Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity


Abstract
1. Introduction
2. The Nuclear Factor κB (NF-κB) Pathway
2.1. The Canonical NF-κB Pathway
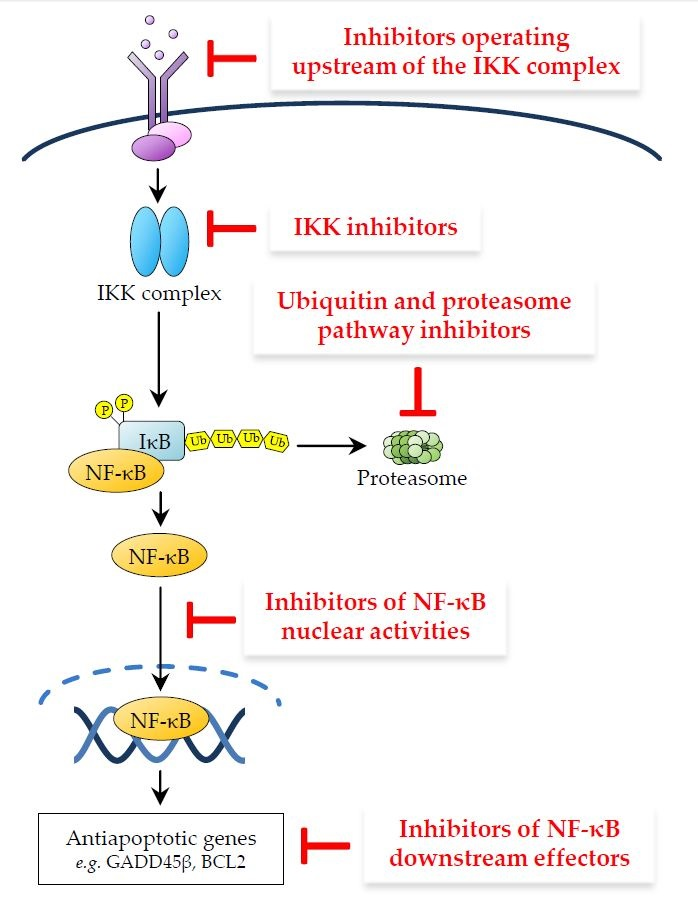
2.1.1. NF-κB Activation by IL-1β Receptor (IL-1βR) and Pattern Recognition Receptors (PRRs)
2.1.2. NF-κB Activation by TNF-R1
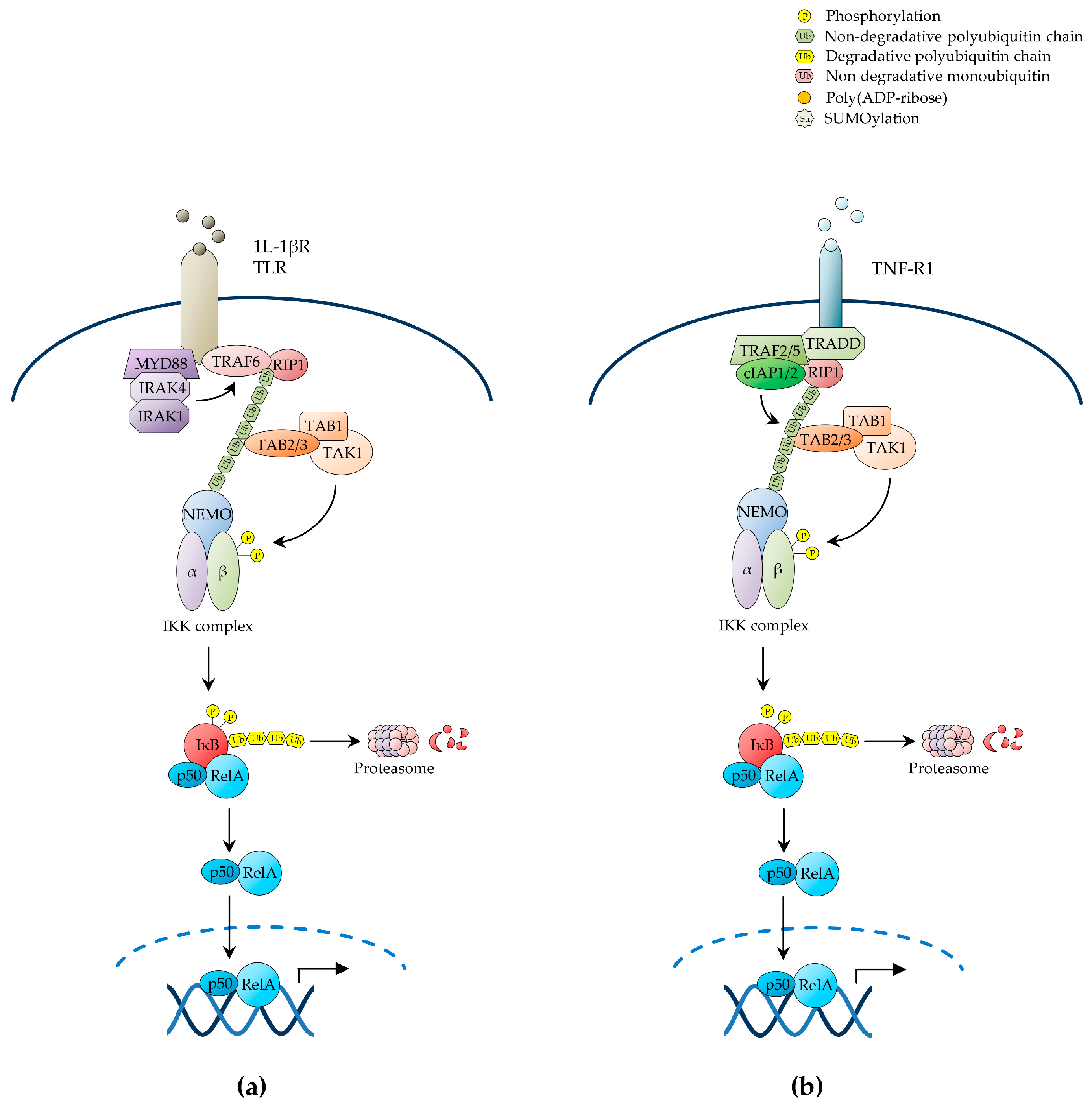
2.1.3. NF-κB Activation by Antigen Receptors
2.1.4. NF-κB Activation by Genotoxic Stress
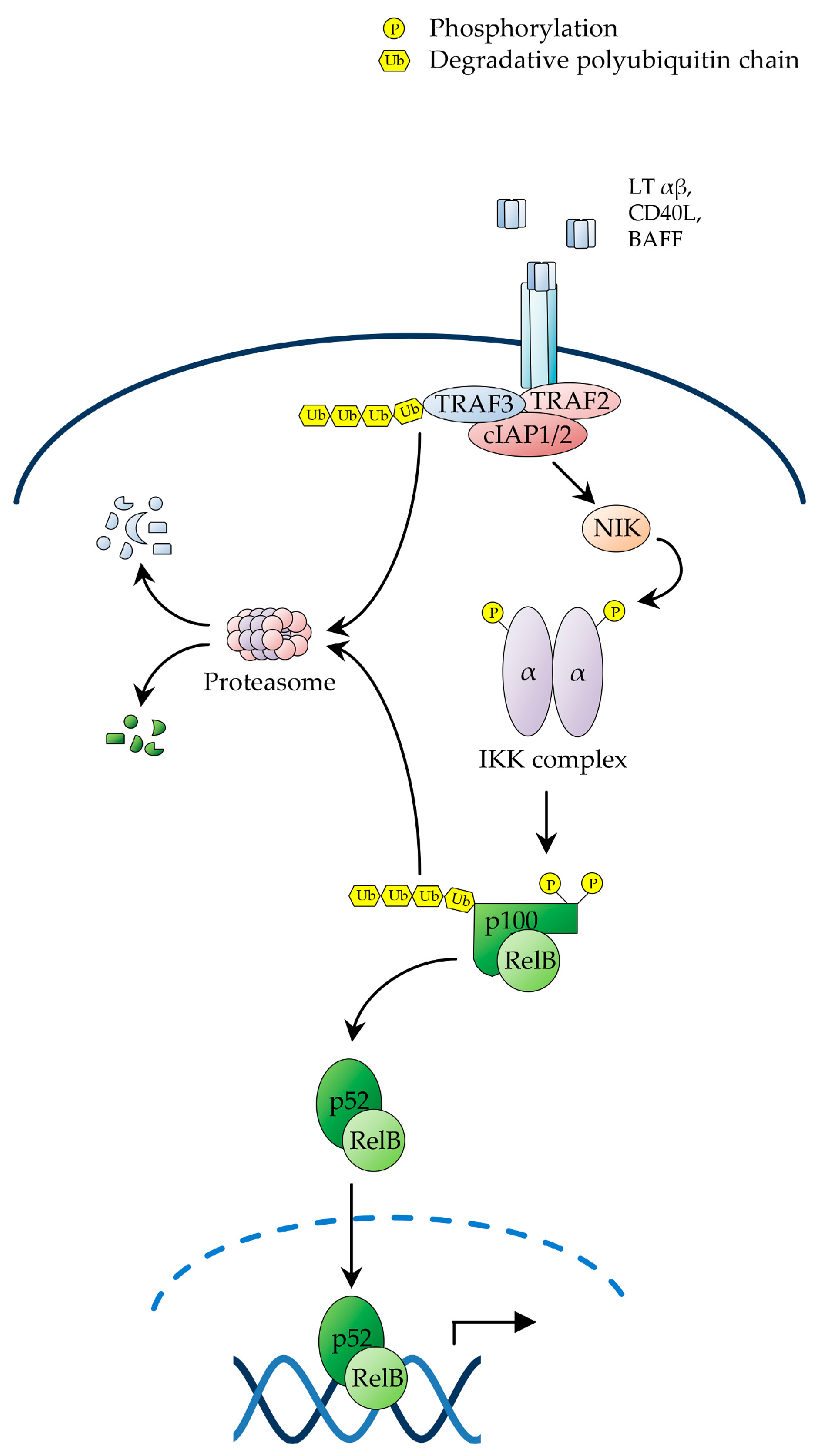
2.2. The Non-Canonical NF-κB Pathway
2.3. Termination of NF-κB Signalling
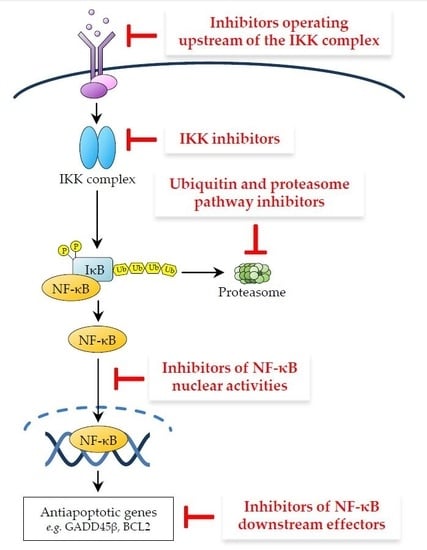
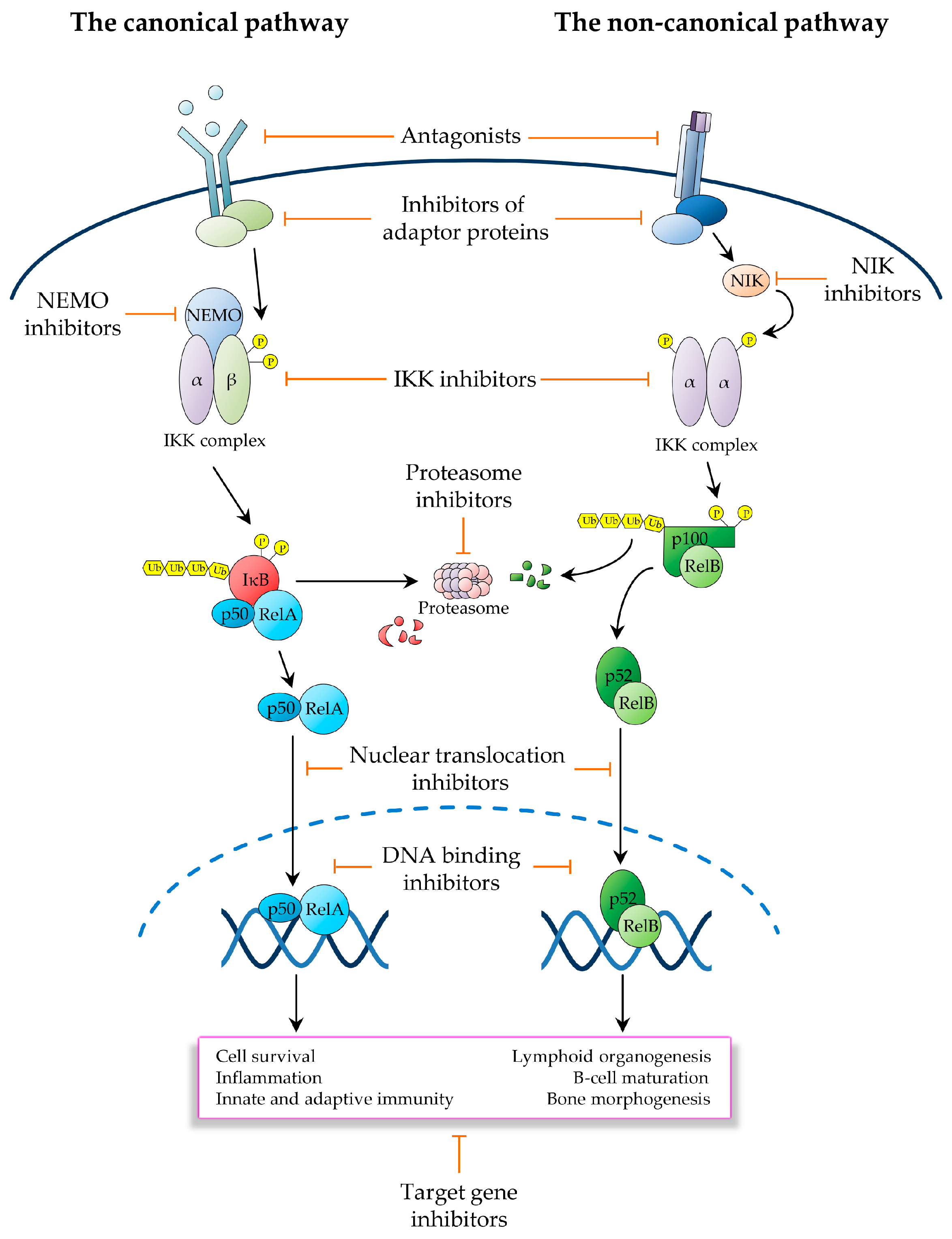
3. Therapeutic Targeting of the NF-κB Pathway in Cancer
3.1. Inhibitors Operating Upstream of the IKK Complex
3.2. IKK Inhibitors
3.3. Ubiquitin and Proteasome Pathway Inhibitors
3.4. Inhibitors of NF-κB Nuclear Activities
3.5. Inhibitors of NF-κB Downstream Effectors
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| A1 | BCL2A1—BCL2-related protein A1 |
| A20 | TNFAIP3 —Tumour necrosis factor α-induced protein 3 |
| ABIN-1 | A20 binding inhibitor of NF-κB |
| ADAP | Adhesion- and degranulation-promoting adaptor protein |
| ALCL | Anaplastic large-cell lymphoma |
| ALL | Acute lymphoblastic leukaemia |
| AML | Acute myeloid leukaemia |
| ARD | Ankyrin repeat domain |
| ATM | Ataxia telangiectasia mutated |
| BAD | Bcl-2-associated death promoter |
| BAFF | B-cell activating factor |
| BAK | BCL-2 antagonist/killer |
| BAX | BCL-2-associated X protein |
| BCL10 | B-cell CLL/lymphoma 10 |
| BCL-2 | B-cell lymphoma 2 |
| Bcl-XL | B-cell lymphoma-extra large |
| BCR | B-cell receptor |
| BH | BCL-2 homology |
| BID | BH3 interacting-domain death agonist |
| BIM | BCL-2-interacting mediator of cell death |
| BIR | Baculovirus IAP repeat |
| CAML | Calcium modulating ligand |
| CARD | Caspase recruitment domain |
| CBM | CARD11-BCL10-MALT1 |
| CD40L | CD40 ligand |
| Cdc25A | Cell division cycle 25A |
| c-FLIP | Cellular FLICE/caspase8-like inhibitory protein |
| c-IAP | Cellular inhibitor of apoptosis |
| CLL | Chronic lymphocytic leukaemia |
| CYLD | Cylindromatosis lysine 63 deubiquitinase |
| DAMP | Damage-associated molecular patterns |
| DHMEQ | Dehydroxymethylepoxyquinomicin |
| DLBCL | Diffuse large B-cell lymphoma |
| DMAPT | Dimethylaminoparthenolide |
| DUB | Deubiquitination enzyme |
| Emi1-FBXO 5 | F-box protein 5 |
| EMT | Epithelial-mesenchymal transition |
| FBXW | F-box and WD repeat domain containing |
| FDA | US Food and Drug Administration |
| FL | Follicular lymphoma |
| GADD45β | Growth arrest and DNA damage inducible β |
| GBM | Glioblastoma multiforme |
| GRR | Glycine rich region |
| GSK3 | Glycogen synthase kinase 3 |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylases |
| HL | Hodgkin’s lymphoma |
| HOIL-1L | RBCK1—RanBP-type and C3HC4-type zinc finger containing 1 |
