HIF Oxygen Sensing Pathways in Lung Biology


Abstract
{kind=link}
{kind=link}
1. Introduction
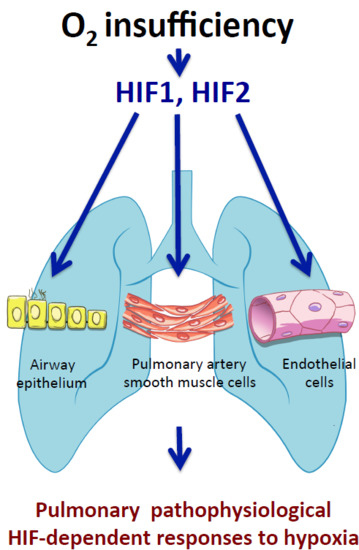
2. The HIF Oxygen Sensing Pathways in the Pulmonary Vasculature
2.1. Chronic Activation of the HIF Pathways Leading to Pulmonary Hypertension
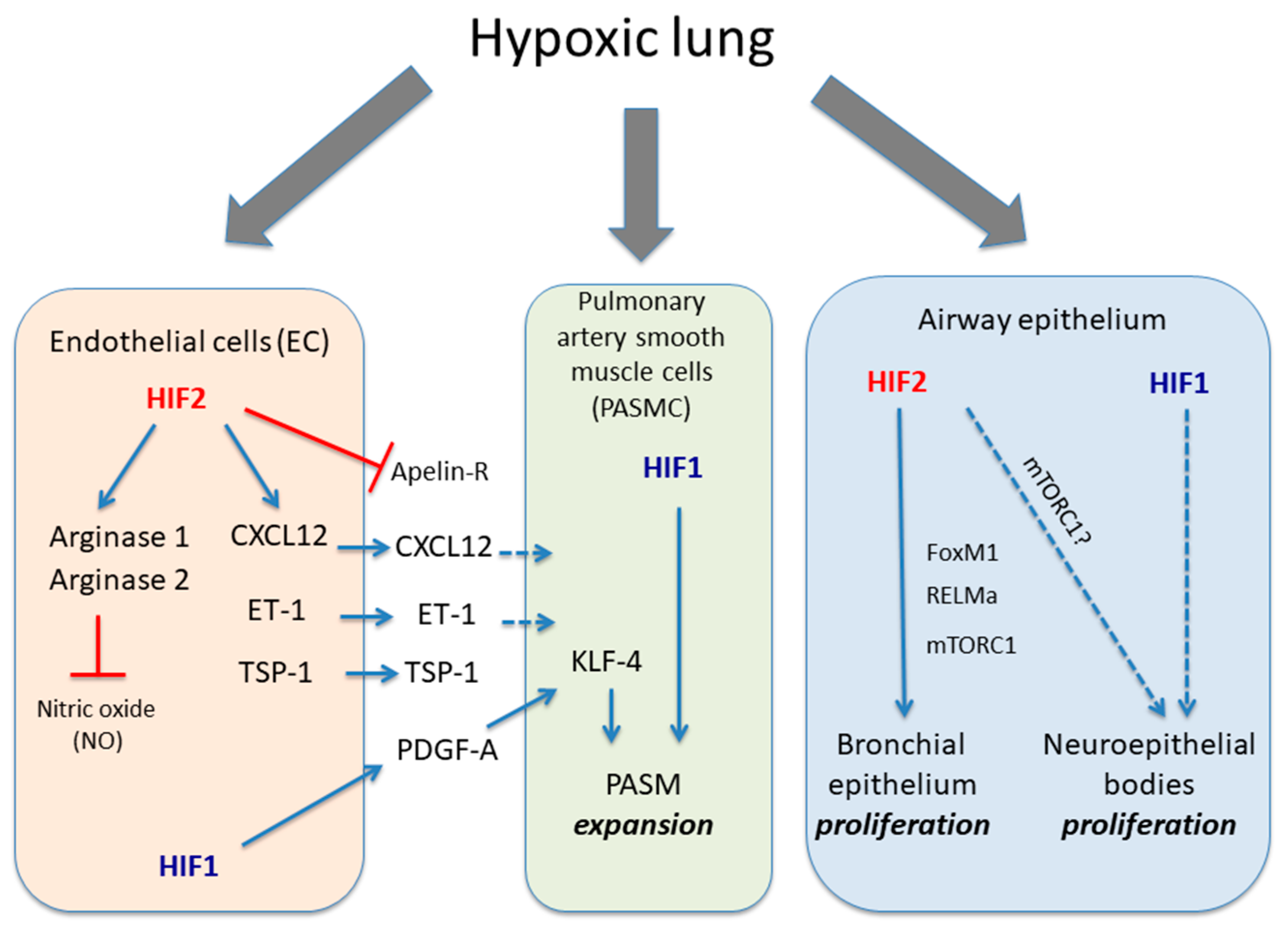
2.2. The HIF1α Activity in Pulmonary Smooth Muscle Cells Associated with Hypoxia-Induced Pulmonary Hypertension
2.3. The Endothelial HIF2 Pathway in Hypoxia-Induced Pulmonary Hypertension
3. Role of HIF Pathway in the Airway Epithelium
The HIF Pathway in Airway Epithelium Proliferation
4. Concluding Remarks and Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 2016, 47, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.K.; López-Barneo, J.; Buckler, K.J.; Archer, S.L. Acute oxygen-sensing mechanisms. N. Engl. J. Med. 2005, 353, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Schermuly, R.T.; Brandes, R.P.; Weissmann, N. Molecular mechanisms of hypoxia-inducible factor-induced pulmonary arterial smooth muscle cell alterations in pulmonary hypertension. J. Physiol. 2016, 594, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Y.; Frid, M.G.; Shimoda, L.A.; Wiener, C.M.; Stenmark, K.; Semenza, G.L. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am. J. Physiol. 1998, 275, L818–L826. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jürgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Hörstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: Implication of natural antisense HIF-1alpha. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Y.; Shimoda, L.A.; Iyer, N.V.; Huso, D.L.; Sun, X.; McWilliams, R.; Beaty, T.; Sham, J.S.; Wiener, C.M.; Sylvester, J.T.; et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J. Clin. Investig. 1999, 103, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Compernolle, V.; Tjwa, M.; Wiesener, M.S.; Maxwell, P.H.; Collen, D.; Carmeliet, P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Investig. 2003, 111, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Dean, H.P. The Pulmonary Circulation. J. Physiol. 1894, 16, 34–158. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D42–D50. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; McLaughlin, V.V.; Dalaan, A.M.; Satoh, T.; Galiè, N. Treatment of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 323–336. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Smith, T.G.; Brooks, J.T.; Balanos, G.M.; Lappin, T.R.; Layton, D.M.; Leedham, D.L.; Liu, C.; Maxwell, P.H.; McMullin, M.F.; McNamara, C.J.; et al. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006, 3, e290. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.P.; Harten, S.K.; Reid, C.D.; Tuddenham, E.G.; Maxwell, P.H. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Kerestes, H.; Percy, M.J.; Pietrofesa, R.; Chen, L.; Khurana, T.S.; Christofidou-Solomidou, M.; Lappin, T.R.; Lee, F.S. