Skipping Multiple Exons to Treat DMD—Promises and Challenges




Abstract
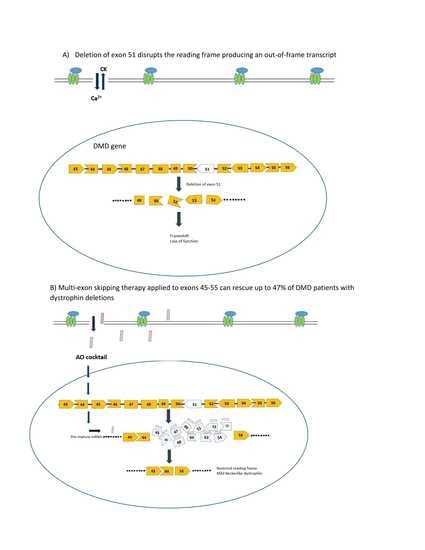
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
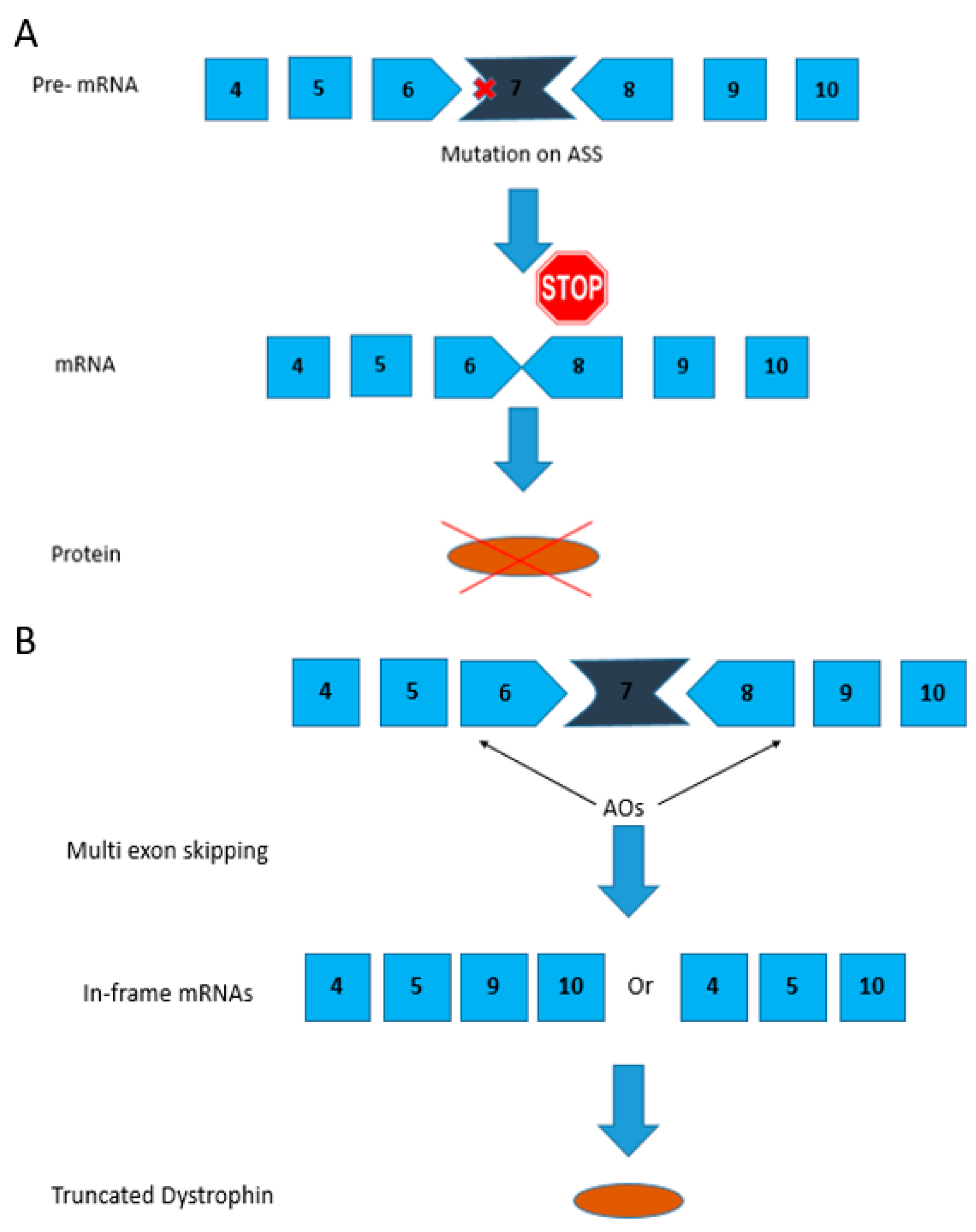
2. Advancements in Multi-Exon Skipping Therapy
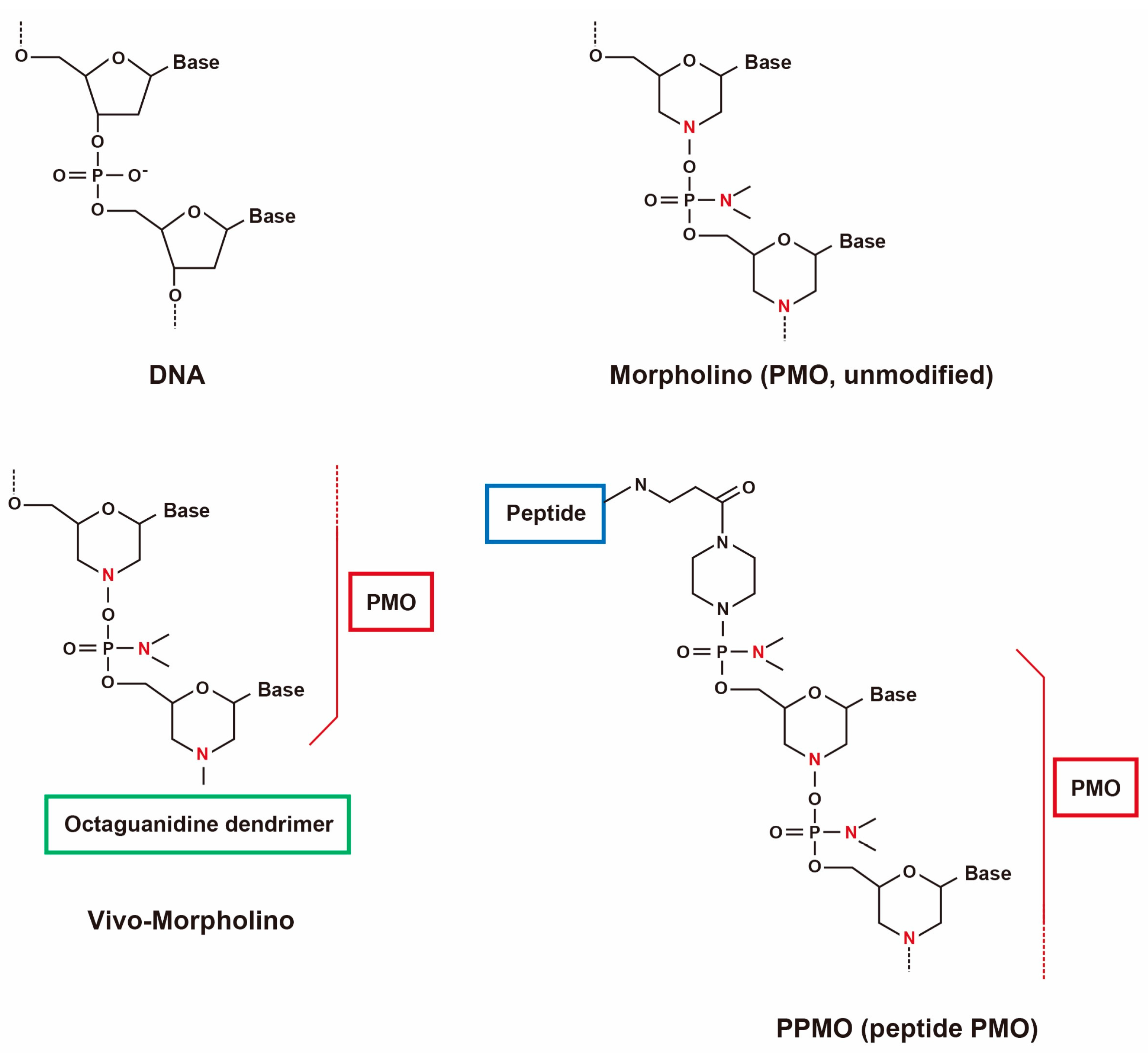
