Metabolic Regulation of Hypoxia-Inducible Transcription Factors: The Role of Small Molecule Metabolites and Iron


Abstract

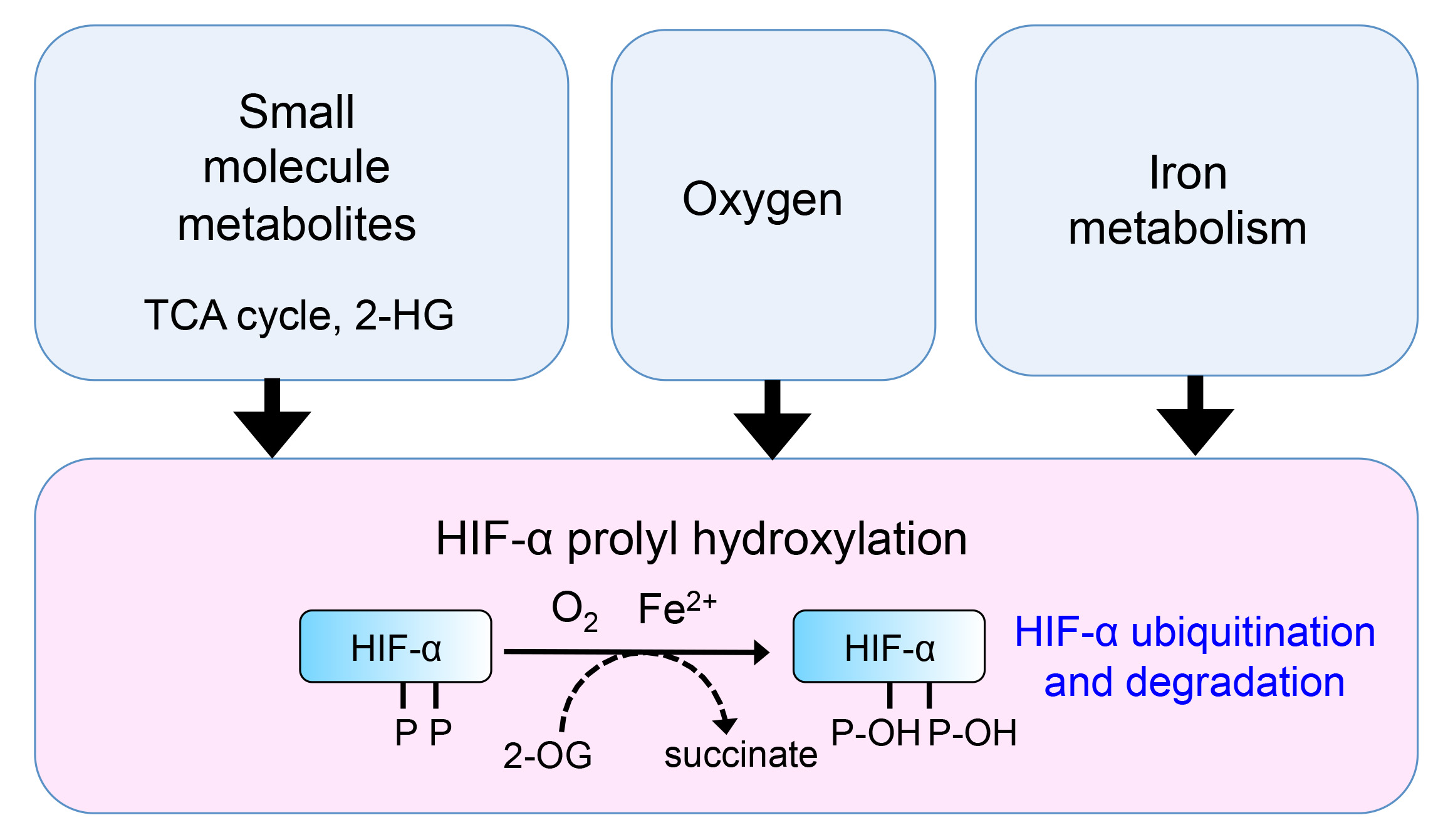
1. Introduction
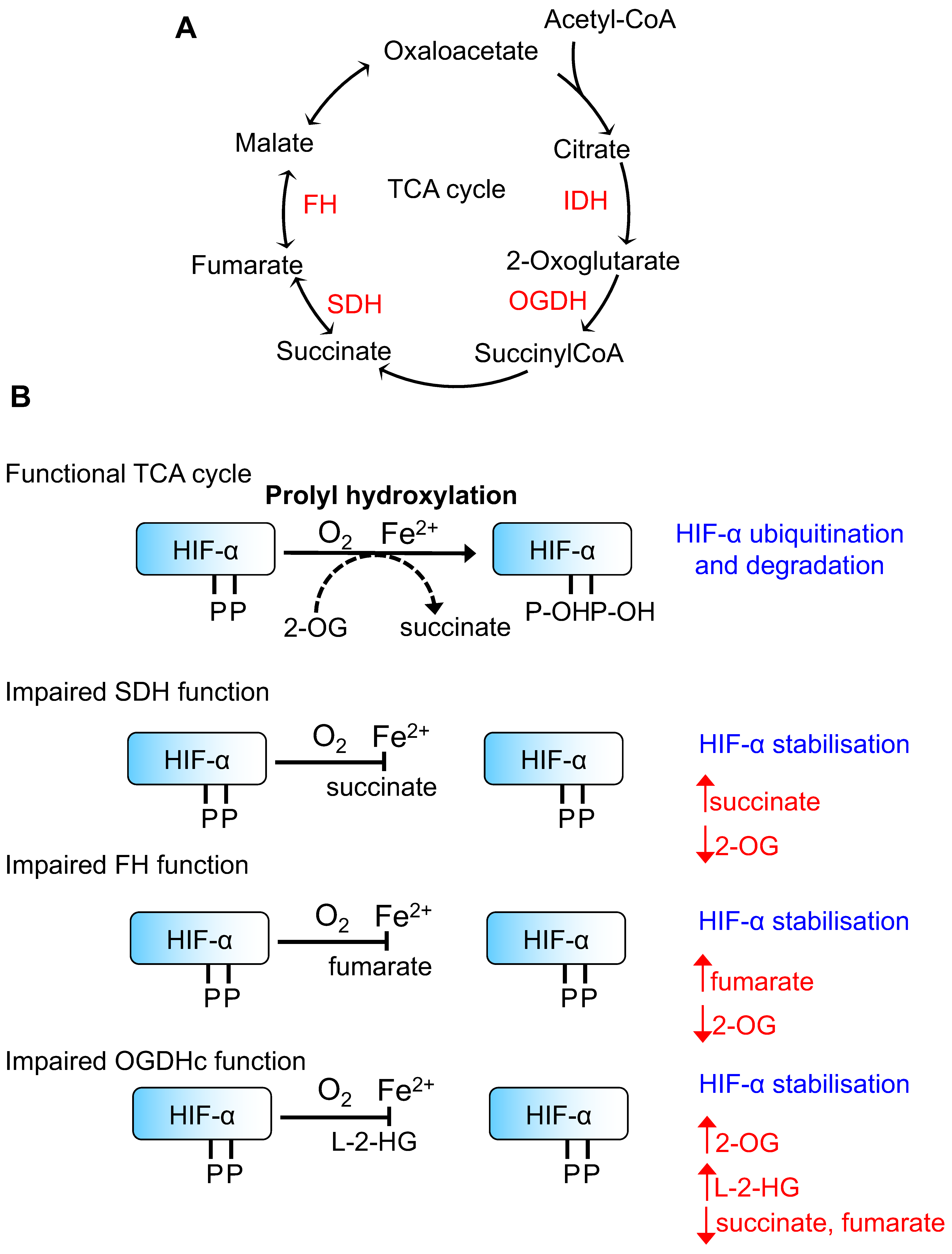
2. Regulation of HIFs by TCA Cycle Metabolites
3. Regulation of HIFs by 2-Hydroxyglutarate
3.1. d-2-HG and the HIF Response
3.2. l-2-HG Activates a HIF Response in Hypoxia or Following the Accumulation of 2-OG
4. Mitochondrial Lipoylation and the HIF Response
5. HIFs and Iron Metabolism
6. Metabolic Control of PHDs and Other 2-OG Dependent Dioxygenases in Disease
6.1. Regulation of Factor Inhibiting HIF by Small Molecule Metabolites
6.2. Metabolic Control of TETs and JHDMs
6.3. 2-Hydroxyglutarate and Cancers
6.4. l-2-Hydroxyglutarate in Cell Fate Decisions
7. Conclusions and Future Directions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G., Jr.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von hippel-lindau protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the hif hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: Evidence for a widespread oxygen-sensing mechanism. Proc. Natl. Acad. Sci. USA 1993, 90, 2423–2427. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von hippel-lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein vhl targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases phd1, phd2, and phd3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.R.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans egl-9 and mammalian homologs define a family of dioxygenases that regulate hif by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: The role of hif-1alpha, hif-2alpha, and other pathways. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Barahona, A.; Villar, D.; Pescador, N.; Amigo, J.; del Peso, L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010, 38, 2332–2345. [Google Scholar] [CrossRef] [PubMed]
- Schodel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of hif-binding sites by chip-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. Hif transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.K.; Walmsley, S.R. The regulation of pulmonary inflammation by the hypoxia-inducible factor-hydroxylase oxygen-sensing pathway. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. 5), S271–S276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Simon, M.C. From stem cells to cancer stem cells: Hif takes the stage. Curr. Opin. Cell Biol. 2012, 24, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.E.; Nickerson, M.L.; Brennan, P.; Toro, J.R.; Jaeger, E.; Rinsky, J.; Han, S.S.; Zaridze, D.; Matveev, V.; Janout, V.; et al. Von hippel-lindau (vhl) inactivation in sporadic clear cell renal cancer: Associations with germline vhl polymorphisms and etiologic risk factors. PLoS Genet. 2011, 7, e1002312. [Google Scholar] [CrossRef] [PubMed]
- Nabi, S.; Kessler, E.R.; Bernard, B.; Flaig, T.W.; Lam, E.T. Renal cell carcinoma: A review of biology and pathophysiology. F1000Res 2018, 7, 307. [Google Scholar] [CrossRef] [PubMed]
- Loenarz, C.; Schofield, C.J. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 2008, 4, 152–156. [Google Scholar] [CrossRef] [PubMed]