| HOIP | RNF31—Ring finger protein 31 |
| IKK | IκB kinase |
| IL-1β | Interleukin 1 β |
| IL-1β | Interleukin 1β |
| IL-1βR | IL-1β receptor |
| IRAK | Interleukin-1 receptor-associated kinase |
| IκB | Nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor |
| JAK | Janus kinase |
| LPS | Lipopolysaccharide |
| LTβ | Lymphotoxin β |
| LUBAC | Linear ubiquitination assembly complex |
| MALT | Mucosa-associated lymphoid tissue |
| MALT1 | Mucosa-associated lymphoid tissue lymphoma translocation protein 1 |
| MAP | Mitogen activated protein |
| MAPKKK | Mitogen activated protein kinase kinase kinase |
| MAVS | Mitochondrial antiviral signalling protein |
| MCL | Mantle-cell lymphoma |
| MCL1 | Myeloid cell leukaemia sequence 1 |
| MDS | Myelodysplastic syndrome |
| MEKK | Mitogen activated protein kinase kinase |
| MM | Multiple myeloma |
| mTOR | Mechanistic target of rapamycin |
| MYD88 | Myeloid differentiation primary response protein 88 |
| NEDD8 | Neural precursor cell-expressed developmentally down-regulated 8 |
| NES | Nuclear export signal |
| NF-κB | Nuclear factor κ B (Nuclear Factor binding to the κ-Light-chain-enhancer B site) |
| NHL | Non-Hodgkin’s lymphoma |
| NIK | NF-κB-inducing kinase |
| NLR | NOD-like receptor |
| NLS | Nuclear localization signals |
| NOD | Nucleotide-binding oligomerisation domain |
| NSCLC | Non-Small Cell Lung Cancer |
| OTUD7B | OTU domain-containing protein 7B |
| PAMP | Pathogen-associated molecular pattern |
| PAR | Poly(ADP-ribose) |
| PARP-1 | Poly(ADP-ribose)-polymerase-1 |
| PDK | Phosphoinoisitide-dependent kinase |
| PDK1 | 3-Phosphoinositide-dependent protein kinase 1 |
| PDLIM2 | PDZ and LIM domain protein 2 |
| PIASy | Protein inhibitor of activated STAT protein y |
| PIDD | p53-induced death domain protein |
| PKC | Protein kinase C |
| PP | Protein phosphatase |
| PTEN | Phosphatase and tensin homolog |
| PUMA | p53 upregulated modulator of apoptosis |
| RANKL | Receptor activator of NF-κB ligand |
| RAS | Rat sarcoma virus oncogene |
| RBX1 | RING-box protein 1 |
| RHD | Rel-homology Domain |
| RIP | Receptor interacting protein |
| RLH | RNA helicase |
| RLR | RIG-I-like receptor |
| SCF | SKP1-Cullin 1-F-box protein |
| Sharpin | SHANK-associated RH domain interacting protein |
| SL | Sesquiterpene lactone |
| SMAC | Second mitochondria-derived activator of caspases |
| SOCS-1 | Suppressor of cytokine signalling-1 |
| STAT | Signal transducer and activator of transcription |
| SUMO | Small ubiquitin-like modifiers |
| TAB | TAK1-associated binding protein |
| TACI | TNFRSF13B—TNF receptor superfamily member 13B |
| TAK1 | Transforming growth factor β-activated kinase 1 |
| T-ALL | T-cell acute lymphoblastic leukaemia |
| TBK1 | TANK-binding kinase |
| TCR | T-cell receptor |
| TLR | Toll-like receptor |
| TME | Tumour microenvironment |
