spider-venom peptides as bioinsecticides. Toxins 2012, 4, 191–227. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Senff, S.; Saez, N.J.; Seshadri, R.; Lau, H.Y.; Bende, N.S.; Undheim, E.A.; Rash, L.D.; Mobli, M.; King, G.F. Production of recombinant disulfide-rich venom peptides for structural and functional analysis via expression in the periplasm of E. coli. PLoS ONE 2013, 8, e63865. [Google Scholar] [CrossRef] [PubMed]
- Pallaghy, P.K.; Nielsen, K.J.; Craik, D.J.; Norton, R.S. A common structural motif incorporating a cystine knot and a triple-stranded β-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994, 3, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Smith, R.; O’Donoghue, S.I.; Nilges, M.; Connor, M.; Howden, M.E.; Christie, M.J.; King, G.F. The structure of a novel insecticidal neurotoxin, ω-atracotoxin-HV1, from the venom of an Australian funnel web spider. Nat. Struct. Biol. 1997, 4, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Herzig, V.; King, G.F. The cystine knot is responsible for the exceptional stability of the insecticidal spider toxin ω-hexatoxin-Hv1a. Toxins 2015, 7, 4366–4380. [Google Scholar] [CrossRef] [PubMed]
- Armishaw, C.J.; Daly, N.L.; Nevin, S.T.; Adams, D.J.; Craik, D.J.; Alewood, P.F. α-Selenoconotoxins, a new class of potent α7 neuronal nicotinic receptor antagonists. J. Biol. Chem. 2006, 281, 14136–14143. [Google Scholar] [CrossRef] [PubMed]
- Nentwig, W. Spider venoms are not suitable insecticides. Toxicon 1993, 31, 233–236. [Google Scholar] [CrossRef]
- Hardy, M.C.; Daly, N.L.; Mobli, M.; Morales, R.A.; King, G.F. Isolation of an orally active insecticidal toxin from the venom of an Australian tarantula. PLoS ONE 2013, 8, e73136. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.K.; Sollod, B.L.; Wikel, S.K.; King, G.F. Orally active acaricidal peptide toxins from spider venom. Toxicon 2006, 47, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Zafar, Y.; Briddon, R.W.; Malik, K.A.; Mukhtar, Z. Spider venom toxin protects plants from insect attack. Transgenic Res. 2006, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Herzig, V.; King, G.F. Dipteran toxicity assays for determining the oral insecticidal activity of venoms and toxins. Toxicon 2018, 150, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 2010, 49, 6545–6548. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Akcan, M.; Kaas, Q.; Daly, N.L.; Craik, D.J. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon 2012, 59, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; Alewood, P.F. Selenopeptide chemistry. J. Pept. Sci. 2008, 14, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, A.D.; Herzig, V.; Windley, M.J.; Dziemborowicz, S.; Mobli, M.; Nicholson, G.M.; Alewood, P.F.; King, G.F. Do vicinal disulfide bridges mediate functionally important redox transformations in proteins? Antioxid. Redox Signal. 2013, 19, 1976–1980. [Google Scholar] [CrossRef] [PubMed]