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J. Biol. Chem. 2013, 288, 17134–17144. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.K.; Waypa, G.B.; Mungai, P.T.; Nielsen, J.M.; Czech, L.; Dudley, V.J.; Beussink, L.; Dettman, R.W.; Berkelhamer, S.K.; Steinhorn, R.H.; et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am. J. Respir. Crit. Care Med. 2014, 189, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Yuan, J.X. Hypoxia-inducible factor-1α in pulmonary arterial smooth muscle cells and hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Saddouk, F.Z.; Ntokou, A.; Mazurek, R.; Greif, D.M. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018, 23, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci. Transl. Med. 2015, 7, 308ra159. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Barnes, E.A.; Alvira, C.M.; Ying, L.; Reddy, S.; Cornfield, D.N. Hypoxia-inducible factor-1α in pulmonary artery smooth muscle cells lowers vascular tone by decreasing myosin light chain phosphorylation. Circ. Res. 2013, 112, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell Biol. 2016, 36, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaço, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zeng, H.; Xie, X.J.; Tao, Y.K.; He, X.; Roman, R.J.; Aschner, J.L.; Chen, J.X. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget 2016, 7, 58848–58861. [Google Scholar] [CrossRef] [PubMed]
- Böhm, F.; Pernow, J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.M.; Richardson, T.; Wang, T.; Mosqueira, M.; Arguiri, E.; Yu, H.; Yu, Q.C.; Solomides, C.C.; Morrisey, E.E.; Khurana, T.S.; et al. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J. Clin. Investig. 2010, 120, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Japp, A.G.; Cruden, N.L.; Amer, D.A.; Li, V.K.; Goudie, E.B.; Johnston, N.R.; Sharma, S.; Neilson, I.; Webb, D.J.; Megson, I.L.; et al. Vascular effects of apelin in vivo in man. J. Am. Coll. Cardiol. 2008, 52, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Davie, N.; Haleen, S.J.; Upton, P.D.; Polak, J.M.; Yacoub, M.H.; Morrell, N.W.; Wharton, J. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 2002, 165, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Kleinz, M.J.; Davenport, A.P. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul. Pept. 2004, 118, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Dhaliwal, R.; Ivanovska, J.; Kantores, C.; McNamara, P.J.; Scott, J.A.; Belik, J.; Jankov, R.P. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L503–L510. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.B.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004, 18, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; XiangLi, X.; Niu, H.; Wang, H.; Jia, P.; Gong, W.; Wu, D.; Qin, W.; Xing, C. Arginase inhibitor attenuates pulmonary artery hypertension induced by hypoxia. Mol. Cell. Biochem. 