| TNF-R | TNF receptor |
| TNFSF | Tumour necrosis factor superfamily member |
| TNF-α | Tumour necrosis factor-α |
| TRADD | TNF receptor-associated death domain protein |
| TRAF | TNF receptor-associated factor |
| TRIM23 | Tripartite motif protein 23 |
| TWEAK | TNF-related weak inducer of apoptosis |
| UBA | Ubiquitin-activating enzyme, E1 |
| UBC | Ubiquitin-conjugating, E2 |
| Ubc/Uev | Ubiquitin-conjugating enzyme/ubiquitin E2 variant |
| UBD | Ubiquitin-binding domain |
| UPP | Ubiquitin-proteasome pathway |
| UPR | Unfolded protein response |
| USP | Ubiquitin-specific peptidase |
| WIP1 | Wild-type p53-induced phosphatase 1 |
| WM | Waldenström’s macroglobulinemia |
| WWOX | WW domain-containing oxidoreductase |
| βTrCP | β-transducin repeat-containing protein |
References
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-κB system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Staudt, L.M. Oncogenic activation of NF-κB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, Y.N.; Glebov, O.K.; Zingone, A.; Keats, J.J.; Bergsagel, P.L.; Kuehl, W.M. Classical and/or alternative NF-κB pathway activation in multiple myeloma. Blood 2010, 115, 3541–3552. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat. Rev. Clin. Oncol. 2014, 11, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dalla-Favera, R. SnapShot: Diffuse large B cell lymphoma. Cancer Cell 2014, 25, 132.e1. [Google Scholar] [CrossRef] [PubMed]
- Bredel, M.; Scholtens, D.M.; Yadav, A.K.; Alvarez, A.A.; Renfrow, J.J.; Chandler, J.P.; Yu, I.L.; Carro, M.S.; Dai, F.; Tagge, M.J.; et al. NFKBIA deletion in glioblastomas. N. Engl. J. Med. 2011, 364, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.E.; Morshed, R.A.; Yamini, B. Nuclear factor-κB in glioblastoma: Insights into regulators and targeted therapy. Neuro-Oncology 2015, 18, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Schaefer, B.C. A new look at T cell receptor signaling to nuclear factor-κB. Trends Immunol. 2013, 34, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xia, Y.; Parker, A.S.; Verma, I.M. IKK biology. Immunol. Rev. 2012, 246, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitin signalling in the NF-κB pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-κB subunits in the control of transcription. Cells 2016, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Dixit, V.M. Signaling to NF-κB: Regulation by ubiquitination. Cold Spring Harb. Perspect. Biol. 2010, 2, a003350. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Ley, S.C. New insights into NF-κB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Dimer-specific regulatory mechanisms within the NF-κB family of transcription factors. Immunol. Rev. 2012, 246, 193–204. [Google Scholar] [CrossRef] [PubMed]
- NF-κB. 2017. Available online: www.nf-kb.org (accessed on 27 January 2017).