- McNeill, L.A.; Hewitson, K.S.; Gleadle, J.M.; Horsfall, L.E.; Oldham, N.J.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. The use of dioxygen by hif prolyl hydroxylase (phd1). Bioorg. Med. Chem. Lett. 2002, 12, 1547–1550. [Google Scholar] [CrossRef]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Skoldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit sdhb cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in sdhd, a mitochondrial complex ii gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Mourad, J.J.; Plouin, P.F.; Corvol, P.; Rotig, A.; Jeunemaitre, X. The r22x mutation of the sdhd gene in hereditary paraganglioma abolishes the enzymatic activity of complex ii in the mitochondrial respiratory chain and activates the hypoxia pathway. Am. J. Hum. Genet. 2001, 69, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A hif1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005, 1, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.H.; Toure, O.; Glenn, G.M.; Pithukpakorn, M.; Neckers, L.; Stolle, C.; Choyke, P.; Grubb, R.; Middelton, L.; Turner, M.L.; et al. Novel mutations in fh and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J. Med. Genet. 2006, 43, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline sdhb mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Yun, J.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. Hif overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of hif stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of krebs cycle intermediates and over-expression of hif1α in tumours which result from germline fh and sdh mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links tca cycle dysfunction to oncogenesis by inhibiting hif-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; Liénard, B.M.R.; McDonough, M.A.; Clifton, I.J.; Butler, D.; Soares, A.S.; Oldham, N.J.; McNeill, L.A.; Schofield, C.J. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J. Biol. Chem. 2007, 282, 3293–3301. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, C.J.; Loenarz, C.; Mecinovic, J.; Yeoh, K.K.; Hindley, N.; Lienard, B.M.; Sobott, F.; Schofield, C.J.; Flashman, E. Application of a proteolysis/mass spectrometry method for investigating the effects of inhibitors on hydroxylase structure. J. Med. Chem. 2009, 52, 2799–2805. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-kg-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of fh and sdh tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (hif) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of hif. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed]
- Abboud, M.I.; McAllister, T.E.; Leung, I.K.H.; Chowdhury, R.; Jorgensen, C.; Domene, C.; Mecinovic, J.; Lippl, K.; Hancock, R.L.; Hopkinson, R.J.; et al. 2-oxoglutarate regulates binding of hydroxylated hypoxia-inducible factor to prolyl hydroxylase domain 2. Chem. Commun. 2018, 54, 3130–3133. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Achouri, Y.; Noel, G.; Vertommen, D.; Rider, M.H.; Veiga-Da-Cunha, M.; Van Schaftingen, E. Identification of a dehydrogenase acting on d-2-hydroxyglutarate. Biochem. J. 2004, 381, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Rzem, R.; Veiga-da-Cunha, M.; Noel, G.; Goffette, S.; Nassogne, M.C.; Tabarki, B.; Scholler, C.; Marquardt, T.; Vikkula, M.; Van Schaftingen, E. A gene encoding a putative fad-dependent l-2-hydroxyglutarate dehydrogenase is mutated in l-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. USA 2004, 101, 16849–16854. [Google Scholar] [CrossRef] [PubMed]