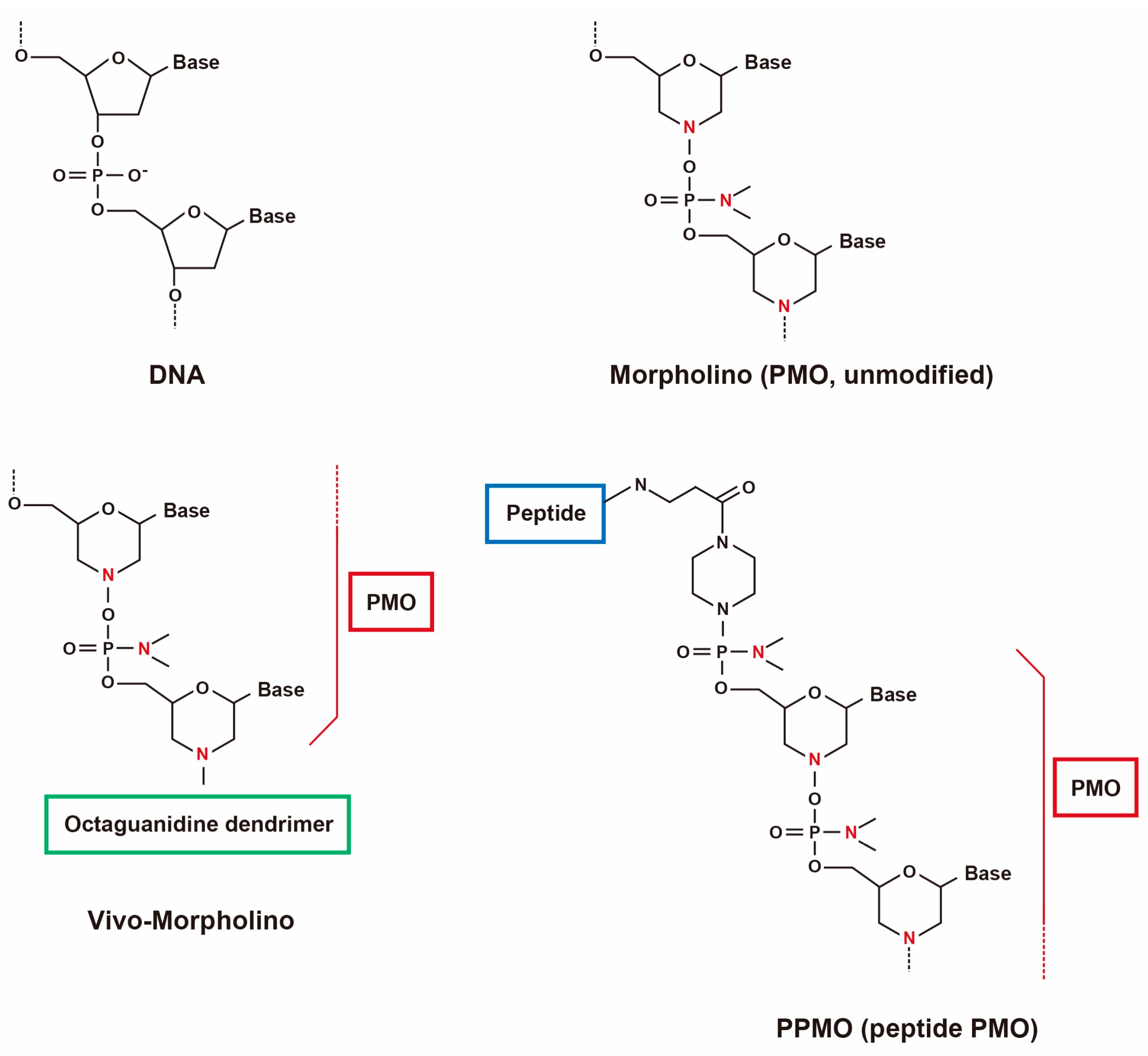
2.1. Use of Antisense Oligonucleotides and Phosphorodiamidate Morpholino Oligomers for Single- and Multi-Exon Skipping
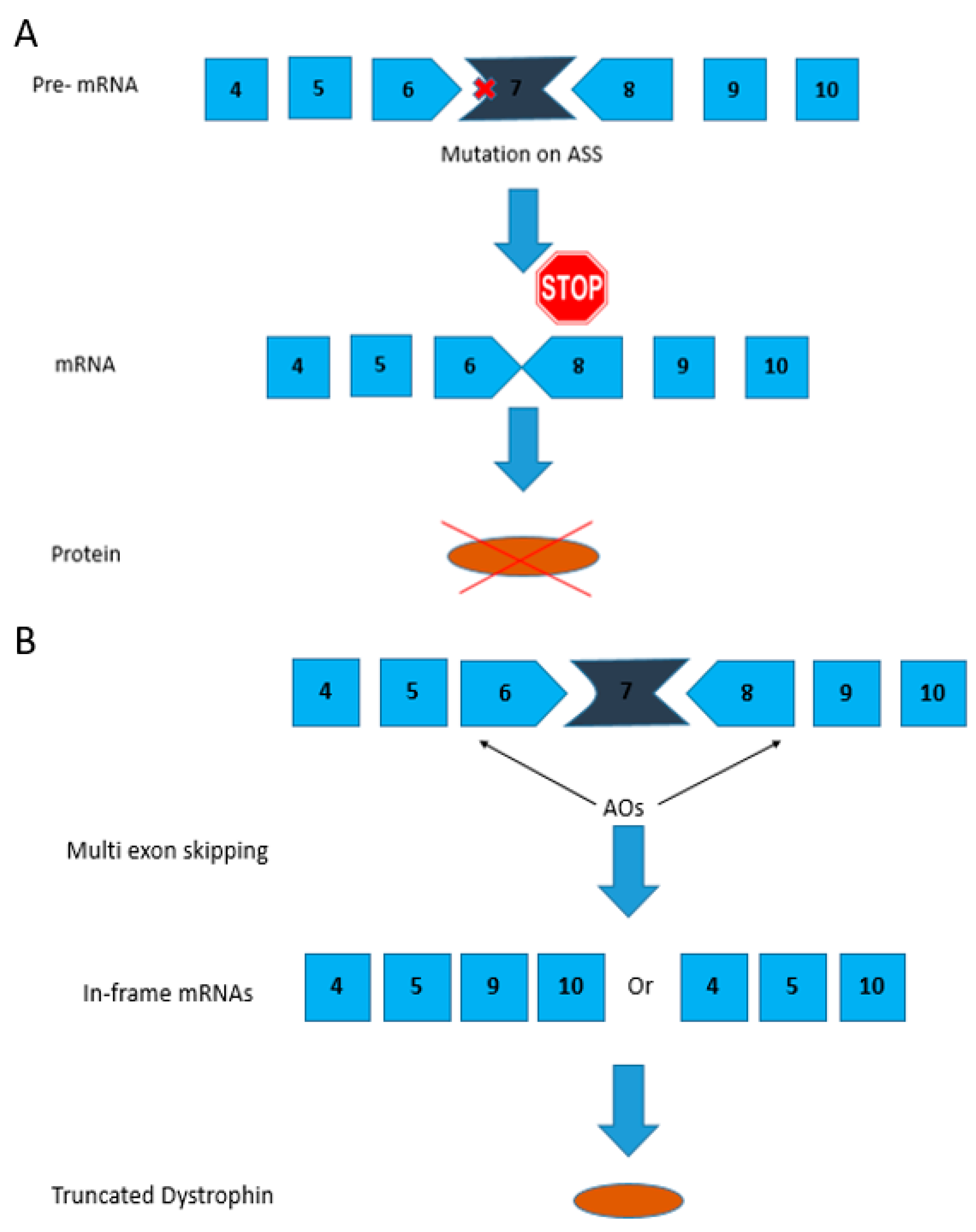
2.2. Exons 6–9 Multi-Exon Skipping Using PMOs in the Canine Model
2.3. Efficacy of Exons 6–9 Multi-Exon Skipping Using Peptide-Conjugated Morpholinos in the Heart of a Dog Model
2.4. Multi-Exon Skipping of Exons 3–9—A Potential Target for Therapy
2.5. Functional Correction of Dystrophin Actin-Binding Domain with DMD Exons 3–9 Deletion Using CRISPR/Cas9
- Generating del. Ex3–9 iPSCs by targeting introns 2 and 9 and the subsequent deletion of exons 3–7.
- Generating del. Ex6–9 iPSCs by targeting introns 5 and 7 and the subsequent deletion of exons 6–9.
- Generating del. Ex7–11 iPSCs by targeting introns 6 and 11 and the subsequent deletion of exons 7–11.