- Häcker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Millman, S.E.; Pagano, M. SCF-mediated degradation of p100 (NF-κB2): Mechanisms and relevance in multiple myeloma. Sci. Signal. 2012, 5, pt14. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good cop, bad cop: The different faces of NF-κB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Sun, L.J. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Spencer, E.; Jiang, J.; Chen, Z.J. Signal-induced ubiquitination of IκBα by the F-box protein Slimb/β-TrCP. Genes Dev. 1999, 13, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Sun, Y. Small RING Finger Proteins RBX1 and RBX2 of SCF E3 Ubiquitin Ligases: The Role in Cancer and as Cancer Targets. Genes Cancer 2010, 1, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Laplantine, E.; Fontan, E.; Chiaravalli, J.; Lopez, T.; Lakisic, G.; Véron, M.; Agou, F.; Israël, A. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009, 28, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Delhase, M.; Hayakawa, M.; Chen, Y.; Karin, M. Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science 1999, 284, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Schmukle, A.C.; Walczak, H. No one can whistle a symphony alone—How different ubiquitin linkages cooperate to orchestrate NF-κB activity. J. Cell Sci. 2012, 125, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK β suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Ju, T.K.; Hung, M.C.; Chen, C.C. Phosphorylation of CBP by IKKα promotes cell growth by switching the binding preference of CBP from p53 to NF-κB. Mol. Cell 2007, 26, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.; Hayden, M.S.; Long, M.; Scott, M.L.; West, A.P.; Zhang, D.; Oeckinghaus, A.; Lynch, C.; Hoffmann, A.; Baltimore, D.; et al. IκBβ acts to inhibit and activate gene expression during the inflammatory response. Nature 2010, 466, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Xu, M.; Skaug, B.; Zeng, W.; Chen, Z.J. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFα and IL-1β. Mol. Cell 2009, 36, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Le Negrate, G. Viral interference with innate immunity by preventing NF-κB activity. Cell. Microbiol. 2012, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Hecker, C.M.; Rozenknop, A.; Nordmeier, R.D.; Rogov, V.; Hofmann, K.; Akira, S.; Dötsch, V.; Dikic, I. Involvement of the ubiquitin-like domain of TBK1/IKK-i kinases in regulation of IFN-inducible genes. EMBO J. 2007, 26, 3451–3462. [Google Scholar] [CrossRef] [PubMed]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Poyet, J.L.; Srinivasula, S.M.; Lin, J.H.; Fernandes-Alnemri, T.; Yamaoka, S.; Tsichlis, P.N.; Alnemri, E.S. Activation of the Iκ B kinases by RIP via IKKgamma /NEMO-mediated oligomerization. J. Biol. Chem. 2000, 275, 37966–37977. [Google Scholar] [CrossRef] [PubMed]
- Scheidereit, C. IκB kinase complexes: Gateways to NF-κB activation and transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalli, J.; Fontan, E.; Fsihi, H.; Coic, Y.M.; Baleux, F.; Véron, M.; Agou, F. Direct inhibition of NF-κB activation by peptide targeting the NOA ubiquitin binding domain of NEMO. Biochem. Pharmacol. 2011, 82, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Deribe, Y.L.; Skånland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 2011, 471, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Charton, J.E.; Pelzer, C.; Hailfinger, S. Antigen receptor signaling to NF-κB via CARMA1, BCL10, and MALT1. Cold Spring Harb. Perspect. Biol. 2010, 2, a003004. [Google Scholar] [CrossRef] [PubMed]
- Turvey, S.E.; Durandy, A.; Fischer, A.; Fung, S.Y.; Geha, R.S.; Gewies, A.; Giese, T.; Greil, J.; Keller, B.; McKinnon, M.L.; et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: Stepping into the limelight of human primary immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Elton, L.; Carpentier, I.; Beyaert, R. MALT1—A universal soldier: Multiple strategies to ensure NF-κB activation and target gene expression. FEBS J. 2015, 282, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Montecalvo, A.; Kane, L.P. Regulation of NF-κB induction by TCR/CD28. Immunol. Res. 2011, 50, 113–117. [Google Scholar] [CrossRef] [PubMed]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Stilmann, M.; Arslan, S.Ç.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Wuerzberger-Davis, S.M.; Wu, Z.H.; Miyamoto, S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell 2003, 115, 565–576. [Google Scholar] [CrossRef]
- Lee, M.H.; Mabb, A.M.; Gill, G.B.; Yeh, E.T.; Miyamoto, S. NF-κB induction of the SUMO protease SENP2: A negative feedback loop to attenuate cell survival response to genotoxic stress. Mol. Cell 2011, 43, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Kracht, M.; Saul, V.V. The intricate interplay between RNA viruses and NF-κB. Biochim. Biophys. Acta 2014, 1843, 2754–2764. [Google Scholar] [CrossRef] [PubMed]
- Deorukhkar, A.; Krishnan, S. Targeting inflammatory pathways for tumor radiosensitization. Biochem. Pharmacol. 2010, 80, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Cusack, J.C., Jr.; Liu, R.; Baldwin, A.S., Jr. Control of inducible chemoresistance: Enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-κB. Nat. Med. 1999, 5, 412–417. [Google Scholar] [PubMed]
- Ahmed, K.M.; Zhang, H.; Park, C.C. NF-κB regulates radioresistance mediated by β1-integrin in three-dimensional culture of breast cancer cells. Cancer Res. 2013, 73, 3737–3748. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-κB to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Namba, H.; Saenko, V.; Ohtsuru, A.; Yamashita, S. Inhibition of nuclear factor-κB cascade potentiates the effect of a combination treatment of anaplastic thyroid cancer cells. J. Clin. Endocrinol. Metab. 2004, 89, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sethi, G. Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 2010, 1805, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.W.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef]