- Mobli, M.; Morgenstern, D.; King, G.F.; Alewood, P.F.; Muttenthaler, M. Site-specific pKa determination of selenocysteine residues in selenovasopressin by using 77Se NMR spectroscopy. Angew. Chem. Int. Ed. 2011, 50, 11952–11955. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; Andersson, A.; de Araujo, A.D.; Dekan, Z.; Lewis, R.J.; Alewood, P.F. Modulating oxytocin activity and plasma stability by disulfide bond engineering. J. Med. Chem. 2010, 53, 8585–8596. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J. Protein folding: Turbo-charged crosslinking. Nat. Chem. 2012, 4, 600–602. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, A.D.; Callaghan, B.; Nevin, S.T.; Daly, N.L.; Craik, D.J.; Moretta, M.; Hopping, G.; Christie, M.J.; Adams, D.J.; Alewood, P.F. Total synthesis of the analgesic conotoxin MrVIB through selenocysteine-assisted folding. Angew. Chem. Int. Ed. 2011, 50, 6527–6529. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; Nevin, S.T.; Grishin, A.A.; Ngo, S.T.; Choy, P.T.; Daly, N.L.; Hu, S.H.; Armishaw, C.J.; Wang, C.I.; Lewis, R.J.; et al. Solving the α-conotoxin folding problem: Efficient selenium-directed on-resin generation of more potent and stable nicotinic acetylcholine receptor antagonists. J. Am. Chem. Soc. 2010, 132, 3514–3522. [Google Scholar] [CrossRef] [PubMed]
- Steiner, A.M.; Woycechowsky, K.J.; Olivera, B.M.; Bulaj, G. Reagentless oxidative folding of disulfide-rich peptides catalyzed by an intramolecular diselenide. Angew. Chem. Int. Ed. 2012, 51, 5580–5584. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Fischer, H.; Dempster, L.; Daly, N.L.; Rosengren, K.J.; Nevin, S.T.; Meunier, F.A.; Adams, D.J.; Craik, D.J. Engineering stable peptide toxins by means of backbone cyclization: Stabilization of the α-conotoxin MII. Proc. Natl. Acad. Sci. USA 2005, 102, 13767–13772. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, E.S.; Armishaw, C.J.; Colgrave, M.L.; Wahlstrom, M.E.; Alewood, P.F.; Daly, N.L.; Craik, D.J. Cyclic MrIA: A stable and potent cyclic conotoxin with a novel topological fold that targets the norepinephrine transporter. J. Med. Chem. 2006, 49, 6561–6568. [Google Scholar] [CrossRef] [PubMed]
- Halai, R.; Callaghan, B.; Daly, N.L.; Clark, R.J.; Adams, D.J.; Craik, D.J. Effects of cyclization on stability, structure, and activity of α-conotoxin RgIA at the α9α10 nicotinic acetylcholine receptor and GABAB receptor. J. Med. Chem. 2011, 54, 6984–6992. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huang, Y.H.; Kaas, Q.; Harvey, P.J.; Wang, C.K.; Tae, H.S.; Adams, D.J.; Craik, D.J. Backbone cyclization of analgesic conotoxin GeXIVA facilitates direct folding of the ribbon isomer. J. Biol. Chem. 2017, 292, 17101–17112. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.; Hayes, J.L.; Sollod, B.; Wen, S.; Wilson, D.T.; Hains, P.G.; Hodgson, W.C.; Broady, K.W.; King, G.F.; Nicholson, G.M. The ω-atracotoxins: Selective blockers of insect M-LVA and HVA calcium channels. Biochem. Pharmacol. 2007, 74, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Bende, N.S.; Alam, M.S.; Tedford, H.W.; Kennedy, R.M.; King, G.F. Methods for deployment of spider-venom peptides as bioinsecticides. In Advances in Insect Physiology: Insect Midgut and Insecticidal Proteins for Insect Control; Dhadialla, T.S., Gill, S.S., Eds.; Academic Press: London, UK, 2014; Volume 7, pp. 389–411. [Google Scholar]