2.6. vPMO-Mediated Multi-Skipping of Exons 45–55 in Mdx52 Mice
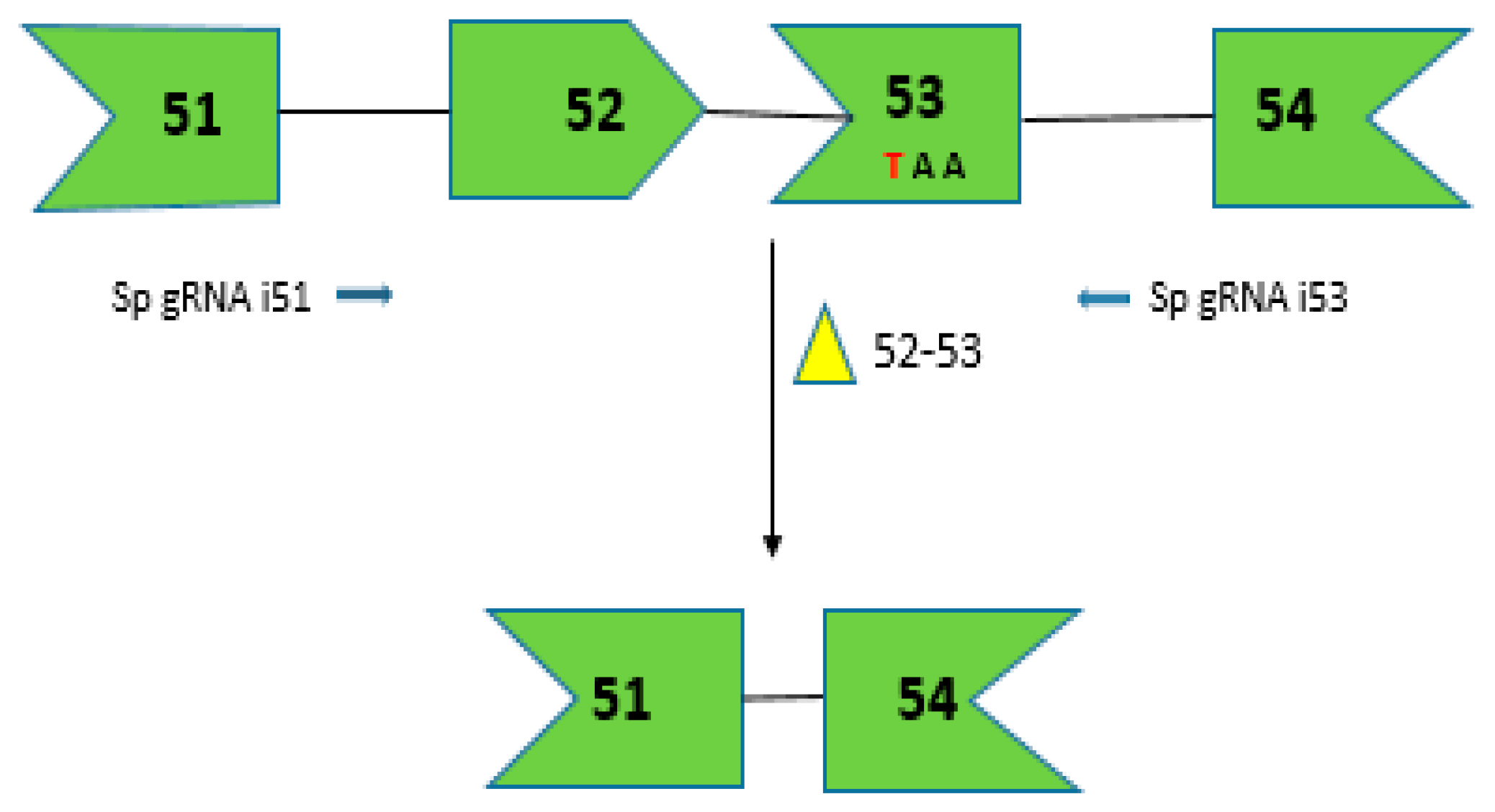
2.7. CRISPR/Cas9 for Multi-Exon Skipping Targeting DMD Exons 52–53
3. Future Implications and Clinical Hurdles
Acknowledgments
Conflicts of Interest
References
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Gardnermedwin, D. Controversies about Duchenne muscular-dystrophy. 1. Neonatal screening. Dev. Med. Child Neurol. 1979, 21, 390–393. [Google Scholar] [CrossRef]
- Duchenne, G.B. The pathology of paralysis with muscular degeneration (paralysie myosclerotique), or paralysis with apparent hypertrophy. Br. Med. J. 1867, 2, 541–542. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Matsumura, K.; Ohlendieck, K.; Ionasescu, V.V.; Tome, F.M.; Nonaka, I.; Burghes, A.H.; Mora, M.; Kaplan, J.C.; Fardeau, M.; Campbell, K.P. The role of the dystrophin-glycoprotein complex in the molecular pathogenesis of muscular dystrophies. Neuromuscul. Disord. NMD 1993, 3, 533–535. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Knudson, C.M.; Campbell, K.P.; Kunkel, L.M. Subcellular fractionation of dystrophin to the triads of skeletal muscle. Nature 1987, 330, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, E.; Samitt, C.E.; Miranda, A.F.; Hays, A.P.; Salviati, G.; DiMauro, S.; Kunkel, L.M.; Hoffman, E.P.; Rowland, L.P. Duchenne muscular dystrophy: Deficiency of dystrophin at the muscle cell surface. Cell 1988, 54, 447–452. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Muller, C.R.; Lindlof, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- Baumbach, L.L.; Chamberlain, J.S.; Ward, P.A.; Farwell, N.J.; Caskey, C.T. Molecular and clinical correlations of deletions leading to Duchenne and Becker muscular dystrophies. Neurology 1989, 39, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Rodino-Klapac, L.R.; Mendell, J.R.; Sahenk, Z. Update on the treatment of Duchenne muscular dystrophy. Curr. Neurol. Neurosci. Rep. 2013, 13, 332. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yokota, T. Translational research in nucleic acid therapies for muscular dystrophies. In Translational Research in Muscular Dystrophy; Takeda, S., Miyagoe-Suzuki, Y., Mori-Yoshimura, M., Eds.; Springer: Tokyo, Japan, 2016; pp. 87–102. [Google Scholar]
- England, S.B.; Nicholson, L.V.; Johnson, M.A.; Forrest, S.M.; Love, D.R.; Zubrzycka-Gaarn, E.E.; Bulman, D.E.; Harris, J.B.; Davies, K.E. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 1990, 343, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Burrow, K.L.; Coovert, D.D.; Klein, C.J.; Bulman, D.E.; Kissel, J.T.; Rammohan, K.W.; Burghes, A.H.; Mendell, J.R.; CIDD Study Group. Dystrophin expression and somatic reversion in prednisone-treated and untreated Duchenne dystrophy. Neurology 1991, 41, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Lu, Q.L.; Morgan, J.E.; Davies, K.E.; Fisher, R.; Takeda, S.; Partridge, T.A. Expansion of revertant fibers in dystrophic mdx muscles reflects activity of muscle precursor cells and serves as an index of muscle regeneration. J. Cell Sci. 2006, 119, 2679–2687. [Google Scholar] [CrossRef] [PubMed]
- Guncay, A.; Yokota, T. Antisense oligonucleotide drugs for Duchenne muscular dystrophy: How far have we come and what does the future hold? Future Med. Chem. 2015, 7, 1631–1635. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nakamura, A.; Yokota, T.; Saito, T.; Okazawa, H.; Nagata, T.; Takeda, S. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Wein, N.; Vulin, A.; Findlay, A.R.; Gumienny, F.; Huang, N.; Wilton, S.D.; Flanigan, K.M. Efficient skipping of single exon duplications in DMD patient-derived cell lines using an antisense oligonucleotide approach. J. Neuromuscul. Dis. 2017, 4, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Echigoya, Y.; Caluseriu, O.; Aoki, Y.; Takeda, S.; Yokota, T. Systemic delivery of morpholinos to skip multiple exons in a dog model of Duchenne muscular dystrophy. Methods Mol. Biol. 2017, 1565, 201–213. [Google Scholar] [PubMed]