- Feng, B.; Cheng, S.; Hsia, C.Y.; King, L.B.; Monroe, J.G.; Liou, H.C. NF-κB inducible genes BCL-X and cyclin E promote immature B-cell proliferation and survival. Cell Immunol. 2004, 232, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Martino, M.; Sutherland, C.L.; Gold, M.R.; Ricciardi-Castagnoli, P. Dendritic cell survival and maturation are regulated by different signaling pathways. J. Exp. Med. 1998, 188, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Z.; Tang, E.; Fan, Z.; McCauley, L.; Franceschi, R.; Guan, K.; Krebsbach, P.H.; Wang, C.Y. Inhibition of osteoblastic bone formation by nuclear factor-κB. Nat. Med. 2009, 15, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Aoki, K.; Saito, H.; D’Acquisto, F.; May, M.J.; Nakamura, I.; Sudo, T.; Kojima, T.; Okamoto, F.; Fukushima, H.; et al. Selective inhibition of NF-κB blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat. Med. 2004, 10, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E. The alternative NF-κB pathway from biochemistry to biology: Pitfalls and promises for future drug development. Biochem. Pharmacol. 2006, 72, 1161–1179. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, A.K.; Savinova, O.V.; Ho, J.Q.; Wang, V.Y.; Vu, D.; Ghosh, G. The 20S proteasome processes NF-κB1 p105 into p50 in a translation-independent manner. EMBO J. 2006, 25, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Zhang, M.; Harhaj, E.W.; Sun, S.C. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 2004, 279, 26243–26250. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. IKKα limits macrophage NF-κB activation and contributes to the resolution of inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Qu, Z.; Yan, P.; Ishikawa, C.; Aqeilan, R.I.; Rabson, A.B.; Xiao, G. The tumor suppressor gene WWOX links the canonical and noncanonical NF-κB pathways in HTLV-I Tax-mediated tumorigenesis. Blood 2011, 117, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Hsu, L.J.; Schultz, L.; Pratt, N.; Mattison, J.; Chang, N.S. Zfra affects TNF-mediated cell death by interacting with death domain protein TRADD and negatively regulates the activation of NF-κB, JNK1, p53 and WOX1 during stress response. BMC Mol. Biol. 2007, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Lai, F.J.; Hsu, L.J.; Lo, C.P.; Cheng, C.L.; Lin, S.R.; Lee, M.H.; Chang, J.Y.; Subhan, D.; Tsai, M.S.; et al. Dramatic co-activation of WWOX/WOX1 with CREB and NF-κB in delayed loss of small dorsal root ganglion neurons upon sciatic nerve transection in rats. PLoS ONE 2009, 4, e7820. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Su, W.P.; Lin, H.P.; Kuo, H.L.; Wei, H.L.; Chang, N.S. Role of WW Domain-containing Oxidoreductase WWOX in Driving T Cell Acute Lymphoblastic Leukemia Maturation. J. Biol. Chem. 2016, 291, 17319–17331. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Huang, S.S.; Chang, N.S. Role of WWOX and NF-κB in lung cancer progression. Transl. Respir. Med. 2013, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Karras, J.R.; Schrock, M.S.; Batar, B.; Huebner, K. Fragile Genes That Are Frequently Altered in Cancer: Players Not Passengers. Cytogenet. Genome Res. 2016, 150, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Jahid, S.; Sun, J.; Gelincik, O.; Blecua, P.; Edelmann, W.; Kucherlapati, R.; Zhou, K.; Jasin, M.; Gümüş, Z.H.; Lipkin, S.M. Inhibition of colorectal cancer genomic copy number alterations and chromosomal fragile site tumor suppressor FHIT and WWOX deletions by DNA mismatch repair. Oncotarget 2017. [Google Scholar] [CrossRef]
- Smida, J.; Xu, H.; Zhang, Y.; Baumhoer, D.; Ribi, S.; Kovac, M.; von Luettichau, I.; Bielack, S.; O’Leary, V.B.; Leib-Mösch, C.; et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int. J. Cancer 2017, 141, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wang, X.M.; Yuan, H.Y.; Liu, Z.H.; Gao, S.; Peng, L. Exploring the mechanism of WWOX growth inhibitory effects on oral squamous cell carcinoma. Oncol. Lett. 2017, 13, 3198–3204. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Brittain, G.C.; Chang, J.H.; Puebla-Osorio, N.; Jin, J.; Zal, A.; Xiao, Y.; Cheng, X.; Chang, M.; Fu, Y.X.; et al. OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature 2013, 494, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Etzrodt, M.; Cortez-Retamozo, V.; Newton, A.; Zhao, J.; Ng, A.; Wildgruber, M.; Romero, P.; Wurdinger, T.; Xavier, R.; Geissmann, F.; et al. Regulation of monocyte functional heterogeneity by miR-146a and Relb. Cell Rep. 2012, 1, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.H.; Jung, J.K.; Lee, H. Nuclear factor-κB inhibitors; a patent review (2006–2010). Expert Opin. Ther. Pat. 2011, 21, 1897–1910. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Baiget, J.; Mackay, S.P. Small-molecule inhibitors of IκB kinase (IKK) and IKK-related kinases. Pharm. Pat. Anal. 2013, 2, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Mody, R. Targeting death receptors: Is this trail still hot? Transl. Pediatr. 2013, 2, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G. Biopharmaceutical benchmarks. Nat. Biotechnol. 2014, 32, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Moskowitz, A.J. Where Do the New Drugs Fit in for Relapsed/Refractory Hodgkin Lymphoma? Curr. Hematol. Malig. Rep. 2017, 12, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Smith, S.M.; Zhang, S.Y.; Lynn Wang, Y. Mechanisms of ibrutinib resistance in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br. J. Haematol. 2015, 170, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Novero, A.; Ravella, P.M.; Chen, Y.; Dous, G.; Liu, D. Ibrutinib for B cell malignancies. Exp. Hematol. Oncol. 2014, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- FDA Website. 2017. Available online: www.fda.gov (accessed on 10 March 2017).