- Kongsuwan, K.; Gough, J.; Kemp, D.; McDevitt, A.; Akhurst, R. Characterization of a new Bacillus thuringiensis endotoxin, Cry47Aa, from strains that are toxic to the Australian sheep blowfly, Lucilia cuprina. FEMS Microbiol. Lett. 2005, 252, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Bende, N.S.; Dziemborowicz, S.; Mobli, M.; Herzig, V.; Gilchrist, J.; Wagner, J.; Nicholson, G.M.; King, G.F.; Bosmans, F. A distinct sodium channel voltage-sensor locus determines the insect selectivity of the spider toxin Dc1a. Nat. Commun. 2014, 5, 4350. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.A.; Grimm, L.L.; Low, C.F.; Morgenstern, D.; Herzig, V.; Zobel-Thropp, P.; Pineda, S.S.; Habib, R.; Dziemborowicz, S.; Fry, B.G.; et al. Weaponization of a hormone: Convergent recruitment of hyperglycemic hormone into the venom of arthropod predators. Structure 2015, 23, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Zlotkin, E.; Fishman, L.; Shapiro, J.P. Oral toxicity to flesh flies of a neurotoxic polypeptide. Arch. Insect Biochem. Physiol. 1992, 21, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Primor, N.; Teitelbaum, Z.; Zlotkin, E. Penetrability of orally toxic protein from cobra venom through the gut of a blowfly. Biochim. Biophys. Acta 1980, 627, 71–81. [Google Scholar] [CrossRef]
- Bloomquist, J.R. Mode of action of atracotoxin at central and peripheral synapses of insects. Invertebr. Neurosci. 2003, 5, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Audsley, N.; Matthews, J.; Nachman, R.J.; Weaver, R.J. Transepithelial flux of an allatostatin and analogs across the anterior midgut of Manduca sexta larvae in vitro. Peptides 2008, 29, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Nachman, R.J.; Teal, P.E.; Strey, A. Enhanced oral availability/pheromonotropic activity of peptidase-resistant topical amphiphilic analogs of pyrokinin/PBAN insect neuropeptides. Peptides 2002, 23, 2035–2043. [Google Scholar] [CrossRef]
- Habibi, J.; Brandt, S.L.; Coudron, T.A.; Wagner, R.M.; Wright, M.K.; Backus, E.A.; Huesing, J.E. Uptake, flow, and digestion of casein and green fluorescent protein in the digestive system of Lygus hesperus Knight. Arch. Insect Biochem. Physiol. 2002, 50, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Modespacher, U.P.; Rudin, W.; Jenni, L.; Hecker, H. Transport of peroxidase through the midgut epithelium of Glossina m. morsitans (Diptera, Glossinidae). Tissue Cell 1986, 18, 429–436. [Google Scholar] [CrossRef]
- Zhu, W.; Vandingenen, A.; Huybrechts, R.; Vercammen, T.; Baggerman, G.; De Loof, A.; Poulos, C.P.; Velentza, A.; Breuer, M. Proteolytic breakdown of the Neb-trypsin modulating oostatic factor (Neb-TMOF) in the hemolymph of different insects and its gut epithelial transport. J. Insect Physiol. 2001, 47, 1235–1242. [Google Scholar] [CrossRef]
- Schnolzer, M.; Alewood, P.; Jones, A.; Alewood, D.; Kent, S.B. In Situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int. J. Pept. Protein Res. 1992, 40, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; Albericio, F.; Dawson, P.E. Methods, setup and safe handling for anhydrous hydrogen fluoride cleavage in Boc solid-phase peptide synthesis. Nat. Protoc. 2015, 10, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Kneller, D.G. SPARKY3; University of California: San Francisco, CA, USA, 2001. [Google Scholar]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Bende, N.S.; Kang, E.; Herzig, V.; Bosmans, F.; Nicholson, G.M.; Mobli, M.; King, G.F. The insecticidal neurotoxin Aps III is an atypical knottin peptide that potently blocks insect voltage-gated sodium channels. Biochem. Pharmacol. 2013, 85, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Hodgson, W.C. Neurotoxic and insecticidal properties of venom from the Australian theraphosid spider Selenotholus foelschei. Neurotoxicology 2008, 29, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Grolleau, F.; Lapied, B. Two distinct low-voltage-activated Ca2+ currents contribute to the pacemaker mechanism in cockroach dorsal unpaired median neurons. J. Neurophysiol. 1996, 76, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.H. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J. Neurosci. Methods 1994, 51, 107–116. [Google Scholar] [CrossRef]