2016, 412, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Labrousse-Arias, D.; Castillo-González, R.; Rogers, N.M.; Torres-Capelli, M.; Barreira, B.; Aragonés, J.; Cogolludo, Á.; Isenberg, J.S.; Calzada, M.J. HIF-2α-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc. Res. 2016, 109, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.M.; Bauer, E.M.; Rogers, N.M.; Yao, M.; Feijoo-Cuaresma, M.; Pilewski, J.M.; Champion, H.C.; Zuckerbraun, B.S.; Calzada, M.J.; Isenberg, J.S. Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc. Res. 2012, 93, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, C.D.; Yu, L.; Al-Ansari, E.; Hales, C.A.; Quinn, D.A. Thrombospondin-1 null mice are resistant to hypoxia-induced pulmonary hypertension. J. Cardiothorac. Surg. 2010, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Compernolle, V.; Brusselmans, K.; Acker, T.; Hoet, P.; Tjwa, M.; Beck, H.; Plaisance, S.; Dor, Y.; Keshet, E.; Lupu, F.; et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002, 8, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.F.; Hellberg, A.K.; Balenger, S.; Depping, R.; Dodd-O, J.; Johns, R.A.; Li, D. Hypoxia-induced mitogenic factor has antiapoptotic action and is upregulated in the developing lung: Coexpression with hypoxia-inducible factor-2alpha. Am. J. Respir. Cell Mol. Biol. 2004, 31, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Torres-Capelli, M.; Marsboom, G.; Li, Q.O.; Tello, D.; Rodriguez, F.M.; Alonso, T.; Sanchez-Madrid, F.; García-Rio, F.; Ancochea, J.; Aragonés, J. Role Of Hif2α Oxygen Sensing Pathway In Bronchial Epithelial Club Cell Proliferation. Sci. Rep. 2016, 6, 25357. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Meng, Q.; Wu, H.; Eid, G.; Zhang, G.; Zhang, X.; Yang, S.; Huang, K.; Lee, T.H.; Corrigan, C.J.; et al. Resistin-like molecule-β is a human airway remodelling mediator. Eur. Respir. J. 2012, 39, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Elorza, A.; Soro-Arnáiz, I.; Meléndez-Rodríguez, F.; Rodríguez-Vaello, V.; Marsboom, G.; de Cárcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordóñez, A.; Serrano-Oviedo, L.; et al. HIF2α acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schmelzle, T.; Hall, M.N. TOR, a central controller of cell growth. Cell 2000, 103, 253–262. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Ustiyan, V.; Wert, S.E.; Ikegami, M.; Wang, I.C.; Kalin, T.V.; Whitsett, J.A.; Kalinichenko, V.V. Foxm1 transcription factor is critical for proliferation and differentiation of Clara cells during development of conducting airways. Dev. Biol. 2012, 370, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.C.; Zhang, Y.; Snyder, J.; Sutherland, M.J.; Burhans, M.S.; Shannon, J.M.; Park, H.J.; Whitsett, J.A.; Kalinichenko, V.V. Increased expression of FoxM1 transcription factor in respiratory epithelium inhibits lung sacculation and causes Clara cell hyperplasia. Dev. Biol. 2010, 347, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Bishop, T.; Ratcliffe, P.J.; Yeger, H.; Cutz, E. Hyperplasia and hypertrophy of pulmonary neuroepithelial bodies, presumed airway hypoxia sensors, in hypoxia-inducible factor prolyl hydroxylase-deficient mice. Hypoxia 2016, 4, 69–80. [Google Scholar] [PubMed]