- Wiestner, A. The role of B-cell receptor inhibitors in the treatment of patients with chronic lymphocytic leukemia. Haematologica 2015, 100, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Jeelall, Y.S.; Ferguson, L.L.; Horikawa, K. Toll-like receptors and cancer: MYD88 mutation and inflammation. Front. Immunol. 2014, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Brenner, L.; Arbeit, R.D.; Sullivan, T. IMO-8400, an Antagonist of Toll-like Receptors 7, 8, and 9, in Development for Genetically Defined B-Cell Lymphomas: Safety and Activity in Phase 1 and Phase 2 Clinical Trials. Blood 2014, 124, 3101. [Google Scholar]
- Srivastava, R.; Geng, D.; Liu, Y.; Zheng, L.; Li, Z.; Joseph, M.A.; McKenna, C.; Bansal, N.; Ochoa, A.; Davila, E. Augmentation of therapeutic responses in melanoma by inhibition of IRAK-1,-4. Cancer Res. 2012, 72, 6209–6216. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Bolanos, L.; Fang, J.; Jerez, A.; Wunderlich, M.; Rigolino, C.; Mathews, L.; Ferrer, M.; Southall, N.; Guha, R.; et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell 2013, 24, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Younger, K.; Gartenhaus, R.; Joseph, A.M.; Hu, F.; Baer, M.R.; Brown, P.; Davila, E. Inhibition of IRAK1/4 sensitizes T cell acute lymphoblastic leukemia to chemotherapies. J. Clin. Investig. 2015, 125, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.Y.L.; Ng, I.O.L.; Lee, T.K.W. Revisiting the Role of TLR/IRAK Signaling and its Therapeutic Potential in Cancer. J. Liver 2015, 5, 1. [Google Scholar] [CrossRef]
- Li, K.; McGee, L.R.; Fisher, B.; Sudom, A.; Liu, J.; Rubenstein, S.M.; Anwer, M.K.; Cushing, T.D.; Shin, Y.; Ayres, M.; et al. Inhibiting NF-κB-inducing kinase (NIK): Discovery, structure-based design, synthesis, structure-activity relationship, and co-crystal structures. Bioorg. Med. Chem. Lett. 2013, 23, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- De Leon-Boenig, G.; Bowman, K.K.; Feng, J.A.; Crawford, T.; Everett, C.; Franke, Y.; Oh, A.; Stanley, M.; Staben, S.T.; Starovasnik, M.A.; et al. The crystal structure of the catalytic domain of the NF-κB inducing kinase reveals a narrow but flexible active site. Structure 2012, 20, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, Y.N.; Brents, L.A.; Li, Z.; Bergsagel, L.P.; McGee, L.R.; Kuehl, M.W. Novel inhibitors are cytotoxic for myeloma cells with NFκB inducing kinase-dependent activation of NFκB. Oncotarget 2014, 5, 4554–4566. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, X.; Jia, L.; Chen, D.; Hou, H.; Rui, L.; Zhao, Y.; Chen, Z. A small-molecule inhibitor of NF-κB-inducing kinase (NIK) protects liver from toxin-induced inflammation, oxidative stress, and injury. FASEB J. 2017, 31, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, C.A.; Mitsuuchi, Y.; Burns, J.M.; Neiman, E.M.; Condon, S.M.; Yu, G.; Seipel, M.E.; Kapoor, G.S.; Laporte, M.G.; Rippin, S.R.; et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-κB activation, and is active in patient-derived xenograft models. Mol. Cancer Ther. 2014, 13, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Bai, L.; Lu, J.; Liu, L.; Yang, C. Targeting Inhibitors of Apoptosis Proteins (IAPs) For New Breast Cancer Therapeutics. J. Mammary Gland Biol. Neoplasia 2012, 17, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Targeting inhibitor of apoptosis proteins for cancer therapy: A double-edge sword? J. Clin. Oncol. 2014, 32, 3190–3191. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.C.; Enzler, T.; Seita, J.; Timmer, A.M.; Lee, C.Y.; Lai, T.Y.; Yu, G.Y.; Lai, L.C.; Temkin, V.; Sinzig, U.; et al. IL-1β-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKβ. Nat Immunol. 2011, 12, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- De Falco, F.; Di Giovanni, C.; Cerchia, C.; De Stefano, D.; Capuozzo, A.; Irace, C.; Iuvone, T.; Santamaria, R.; Carnuccio, R.; Lavecchia, A. Novel non-peptide small molecules preventing IKKβ/NEMO association inhibit NF-κB activation in LPS-stimulated J774 macrophages. Biochem. Pharmacol. 2016, 104, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory κB Kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Bhattacharjee, A.; Ali, A.; Mandal, N.C.; Mandal, S.C.; Pal, M. Chronic inflammation and cancer: Potential chemoprevention through nuclear factor κB and p53 mutual antagonism. J. Inflamm. 2014, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I κB kinase that binds at an allosteric site of the enzyme and blocks NF-κB-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, K.W.; Shuster, D.J.; Gillooly, K.M.; Dambach, D.M.; Pattoli, M.A.; Lu, P.; Zhou, X.D.; Qiu, Y.; Zusi, F.C.; Burke, J.R. A highly selective inhibitor of IκB kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003, 48, 2652–2659. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Quast, S.A.; Plötz, M.; Kammermeier, A.; Eberle, J. Sensitization of melanoma cells for TRAIL-induced apoptosis by BMS-345541 correlates with altered phosphorylation and activation of Bax. Cell Death Dis. 2013, 4, e477. [Google Scholar] [CrossRef] [PubMed]
- MacMaster, J.F.; Dambach, D.M.; Lee, D.B.; Berry, K.K.; Qiu, Y.; Zusi, F.C.; Burke, J.R. An inhibitor of IκB kinase, BMS-345541, blocks endothelial cell adhesion molecule expression and reduces the severity of dextran sulfate sodium-induced colitis in mice. Inflamm. Res. 2003, 52, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Atta-ur-Rahman. Studies in Natural Products Chemistry; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 54, pp. 1–414. [Google Scholar]
- Grothe, K.; Flechsenhar, K.; Paehler, T.; Ritzeler, O.; Beninga, J.; Saas, J.; Herrmann, M.; Rudolphi, K. IκB kinase inhibition as a potential treatment of osteoarthritis—Results of a clinical proof-of-concept study. Osteoarthr. Cartil. 2017, 25, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Agou, F.; Courtois, G.; Chiaravalli, J.; Baleux, F.; Coïc, Y.M.; Traincard, F.; Israël, A.; Véron, M. Inhibition of NF-κB activation by peptides targeting NF-κB essential modulator (NEMO) oligomerization. J. Biol. Chem. 2004, 279, 54248–54257. [Google Scholar] [CrossRef] [PubMed]
- Vincendeau, M.; Hadian, K.; Messias, A.C.; Brenke, J.K.; Halander, J.; Griesbach, R.; Greczmiel, U.; Bertossi, A.; Stehle, R.; Nagel, D.; et al. Inhibition of Canonical NF-κB Signaling by a Small Molecule Targeting NEMO-Ubiquitin Interaction. Sci. Rep. 2016, 6, 18934. [Google Scholar] [CrossRef] [PubMed]
- Clément, J.F.; Meloche, S.; Servant, M.J. The IKK-related kinases: From innate immunity to oncogenesis. Cell Res. 2008, 18, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Beyaert, R. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. 2012, 76, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, J.L.; Baltimore, D. NF-κB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.T.; Liao, S.M.; Maniatis, T. IKKε is part of a novel PMA-inducible IκB kinase complex. Mol. Cell 2000, 5, 513–522. [Google Scholar] [CrossRef]
- Adli, M.; Baldwin, A.S. IKK-i/IKKepsilon controls constitutive, cancer cell-associated NF-κB activity via regulation of Ser-536 p65/RelA phosphorylation. J. Biol. Chem. 2006, 281, 26976–26984. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.R.; Hahn, W.C. Emerging roles for the non-canonical IKKs in cancer. Oncogene 2011, 30, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. IKKepsilon: A bridge between obesity and inflammation. Cell 2009, 138, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Welsh, E.A.; Oguz, U.; Fang, B.; Bai, Y.; Kinose, F.; Bronk, C.; Remsing Rix, L.L.; Beg, A.A.; Rix, U.; et al. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc. Natl. Acad. Sci. USA 2013, 110, 12414–12419. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Liu, J.C.; Chung, P.E.; Egan, S.E.; Zacksenhaus, E. Targeting HER2(+) breast cancer: The TBK1/IKKε axis. Oncoscience 2014, 1, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Barbie, T.U.; Alexe, G.; Aref, A.R.; Li, S.; Zhu, Z.; Zhang, X.; Imamura, Y.; Thai, T.C.; Huang, Y.; Bowden, M.; et al. Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J. Clin. Investig. 2014, 124, 5411–5423. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.I.; Wu, J.M.; Polokoff, M.A.; Kochanny, M.J.; Dinter, H.; Zhu, D.; Biroc, S.L.; Alicke, B.; Bryant, J.; Yuan, S.; et al. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2005, 280, 19867–19874. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Plater, L.; Peggie, M.; Cohen, P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IκB kinase epsilon: A distinct upstream kinase mediates Ser-172 phosphorylation and activation. J. Biol. Chem. 2009, 284, 14136–14146. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yang, Y.; Yin, D.Q.; Hong, S.; Son, Y.J.; Kim, J.H.; Cho, J.Y. TBK1 inhibitors: A review of patent literature (2011–2014). Expert Opin. Ther. Pat. 2015, 25, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Domainex. 2017. Available online: http://www.domainex.co.uk/drug-discovery-pipeline/inflammatory-diseases (accessed on 8 March 2017).