- Bezanilla, F.; Armstrong, C.M. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 1977, 70, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Wicher, D.; Penzlin, H. Ca2+ currents in central insect neurons: Electrophysiological and pharmacological properties. J. Neurophysiol. 1997, 77, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Goldin, A.L. Use-dependent potentiation of the Nav1.6 sodium channel. Biophys. J. 2004, 87, 3862–3872. [Google Scholar] [CrossRef] [PubMed]
- Audsley, N.; Weaver, R.J.; Edwards, J.P. Enzyme linked immunosorbent assay for Manduca sexta allatostatin (Mas-AS), isolation and measurement of Mas-AS immunoreactive peptide in Lacanobia oleracea. Insect Biochem. Mol. Biol. 1998, 28, 775–784. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Folding Efficiency | Correctly Folded Toxin | |
|---|---|---|---|
| With 1 Equivalent GSH, pH 7.2 | In Buffer, pH 7.2 | ||
| native Hv1a | 48% | 85% | 100% |
| 1,4-SeSe-Hv1a | 55% | 85% | 100% |
| 2,5-SeSe-Hv1a | 38% | 82% | 100% |
| 3,6-SeSe-Hv1a | 27% | 60% | 95% |
| Toxin | ω-Hv1a | 1,4-SeSe-ω-Hv1a | 2,5-SeSe-ω-Hv1a | 3,6-SeSe-ω-Hv1a | Cyclic-ω-Hv1a |
|---|---|---|---|---|---|
| LD50 (pmol/g) | 499 ± 53 | 407 ± 22 | 455 ± 35 | 446 ± 12 | 293 ± 18 |
| Concentration of Hv1a or Analog (µM) | Lumen to Hemolymph Flux (pmol/cm2/h) | ||
|---|---|---|---|
| Hv1a | Cyclic Hv1a | 1,4-SeSe-Hv1a | |
| 0.1 | 11.8 ± 2.7 | 0.14 ± 0.03 | — |
| 1.0 | 41 ± 11 | 1.7 ± 0.6 | — |
| 10 | 351 ± 32 | 94 ± 16 | — |
| 10 | 287 ± 23 | — | 227 ± 14 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herzig, V.; De Araujo, A.D.; Greenwood, K.P.; Chin, Y.K.-Y.; Windley, M.J.; Chong, Y.; Muttenthaler, M.; Mobli, M.; Audsley, N.; Nicholson, G.M.; et al. Evaluation of Chemical Strategies for Improving the Stability and Oral Toxicity of Insecticidal Peptides. Biomedicines 2018, 6, 90. https://doi.org/10.3390/biomedicines6030090
Herzig V, De Araujo AD, Greenwood KP, Chin YK-Y, Windley MJ, Chong Y, Muttenthaler M, Mobli M, Audsley N, Nicholson GM, et al. Evaluation of Chemical Strategies for Improving the Stability and Oral Toxicity of Insecticidal Peptides. Biomedicines. 2018; 6(3):90. https://doi.org/10.3390/biomedicines6030090
Chicago/Turabian StyleHerzig, Volker, Aline Dantas De Araujo, Kathryn P. Greenwood, Yanni K.-Y. Chin, Monique J. Windley, Youmie Chong, Markus Muttenthaler, Mehdi Mobli, Neil Audsley, Graham M. Nicholson, and et al. 2018. "Evaluation of Chemical Strategies for Improving the Stability and Oral Toxicity of Insecticidal Peptides" Biomedicines 6, no. 3: 90. https://doi.org/10.3390/biomedicines6030090
APA StyleHerzig, V., De Araujo, A. D., Greenwood, K. P., Chin, Y. K.-Y., Windley, M. J., Chong, Y., Muttenthaler, M., Mobli, M., Audsley, N., Nicholson, G. M., Alewood, P. F., & King, G. F. (2018). Evaluation of Chemical Strategies for Improving the Stability and Oral Toxicity of Insecticidal Peptides. Biomedicines, 6(3), 90. https://doi.org/10.3390/biomedicines6030090