- Yokota, T.; Duddy, W.; Echigoya, Y.; Kolski, H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: Too many mutations, too few patients? Expert Opin. Biol. Ther. 2012, 12, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Foster, H.; Popplewell, L.; Dickson, G. Genetic therapeutic approaches for Duchenne muscular dystrophy. Hum. Gene Ther. 2012, 23, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Yokota, T. Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotides. Nucleic Acid Ther. 2014, 24, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Banks, G.B.; Judge, L.M.; Allen, J.M.; Chamberlain, J.S. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010, 6, e1000958. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2, e95918. [Google Scholar] [CrossRef] [PubMed]
- Touznik, A.; Lee, J.J.; Yokota, T. New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Expert Opin. Biol. Ther. 2014, 14, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2007, 26, 179–184. [Google Scholar]
- Nakamura, A.; Fueki, N.; Shiba, N.; Motoki, H.; Miyazaki, D.; Nishizawa, H.; Echigoya, Y.; Yokota, T.; Aoki, Y.; Takeda, S. Deletion of exons 3–9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 2016, 61, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, V.; Giliberto, F.; Muniz, G.M.; Francipane, L.; Marzese, D.M.; Mampel, A.; Roque, M.; Frechtel, G.D.; Szijan, I. Asymptomatic Becker muscular dystrophy in a family with a multiexon deletion. Muscle Nerve 2009, 39, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Yoshida, K.; Fukushima, K.; Ueda, H.; Urasawa, N.; Koyama, J.; Yazaki, Y.; Yazaki, M.; Sakai, T.; Haruta, S.; et al. Follow-up of three patients with a large in-frame deletion of exons 45–55 in the Duchenne muscular dystrophy (DMD) gene. J. Clin. Neurosci. 2008, 15, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Beroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial dmd protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Toh, Z.Y.; Thandar Aung-Htut, M.; Pinniger, G.; Adams, A.M.; Krishnaswarmy, S.; Wong, B.L.; Fletcher, S.; Wilton, S.D. Deletion of dystrophin in-frame exon 5 leads to a severe phenotype: Guidance for exon skipping strategies. PLoS ONE 2016, 11, e0145620. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.R.; Wein, N.; Kaminoh, Y.; Taylor, L.E.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. Clinical phenotypes as predictors of the outcome of skipping around dmd exon 45. Ann. Neurol. 2015, 77, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Shiba, N.; Miyazaki, D.; Nishizawa, H.; Inaba, Y.; Fueki, N.; Maruyama, R.; Echigoya, Y.; Yokota, T. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J. Hum. Genet. 2017, 62, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Aoki, Y.; Miskew, B.; Panesar, D.; Touznik, A.; Nagata, T.; Tanihata, J.; Nakamura, A.; Nagaraju, K.; Yokota, T. Long-term efficacy of systemic multiexon skipping targeting dystrophin exons 45–55 with a cocktail of vivo-morpholinos in mdx52 mice. Mol. Ther. Nucleic Acids 2015, 4, e225. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 13763–13768. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Lu, Q.L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Devos, S.L.; Miller, T.M. Antisense oligonucleotides: Treating neurodegeneration at the level of RNA. Neurotherapeutics 2013, 10, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.G.; Wood, M.J. Splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Yokota, T. Antisense therapy in neurology. J. Pers. Med. 2013, 3, 144–176. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.; Devi, G.R.; Iversen, P.L. Neutrally charged phosphorodiamidate morpholino antisense oligomers: Uptake, efficacy and pharmacokinetics. Curr. Pharm. Biotechnol. 2004, 5, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Mizobe, Y.; Takizawa, H.; Hara, Y.; Yokota, T.; Takeda, S.; Aoki, Y. Exon skipping therapy using phosphorodiamidate morpholino oligomers in the mdx52 mouse model of Duchenne muscular dystrophy. Methods Mol. Biol. 2018, 1687, 123–141. [Google Scholar] [PubMed]
- Yokota, T.; Hoffman, E.; Takeda, S. Antisense oligo-mediated multiple exon skipping in a dog model of Duchenne muscular dystrophy. Methods Mol. Biol. 2011, 709, 299–312. [Google Scholar] [PubMed]
- Goyenvalle, A.; Babbs, A.; Powell, D.; Kole, R.; Fletcher, S.; Wilton, S.D.; Davies, K.E. Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Bronson, A.; Levin, A.A.; Takeda, S.; Yokota, T.; Baudy, A.R.; Connor, E.M. Restoring dystrophin expression in Duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am. J. Pathol. 2011, 179, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.S.; Hogarth, M.W.; Boehler, J.F.; Nearing, M.; Vila, M.C.; Heredia, R.; Fiorillo, A.A.; Zhang, A.; Hathout, Y.; Hoffman, E.P.; et al. Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat. Commun. 2017, 8, 941. [Google Scholar] [CrossRef] [PubMed]
- Anthony, K.; Feng, L.; Arechavala-Gomeza, V.; Guglieri, M.; Straub, V.; Bushby, K.; Cirak, S.; Morgan, J.; Muntoni, F. Exon skipping quantification by quantitative reverse-transcription polymerase chain reaction in Duchenne muscular dystrophy patients treated with the antisense oligomer eteplirsen. Hum. Gene Ther. Methods 2012, 23, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.; Yokota, T. Immortalized muscle cell model to test the exon skipping efficacy for Duchenne muscular dystrophy. J. Pers. Med. 2017, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokota, T.; Wood, M.J. Development of multiexon skipping antisense oligonucleotide therapy for Duchenne muscular dystrophy. BioMed Res. Int. 2013, 2013, 402369. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Fletcher, S.; Neuman, B.W.; McClorey, G.; Stein, D.A.; Abes, S.; Wilton, S.D.; Buchmeier, M.J.; Lebleu, B.; Iversen, P.L. Cell-penetrating peptide-morpholino conjugates alter pre-mRNA splicing of DMD (Duchenne muscular dystrophy) and inhibit murine coronavirus replication in vivo. Biochem. Soc. Trans. 2007, 35, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M. In vivo delivery of morpholino oligos by cell-penetrating peptides. Curr. Pharm. Des. 2013, 19, 2963–2969. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Nakamura, A.; Nagata, T.; Saito, T.; Kobayashi, M.; Aoki, Y.; Echigoya, Y.; Partridge, T.; Hoffman, E.P.; Takeda, S. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012, 22, 306–315. [Google Scholar] [PubMed]