- Richards, B.; Cronin, M.; Seager, N.; Niederjohn, J.; Baichwal, V.; Chan, A.; Robinson, R.; Hess, M.; Davis, T.; Papac, D.; et al. Cellular and In Vivo Properties of MPI-0485520, a Novel and Potent Small Molecule Inhibitor of IKKe. FASEB J. 2010, 24. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.L.; Ciechanover, A. Targeting proteins for destruction by the ubiquitin system: Implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 73–96. [Google Scholar] [CrossRef] [PubMed]
- Bulatov, E.; Ciulli, A. Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: Structure, assembly and small-molecule modulation. Biochem. J. 2015, 467, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Lin, H.K.; Sun, S.C.; Zhang, S. Targeting ubiquitination for cancer therapies. Future Med. Chem. 2015, 7, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef]
- Goldberg, A.L. Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 2012, 199, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.M.; Tepper, J.E.; Baldwin, A.S., Jr.; Liu, R.; Adams, J.; Elliott, P.; Cusack, J.C., Jr. Enhancement of radiosensitivity by proteasome inhibition: Implications for a role of NF-κB. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 183–193. [Google Scholar] [CrossRef]
- Goel, A.; Dispenzieri, A.; Greipp, P.R.; Witzig, T.E.; Mesa, R.A.; Russell, S.J. PS-341-mediated selective targeting of multiple myeloma cells by synergistic increase in ionizing radiation-induced apoptosis. Exp. Hematol. 2005, 33, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Goktas, S.; Baran, Y.; Ural, A.U.; Yazici, S.; Aydur, E.; Basal, S.; Avcu, F.; Pekel, A.; Dirican, B.; Beyzadeoglu, M. Proteasome inhibitor bortezomib increases radiation sensitivity in androgen independent human prostate cancer cells. Urology 2010, 75, 793–798. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Raftery, L.; Calvo, B.F.; Chakravarthy, A.B.; Ivanova, A.; Myers, M.O.; Kim, H.J.; Chan, E.; Wise, P.E.; Caskey, L.S.; et al. A phase I study of bortezomib in combination with standard 5-fluorouracil and external-beam radiation therapy for the treatment of locally advanced or metastatic rectal cancer. Clin. Colorectal Cancer 2010, 9, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.; Brown, C.O.; Schibler, J.; Goel, A. Combination chemotherapy increases cytotoxicity of multiple myeloma cells by modification of nuclear factor (NF)-κB activity. Exp. Hematol. 2013, 41, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Foster, N.R.; Meyers, J.P.; Thomas, S.P.; Northfelt, D.W.; Rowland, K.M., Jr.; Mattar, B.; Johnson, D.B.; Molina, J.R.; Mandrekar, S.J.; et al. A phase I/II study of bortezomib in combination with paclitaxel, carboplatin, and concurrent thoracic radiation therapy for non-small-cell lung cancer: North Central Cancer Treatment Group (NCCTG)-N0321. J. Thorac. Oncol. 2015, 10, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Ixazomib: First Global Approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.C.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Results of a clinical study of pevonedistat (pev), a first-in-class NEDD8-activating enzyme (NAE) inhibitor, combined with azacitidine (aza) in older patients (pts) with acute myeloid leukemia (AML). In Proceedings of the 58th American Society of Hematology Annual Meeting & Exposition, San Diego, CA, USA, 3 December 2016. [Google Scholar]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-Neriah, Y. Ubiquitination and degradation of the inhibitors of NF-κB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000166. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Fujiwara, H.; Furuichi, Y.; Tanaka, K.; Shimbara, N. A novel small-molecule inhibitor of NF-κB signaling. Biochem. Biophys. Res. Commun. 2008, 368, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, Z.J. The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 2011, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G. Improving the therapeutic index in myeloma. Blood 2010, 116, 4733–4734. [Google Scholar] [CrossRef] [PubMed]
- Horie, R. Molecularly-targeted Strategy and NF-κB in lymphoid malignancies. J. Clin. Exp. Hematop. 2013, 53, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Brassesco, M.S.; Roberto, G.M.; Morales, A.G.; Oliveira, J.C.; Delsin, L.E.; Pezuk, J.A.; Valera, E.T.; Carlotti, C.G., Jr.; Rego, E.M.; De Oliveira, H.F.; et al. Inhibition of NF-κB by Dehydroxymethylepoxyquinomicin Suppresses Invasion and Synergistically Potentiates Temozolomide and γ-Radiation Cytotoxicity in Glioblastoma Cells. Chemother. Res. Pract. 2013, 2013, 593020. [Google Scholar] [CrossRef] [PubMed]
- Kozakai, N.; Kikuchi, E.; Hasegawa, M.; Suzuki, E.; Ide, H.; Miyajima, A.; Horiguchi, Y.; Nakashima, J.; Umezawa, K.; Shigematsu, N.; et al. Enhancement of radiosensitivity by a unique novel NF-κB inhibitor, DHMEQ, in prostate cancer. Br. J. Cancer 2012, 107, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Nishio, H.; Yaguchi, T.; Sugiyama, J.; Sumimoto, H.; Umezawa, K.; Iwata, T.; Susumu, N.; Fujii, T.; Kawamura, N.; Kobayashi, A.; et al. Immunosuppression through constitutively activated NF-κB signalling in human ovarian cancer and its reversal by an NF-κB inhibitor. Br. J. Cancer 2014, 110, 2965–2974. [Google Scholar] [CrossRef] [PubMed]
- Verzella, D.; Fischietti, M.; Capece, D.; Vecchiotti, D.; Del Vecchio, F.; Cicciarelli, G.; Mastroiaco, V.; Tessitore, A.; Alesse, E.; Zazzeroni, F. Targeting the NF-κB pathway in prostate cancer: A promising therapeutic approach? Curr. Drug Targets. 2016, 17, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov. Today 2010, 15, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, R.; Kusumanchi, P.; Appaiah, H.; Cheng, L.; Crooks, P.; Neelakantan, S.; Peat, T.; Klaunig, J.; Matthews, W.; Nakshatri, H.; et al. A water soluble parthenolide analogue suppresses in vivo tumor growth of two tobacco associated cancers, lung and bladder cancer, by targeting NF-κB and generating reactive oxygen species. Int. J. Cancer 2011, 128, 2481–2494. [Google Scholar] [CrossRef] [PubMed]
- Adis Insight. 2017. Available online: http://adisinsight.springer.com/search (accessed on 25 January 2017).
- NF-kB Target Genes. 2017. Available online: https://www.bu.edu/nf-kb/gene-resources/target-genes/ (accessed on 25 January 2017).