- Kranendijk, M.; Struys, E.A.; Salomons, G.S.; Van der Knaap, M.S.; Jakobs, C. Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 2012, 35, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Moroni, I.; Bugiani, M.; D’Incerti, L.; Maccagnano, C.; Rimoldi, M.; Bissola, L.; Pollo, B.; Finocchiaro, G.; Uziel, G. l-2-hydroxyglutaric aciduria and brain malignant tumors: A predisposing condition? Neurology 2004, 62, 1882–1884. [Google Scholar] [CrossRef] [PubMed]
- Ozisik, P.A.; Akalan, N.; Palaoglu, S.; Topcu, M. Medulloblastoma in a child with the metabolic disease l-2-hydroxyglutaric aciduria. Pediatr. Neurosurg. 2002, 37, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Su, S.M. Isocitrate dehydrogenase mutation and (r)-2-hydroxyglutarate: From basic discovery to therapeutics development. Annu. Rev. Biochem. 2017, 86, 305–331. [Google Scholar] [CrossRef] [PubMed]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the idh1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated idh1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Kickingereder, P.; Sahm, F.; Radbruch, A.; Wick, W.; Heiland, S.; Deimling, A.; Bendszus, M.; Wiestler, B. Idh mutation status is associated with a distinct hypoxia/angiogenesis transcriptome signature which is non-invasively predictable with rcbv imaging in human glioma. Sci. Rep. 2015, 5, 16238. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (r)-enantiomer of 2-hydroxyglutarate linked to egln activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in idh1 dominantly inhibit idh1 catalytic activity and induce hif-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Ohka, F.; Ito, M.; Ranjit, M.; Senga, T.; Motomura, A.; Motomura, K.; Saito, K.; Kato, K.; Kato, Y.; Wakabayashi, T.; et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with idh1 mutation. Tumour Biol. 2014, 35, 5911–5920. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. d-2-hydroxyglutarate produced by mutant idh1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Tarhonskaya, H.; Rydzik, A.M.; Leung, I.K.H.; Loik, N.D.; Chan, M.C.; Kawamura, A.; McCullagh, J.S.O.; Claridge, T.D.W.; Flashman, E.; Schofield, C.J. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat. Commun. 2014, 5, 3423. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-mediated increases in l-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; DeMatteo, R.G.; Venneti, S.; Finley, L.W.S.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Burr, S.P.; Costa, A.S.H.; Grice, G.L.; Timms, R.T.; Lobb, I.T.; Freisinger, P.; Dodd, R.B.; Dougan, G.; Lehner, P.J.; Frezza, C.; et al. Mitochondrial protein lipoylation and the 2-oxoglutarate dehydrogenase complex controls hif1a stability in aerobic conditions. Cell Metab. 2016, 24, 740–752. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Wang, B.; Liu, H.; Shah, H.; Carmona-Fontaine, C.; Rustenburg, A.S.; Salah, S.; Gunner, M.R.; Chodera, J.D.; Cross, J.R.; et al. l-2-hydroxyglutarate production arises from noncanonical enzyme function at acidic ph. Nat. Chem. Biol. 2017, 13, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.M.; Schafer, X.; Fu, D.; Nehrke, K.; Munger, J.; Brookes, P.S. Acidic ph is a metabolic switch for 2-hydroxyglutarate generation and signaling. J. Biol. Chem. 2016, 291, 20188–20197. [Google Scholar] [CrossRef] [PubMed]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8+ t-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Briggs, K.J.; Koivunen, P.; Cao, S.; Backus, K.M.; Olenchock, B.A.; Patel, H.; Zhang, Q.; Signoretti, S.; Gerfen, G.J.; Richardson, A.L.; et al. Paracrine induction of hif by glutamate in breast cancer: Egln1 senses cysteine. Cell 2016, 166, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mtor and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Araújo, W.L.; Trofimova, L.; Mkrtchyan, G.; Steinhauser, D.; Krall, L.; Graf, A.; Fernie, A.R.; Bunik, V.I. On the role of the mitochondrial 2-oxoglutarate dehydrogenase complex in amino acid metabolism. Amino Acids 2013, 44, 683–700. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Feichtinger, R.G.; Tort, F.; Ribes, A.; Sperl, W. Lipoic acid biosynthesis defects. J. Inherit. Metab. Dis. 2014, 37, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, S.; Patel, M.S.; Jordan, F. Nuclear magnetic resonance approaches in the study of 2-oxo acid dehydrogenase multienzyme complexes-a literature review. Molecules 2013, 18, 11873–11903. [Google Scholar] [CrossRef] [PubMed]
- Soreze, Y.; Boutron, A.; Habarou, F.; Barnerias, C.; Nonnenmacher, L.; Delpech, H.; Mamoune, A.; Chrétien, D.; Hubert, L.; Bole-Feysot, C.; et al. Mutations in human lipoyltransferase gene lipt1 cause a leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase. Orphanet J. Rare Dis. 2013, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Paredes, F.; Sheldon, K.; Lassègue, B.; Williams, H.C.; Faidley, E.A.; Benavides, G.A.; Torres, G.; Sanhueza-Olivares, F.; Yeligar, S.M.; Griendling, K.K.; et al. Poldip2 is an oxygen-sensitive protein that controls pdh and αkgdh lipoylation and activation to support metabolic adaptation in hypoxia and cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Hernandes, M.S.; Lassegue, B.; Griendling, K.K. Polymerase delta-interacting protein 2: A multifunctional protein. J. Cardiovasc. Pharmacol. 2017, 69, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Shvetsova, A.N.; Mennerich, D.; Keratar, J.M.; Hiltunen, J.K.; Kietzmann, T. Non-electron transfer chain mitochondrial defects differently regulate hif-1alpha degradation and transcription. Redox Biol. 2017, 12, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondrial regulation of oxygen sensing. Adv. Exp. Med. Biol. 2010, 661, 339–354. [Google Scholar] [PubMed]
- Kietzmann, T.; Gorlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol. 2005, 16, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Greco, T.M.; Oberstein, A.; Budayeva, H.G.; Chakrabarti, R.; Rowland, E.A.; Kang, Y.; Shenk, T.; Cristea, I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Rowland, E.A.; Greco, T.M.; Snowden, C.K.; McCabe, A.L.; Silhavy, T.J.; Cristea, I.M. Sirtuin lipoamidase activity is conserved in bacteria as a regulator of metabolic enzyme complexes. MBio 2017, 8, e01096-17. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Koury, S.T.; Nejfelt, M.K.; Gearhart, J.D.; Antonarakis, S.E. Cell-type-specific and hypoxia-inducible expression of the human erythropoietin gene in transgenic mice. Proc. Natl. Acad. Sci. USA 1991, 88, 8725–8729. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J. Effect of hypoxic exposure on iron absorption in heterozygous hypotransferrinaemic mice. Ann. Hematol. 1992, 65, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Kvietikova, I.; Gassmann, M.; Wenger, R.H. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J. Biol. Chem. 1997, 272, 20055–20062. [Google Scholar] [CrossRef] [PubMed]