- Echigoya, Y.; Nakamura, A.; Nagata, T.; Urasawa, N.; Lim, K.R.Q.; Trieu, N.; Panesar, D.; Kuraoka, M.; Moulton, H.M.; Saito, T.; et al. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 4213–4218. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cloer, C.; Lu, P.; Milazi, S.; Shaban, M.; Shah, S.N.; Marston-Poe, L.; Moulton, H.M.; Lu, Q.L. Exon skipping restores dystrophin expression, but fails to prevent disease progression in later stage dystrophic dko mice. Gene Ther. 2014, 21, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Crisp, A.; Yin, H.; Goyenvalle, A.; Betts, C.; Moulton, H.M.; Seow, Y.; Babbs, A.; Merritt, T.; Saleh, A.F.; Gait, M.J.; et al. Diaphragm rescue alone prevents heart dysfunction in dystrophic mice. Hum. Mol. Genet. 2011, 20, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Wu, B.; Jearawiriyapaisarn, N.; Sazani, P.; Lu, Q.L.; Kole, R. Peptide-morpholino conjugate: A promising therapeutic for Duchenne muscular dystrophy. Ann. N. Y. Acad. Sci. 2009, 1175, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Bao, B.; Echigoya, Y.; Yokota, T. Dystrophin-deficient large animal models: Translational research and exon skipping. Am. J. Transl. Res. 2015, 7, 1314–1331. [Google Scholar] [PubMed]
- Miskew Nichols, B.; Aoki, Y.; Kuraoka, M.; Lee, J.J.; Takeda, S.; Yokota, T. Multi-exon skipping using cocktail antisense oligonucleotides in the canine X-linked muscular dystrophy. J. Vis. Exp. JoVE 2016, 111, 53776. [Google Scholar] [CrossRef] [PubMed]
- Al-Attar, S.; Westra, E.R.; van der Oost, J.; Brouns, S.J. Clustered regularly interspaced short palindromic repeats (CRISPRs): The hallmark of an ingenious antiviral defense mechanism in prokaryotes. Biol. Chem. 2011, 392, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.Q.; Fisher, A.L.; Huang, H.C.; Xie, Z.J. CRISPR-mediated genome editing and human diseases. Genes Dis. 2016, 3, 244–251. [Google Scholar] [CrossRef]
- Miyazaki, D.; Yoshida, K.; Fukushima, K.; Nakamura, A.; Suzuki, K.; Sato, T.; Takeda, S.; Ikeda, S. Characterization of deletion breakpoints in patients with dystrophinopathy carrying a deletion of exons 45–55 of the Duchenne muscular dystrophy (DMD) gene. J. Hum. Genet. 2009, 54, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokota, T.; Saito, T.; Nakamura, A.; Takeda, S.I. Exon 51 Skipping by Morpholinos Can Restore Dystrophin Expression, Muscle Pathology and Function in Exon 52-Deficient mdx52 Mice. In Molecular Therapy; Nature Publishing Group: New York, NY, USA, 2009; p. S386. [Google Scholar]
- Pearson-White, S.H. DMD(mdx3cv) and DMD(mdx4cv) dystrophin mutations in mice: Rapid polymerase chain reaction genotyping. Neuromuscul. Disord. NMD 2002, 12, 366–370. [Google Scholar] [CrossRef]
- Pramono, Z.A.; Takeshima, Y.; Alimsardjono, H.; Ishii, A.; Takeda, S.; Matsuo, M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 1996, 226, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Takeda, S.; Lu, Q.L.; Partridge, T.A.; Nakamura, A.; Hoffman, E.P. A renaissance for antisense oligonucleotide drugs in neurology: Exon skipping breaks new ground. Arch. Neurol. 2009, 66, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Blankinship, M.J.; Allen, J.M.; Crawford, R.W.; Meuse, L.; Miller, D.G.; Russell, D.W.; Chamberlain, J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004, 10, 828–834. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslesh, T.; Maruyama, R.; Yokota, T. Skipping Multiple Exons to Treat DMD—Promises and Challenges. Biomedicines 2018, 6, 1. https://doi.org/10.3390/biomedicines6010001
Aslesh T, Maruyama R, Yokota T. Skipping Multiple Exons to Treat DMD—Promises and Challenges. Biomedicines. 2018; 6(1):1. https://doi.org/10.3390/biomedicines6010001
Chicago/Turabian StyleAslesh, Tejal, Rika Maruyama, and Toshifumi Yokota. 2018. "Skipping Multiple Exons to Treat DMD—Promises and Challenges" Biomedicines 6, no. 1: 1. https://doi.org/10.3390/biomedicines6010001
APA StyleAslesh, T., Maruyama, R., & Yokota, T. (2018). Skipping Multiple Exons to Treat DMD—Promises and Challenges. Biomedicines, 6(1), 1. https://doi.org/10.3390/biomedicines6010001