- Cutz, E.; Pan, J.; Yeger, H.; Domnik, N.J.; Fisher, J.T. Recent advances and contraversies on the role of pulmonary neuroepithelial bodies as airway sensors. Semin. Cell Dev. Biol. 2013, 24, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Gosney, J.R. Pulmonary neuroendocrine cells in species at high altitude. Anat. Rec. 1993, 236, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Livermore, S.; Pan, J.; Yeger, H.; Ratcliffe, P.; Bishop, T.; Cutz, E. Augmented 5-HT Secretion in Pulmonary Neuroepithelial Bodies from PHD1 Null Mice. Adv. Exp. Med. Biol. 2015, 860, 309–313. [Google Scholar] [PubMed]
- López-Barneo, J.; González-Rodríguez, P.; Gao, L.; Fernández-Agüera, M.C.; Pardal, R.; Ortega-Sáenz, P. Oxygen sensing by the carotid body: Mechanisms and role in adaptation to hypoxia. Am. J. Physiol. Cell Physiol. 2016, 310, C629–C642. [Google Scholar] [CrossRef] [PubMed]
- Hodson, E.J.; Nicholls, L.G.; Turner, P.J.; Llyr, R.; Fielding, J.W.; Douglas, G.; Ratnayaka, I.; Robbins, P.A.; Pugh, C.W.; Buckler, K.J.; et al. Regulation of ventilatory sensitivity and carotid body proliferation in hypoxia by the PHD2/HIF-2 pathway. J. Physiol. 2016, 594, 1179–1195. [Google Scholar] [CrossRef] [PubMed]
- Macias, D.; Cowburn, A.S.; Torres-Torrelo, H.; Ortega-Sáenz, P.; López-Barneo, J.; Johnson, R.S. HIF-2α is essential for carotid body development and function. Elife 2018, 7, e34681. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Skinner, J.T.; Champion, H.C.; Crow, M.T.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L582–L593. [Google Scholar] [CrossRef] [PubMed]
- Johns, R.A.; Takimoto, E.; Meuchel, L.W.; Elsaigh, E.; Zhang, A.; Heller, N.M.; Semenza, G.L.; Yamaji-Kegan, K. Hypoxia-Inducible Factor 1α Is a Critical Downstream Mediator for Hypoxia-Induced Mitogenic Factor (FIZZ1/RELMα)-Induced Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 134–144. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urrutia, A.A.; Aragonés, J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines 2018, 6, 68. https://doi.org/10.3390/biomedicines6020068
Urrutia AA, Aragonés J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines. 2018; 6(2):68. https://doi.org/10.3390/biomedicines6020068
Chicago/Turabian StyleUrrutia, Andrés A., and Julián Aragonés. 2018. "HIF Oxygen Sensing Pathways in Lung Biology" Biomedicines 6, no. 2: 68. https://doi.org/10.3390/biomedicines6020068
APA StyleUrrutia, A. A., & Aragonés, J. (2018). HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines, 6(2), 68. https://doi.org/10.3390/biomedicines6020068