- Tacchini, L.; Gammella, E.; De Ponti, C.; Recalcati, S.; Cairo, G. Role of hif-1 and nf-kappab transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J. Biol. Chem. 2008, 283, 20674–20686. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Qu, A.; Anderson, E.R.; Matsubara, T.; Martin, A.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 2011, 140, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Investig. 2012, 122, 4635–4644. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J.; McKie, A.T. Iron and oxygen sensing: A tale of 2 interacting elements? Metall. Integr. Biomet. Sci. 2015, 7, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors hif in iron metabolism. Cell Cycle 2008, 7, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.A.; Song, H.K.; Lee, S.-H.; Chung, B.H.; Lim, H.M.; Lee, M.K. Differential in vitro and cellular effects of iron chelators for hypoxia inducible factor hydroxylases. J. Cell. Biochem. 2013, 114, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Hirsila, M.; Koivunen, P.; Xu, L.; Seeley, T.; Kivirikko, K.I.; Myllyharju, J. Effect of desferrioxamine and metals on the hydroxylases in the oxygen sensing pathway. FASEB J. 2005, 19, 1308–1310. [Google Scholar] [CrossRef] [PubMed]
- Frise, M.C.; Cheng, H.-Y.; Nickol, A.H.; Curtis, M.K.; Pollard, K.A.; Roberts, D.J.; Ratcliffe, P.J.; Dorrington, K.L.; Robbins, P.A. Clinical iron deficiency disturbs normal human responses to hypoxia. J. Clin. Investig. 2016, 126, 2139–2150. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Trowbridge, I.S.; Harris, A.L. Effects of transferrin receptor blockade on cancer cell proliferation and hypoxia-inducible factor function and their differential regulation by ascorbate. Cancer Res. 2006, 66, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Miles, A.L.; Burr, S.P.; Grice, G.L.; Nathan, J.A. The vacuolar-atpase complex and assembly factors, tmem199 and ccdc115, control hif1alpha prolyl hydroxylation by regulating cellular iron levels. Elife 2017, 6, 22693. [Google Scholar] [CrossRef] [PubMed]
- Nandal, A.; Ruiz, J.C.; Subramanian, P.; Ghimire-Rijal, S.; Sinnamon, R.A.; Stemmler, T.L.; Bruick, R.K.; Philpott, C.C. Activation of the hif prolyl hydroxylase by the iron chaperones pcbp1 and pcbp2. Cell Metab. 2011, 14, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Glasner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Siegert, I.; Schödel, J.; Nairz, M.; Schatz, V.; Dettmer, K.; Dick, C.; Kalucka, J.; Franke, K.; Ehrenschwender, M.; Schley, G.; et al. Ferritin-mediated iron sequestration stabilizes hypoxia-inducible factor-1α upon lps activation in the presence of ample oxygen. Cell Rep. 2015, 13, 2048–2055. [Google Scholar] [CrossRef] [PubMed]