- Bennett, J.; Moretti, M.; Thotakura, A.K.; Tornatore, L.; Franzoso, G. The Regulation of the JNK Cascade and Programmed Cell Death by NF-κB: Mechanisms and Functions. In Trends in Stem Cell. Proliferation and Cancer Research; Springer: Dordrecht, The Netherlands, 2013. [Google Scholar]
- Jin, R.; De Smaele, E.; Zazzeroni, F.; Nguyen, D.U.; Papa, S.; Jones, J.; Cox, C.; Gelinas, C.; Franzoso, G. Regulation of the GADD45β promoter by NF-κB. DNA Cell Biol. 2002, 21, 491–503. [Google Scholar] [CrossRef] [PubMed]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of GADD45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J., Jr. GADD45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 2013, 793, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A. GADD45 proteins: Key players of repair-mediated DNA demethylation. Adv. Exp. Med. Biol. 2013, 793, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Z.; Liu, Y. GADD45 proteins: Roles in cellular senescence and tumor development. Exp. Biol. Med. 2014, 239, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvarez, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T.; et al. GADD45 β mediates the NF-κ B suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Døssing, M.G.; Papa, S.; Franzoso, G.; Billestrup, N.; Mandrup-Poulsen, T. Growth arrest- and DNA-damage-inducible 45β gene inhibits c-Jun N-terminal kinase and extracellular signal-regulated kinase and decreases IL-1β-induced apoptosis in insulin-producing INS-1E cells. Diabetologia 2006, 49, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, L.; Sandomenico, A.; Raimondo, D.; Low, C.; Rocci, A.; Tralau-Stewart, C.; Capece, D.; D’Andrea, D.; Bua, M.; Boyle, E.; et al. Cancer-selective targeting of the NF-κB survival pathway with GADD45β/MKK7 inhibitors. Cancer Cell 2014, 26, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, L.; Acton, G.; Adams, N.; Campbell, E.A.; Kelly, J.; Szydlo, R.M.; Tarbit, M.; Bannoo, S.; D’Andrea, D.; Capece, D.; et al. Cancer-Selective Targeting of the NF-κB Survival Pathway in Multiple Myeloma with the GADD45β/MKK7 Inhibitor, DTP3. Blood 2015, 126, 868. [Google Scholar]
- Tang, G.; Minemoto, Y.; Dibling, B.; Purcell, N.H.; Li, Z.; Karin, M.; Lin, A. Inhibition of JNK activation through NF-κB target genes. Nature 2001, 414, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Monti, S.M.; Vitale, R.M.; Bubici, C.; Jayawardena, S.; Alvarez, K.; De Smaele, E.; Dathan, N.; Pedone, C.; Ruvo, M.; et al. Insights into the structural basis of the GADD45β-mediated inactivation of the JNK kinase, MKK7/JNKK2. J. Biol. Chem. 2007, 282, 19029–19041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ahsan, M.H.; Zhu, L.; Sambucetti, L.C.; Purchio, A.F.; West, D.B. NF-κB and not the MAPK signaling pathway regulates GADD45β expression during acute inflammation. J. Biol. Chem. 2005, 280, 21400–21408. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Fu, Y.X.; Bubici, C.; Alvarez, K.; Dean, K.; Christiansen, P.A.; Anders, R.A.; Franzoso, G. Gadd45β promotes hepatocyte survival during liver regeneration in mice by modulating JNK signaling. J. Clin. Investig. 2008, 118, 1911–1923. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Ferrandino, A.F.; Flavell, R.A. GADD45β is important for perpetuating cognate and inflammatory signals in T cells. Nat. Immunol. 2004, 5, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Whipping NF-κB to Submission via GADD45 and MKK7. Cancer Cell 2014, 26, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Vaccarezza, M.; Vitale, M. Harnessing downstream NF-κB signalling to achieve apoptosis-inducing anti-cancer-specific activity. Cell Death Dis. 2016, 7, e2306. [Google Scholar] [CrossRef] [PubMed]
- Catz, S.D.; Johnson, J.L. Transcriptional regulation of bcl-2 by nuclear factor κB and its significance in prostate cancer. Oncogene 2001, 20, 7342–7351. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Bougie, P.; Amiot, M. Apoptotic machinery diversity in multiple myeloma molecular subtypes. Front. Immunol. 2013, 4, 467. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M. Targeting BCL2-Proteins for the Treatment of Solid Tumours. Adv. Med. 2014, 2014, 943648. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Prasad, S.; Tyagi, A.K.; Deb, L.; Huang, J.; Karelia, D.N.; Amin, S.G.; Aggarwal, B.B. Targeting Cell Survival Proteins for Cancer Cell Death. Pharmaceuticals 2016, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, F.; Demir, S.; Debatin, K.M.; Meyer, L.H. Synergistic Activity of ABT-199 with Conventional Chemotherapy and Dinaciclib in B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 2015, 126, 2631. [Google Scholar]
- Fox, J.L.; MacFarlane, M. Targeting cell death signalling in cancer: Minimising “Collateral damage”. Br. J. Cancer 2016, 115, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.A.; Claxton, D.F. Therapeutic inhibition of BCL-2 and related family members. Expert Opin. Investig. Drugs 2017, 26, 293–301. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Target | Cancer Type | Ongoing Clinical Trials |
|---|---|---|---|
| Upstream IKKs complex | |||
| Brentuximab (Vedotin) | CD30 | HL, Anaplastic large cell lymphoma, etc. | NCT01657331, NCT02462538, NCT01807598, NCT02939014, NCT03007030, NCT02169505, NCT01900496, etc. |
| Ibrutinib (PCI-32765) | BTK | MCL, CLL, WM, DLBCL, FL, MM, and NSCLC, etc. | NCT02801578, NTC0275689, NCT02943473, NCT02321540, NCT02558816, NCT02420912, NCT02315768, NCT02451111, NCT02356458, etc. |
| IMO-8400 | TLR 7, 8, and 9 | WM, DLBCL | NCT02252146 |
| LCL-161 | cIAPs | Ovarian cancer, MM | NCT02649673, NCT02890069, NCT01955434 |
| Birinapant (TL32711) | cIAPs | Solid tumours and high grade serous carcinomas | NCT02587962, NCT02756130 |
| Ubiquitin proteasome pathway | |||
| Bortezomib | Proteasome | AML, lymphoma, MDS, neuroblastoma, ALL, etc. | NCT02308280, NCT02535806, NCT01736943, NCT01534260, NCT02613598, NCT02356458, NCT01241708, NCT03016988, NCT02139397, NCT02237261, etc. |
| Carfizomib | Proteasome | MM, neuroendocrine cancer, NHL, DLBCL, MCL, FL, peripheral T-cell lymphoma, HL, T-cell NHL, solid tumours, leukaemia, etc. | NCT02302495, NCT02572492, NCT02318784, NCT02142530, NCT02867618, NCT01738594, NCT02512926, etc. |
| Ixazomib (MNL-9708) | Proteasome | Glioblastoma, MM, lymphoma, amyloidosis, solid tumours, B-cell lymphoma, lymphoma, etc. | NCT02630030, NCT02924272, NCT02942095, NCT02312258, NCT02477215, NCT02898259, etc. |
| MLN4924 (Pevonedistat) | NEDD8 | AML, solid tumours, chronic myelomonocytic leukaemia, MDS | NCT01814826, NCT02782468, NCT02610777, NCT03009240, NCT03057366 |
| NF-κB target genes | |||
| DTP3 | Gadd45β/MKK7 | MM | MR/L005069/1 |
| ABT-199 | BCL-2 | CLL, WM, MCL, AML, NHL, DLBCL, FL, MM, MDS, etc. | NCT02677324, NCT02471391, NCT02558816, NCT02203773, NCT02055820, NCT03136497, NCT03128879, NCT02427451, etc. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5, 50. https://doi.org/10.3390/biomedicines5030050
Begalli F, Bennett J, Capece D, Verzella D, D’Andrea D, Tornatore L, Franzoso G. Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines. 2017; 5(3):50. https://doi.org/10.3390/biomedicines5030050
Chicago/Turabian StyleBegalli, Federica, Jason Bennett, Daria Capece, Daniela Verzella, Daniel D’Andrea, Laura Tornatore, and Guido Franzoso. 2017. "Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity" Biomedicines 5, no. 3: 50. https://doi.org/10.3390/biomedicines5030050
APA StyleBegalli, F., Bennett, J., Capece, D., Verzella, D., D’Andrea, D., Tornatore, L., & Franzoso, G. (2017). Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines, 5(3), 50. https://doi.org/10.3390/biomedicines5030050