- Kozik, P.; Hodson, N.A.; Sahlender, D.A.; Simecek, N.; Soromani, C.; Wu, J.; Collinson, L.M.; Robinson, M.S. A human genome-wide screen for regulators of clathrin-coated vesicle formation reveals an unexpected role for the v-atpase. Nat. Cell Biol. 2013, 15, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies ncoa4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- McNeill, L.A.; Flashman, E.; Buck, M.R.G.; Hewitson, K.S.; Clifton, I.J.; Jeschke, G.; Claridge, T.D.W.; Ehrismann, D.; Oldham, N.J.; Schofield, C.J. Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol. BioSyst. 2005, 1, 321. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. Fih-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Tarhonskaya, H.; Hardy, A.P.; Howe, E.A.; Loik, N.D.; Kramer, H.B.; McCullagh, J.S.; Schofield, C.J.; Flashman, E. Kinetic investigations of the role of factor inhibiting hypoxia-inducible factor (fih) as an oxygen sensor. J. Biol. Chem. 2015, 290, 19726–19742. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Lancaster, D.E.; Stolze, I.P.; Hewitson, K.S.; McDonough, M.A.; Coleman, M.L.; Coles, C.H.; Yu, X.; Hay, R.T.; Ley, S.C.; et al. Posttranslational hydroxylation of ankyrin repeats in ikappab proteins by the hypoxia-inducible factor (hif) asparaginyl hydroxylase, factor inhibiting hif (fih). Proc. Natl. Acad. Sci. USA 2006, 103, 14767–14772. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Linke, S.; Dias, J.M.; Zheng, X.; Gradin, K.; Wallis, T.P.; Hamilton, B.R.; Gustafsson, M.; Ruas, J.L.; Wilkins, S.; et al. Interaction with factor inhibiting hif-1 defines an additional mode of cross-coupling between the notch and hypoxia signaling pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 3368–3373. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, S.E.; Karttunen, S.; Hampton-Smith, R.J.; Murchland, I.; Chapman-Smith, A.; Peet, D.J. Factor inhibiting hif (fih) recognizes distinct molecular features within hypoxia-inducible factor-alpha (hif-alpha) versus ankyrin repeat substrates. J. Biol. Chem. 2012, 287, 8769–8781. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and inflammation in cancer, focus on hif and nf-kappab. Biomedicines 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.E., 3rd; Wu, Y.; Smith, K.; Charles, P.; Powers, K.; Wang, H.; Patterson, C. Asb4 is a hydroxylation substrate of fih and promotes vascular differentiation via an oxygen-dependent mechanism. Mol. Cell Biol. 2007, 27, 6407–6419. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.; Crifo, B.; Halligan, D.N.; et al. Fih regulates cellular metabolism through hydroxylation of the deubiquitinase otub1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [PubMed]
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. Tet enzymes, variants, and differential effects on function. Front. Cell Dev. Biol. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. Tet-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Markolovic, S.; Leissing, T.M.; Chowdhury, R.; Wilkins, S.E.; Lu, X.; Schofield, C.J. Structure-function relationships of human jmjc oxygenases-demethylases versus hydroxylases. Curr. Opin. Struct. Biol. 2016, 41, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Muller, U. Mutations in sdhc cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Briere, J.J.; Libe, R.; Vescovo, L.; Riviere, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Benit, P.; Tzagoloff, A.; et al. Sdha is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. Sdh mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Goncalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Ternette, N.; Yang, M.; Laroyia, M.; Kitagawa, M.; O’Flaherty, L.; Wolhulter, K.; Igarashi, K.; Saito, K.; Kato, K.; Fischer, R.; et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013, 3, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J., III. Chromatin succinylation correlates with active gene expression and is perturbed by defective tca cycle metabolism. iScience 2018, 2, 63–75. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic idh1 and idh2 mutations result in a hypermethylation phenotype, disrupt tet2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. Idh mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G., Jr. (r)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Anso, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chawla, G.; Hurlburt, A.J.; Sterrett, M.C.; Zaslaver, O.; Cox, J.; Karty, J.A.; Rosebrock, A.P.; Caudy, A.A.; Tennessen, J.M. Drosophila larvae synthesize the putative oncometabolite l-2-hydroxyglutarate during normal developmental growth. Proc. Natl. Acad. Sci. USA 2017, 114, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 2016, 166, 1324–1337.e11. [Google Scholar] [CrossRef] [PubMed]
- Salamanca-Cardona, L.; Shah, H.; Poot, A.J.; Correa, F.M.; Di Gialleonardo, V.; Lui, H.; Miloushev, V.Z.; Granlund, K.L.; Tee, S.S.; Cross, J.R.; et al. In vivo imaging of glutamine metabolism to the oncometabolite 2-hydroxyglutarate in idh1/2 mutant tumors. Cell Metab. 2017, 26, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Eckardt, K.U. Hif prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol. 2016, 12, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.L.; Leissing, T.M.; Abboud, M.I.; Thinnes, C.C.; Atasoylu, O.; Holt-Martyn, J.P.; Zhang, D.; Tumber, A.; Lippl, K.; Lohans, C.T.; et al. Molecular and cellular mechanisms of hif prolyl hydroxylase inhibitors in clinical trials. Chem. Sci. 2017, 8, 7651–7668. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Metabolite | PHD1 IC50 | PHD2 IC50 | PHD3 IC50 |
|---|---|---|---|
| Oxaloacetate [34] | 1 mM | 3.8 mM | 1.2 mM |
| Citrate [34] | 6.3 mM | 4.8 mM | 550 µM |
| Succinate [34] | 830 µM | 510 µM | 570 µM |
| Fumarate [34] | 120 µM | 80 µM | 60 µM |
| l-2-hydroxyglutarate [36] | 420 µM | ||
| d-2-hydroxyglutarate [36] | 7.3 mM |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailey, P.S.J.; Nathan, J.A. Metabolic Regulation of Hypoxia-Inducible Transcription Factors: The Role of Small Molecule Metabolites and Iron. Biomedicines 2018, 6, 60. https://doi.org/10.3390/biomedicines6020060
Bailey PSJ, Nathan JA. Metabolic Regulation of Hypoxia-Inducible Transcription Factors: The Role of Small Molecule Metabolites and Iron. Biomedicines. 2018; 6(2):60. https://doi.org/10.3390/biomedicines6020060
Chicago/Turabian StyleBailey, Peter S. J., and James A. Nathan. 2018. "Metabolic Regulation of Hypoxia-Inducible Transcription Factors: The Role of Small Molecule Metabolites and Iron" Biomedicines 6, no. 2: 60. https://doi.org/10.3390/biomedicines6020060
APA StyleBailey, P. S. J., & Nathan, J. A. (2018). Metabolic Regulation of Hypoxia-Inducible Transcription Factors: The Role of Small Molecule Metabolites and Iron. Biomedicines, 6(2), 60. https://doi.org/10.3390/biomedicines6020060