Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus

Abstract
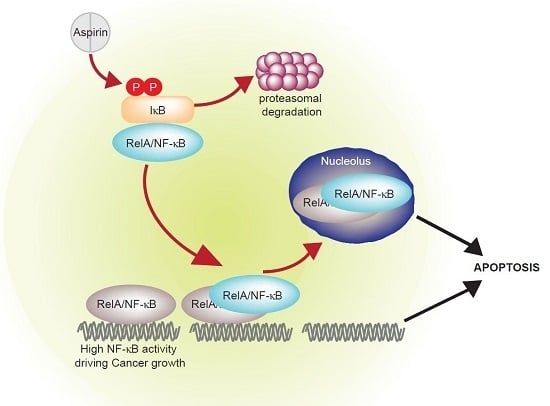
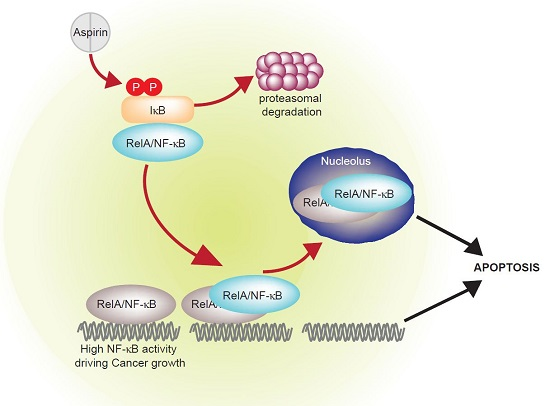{kind=link}
{kind=link}
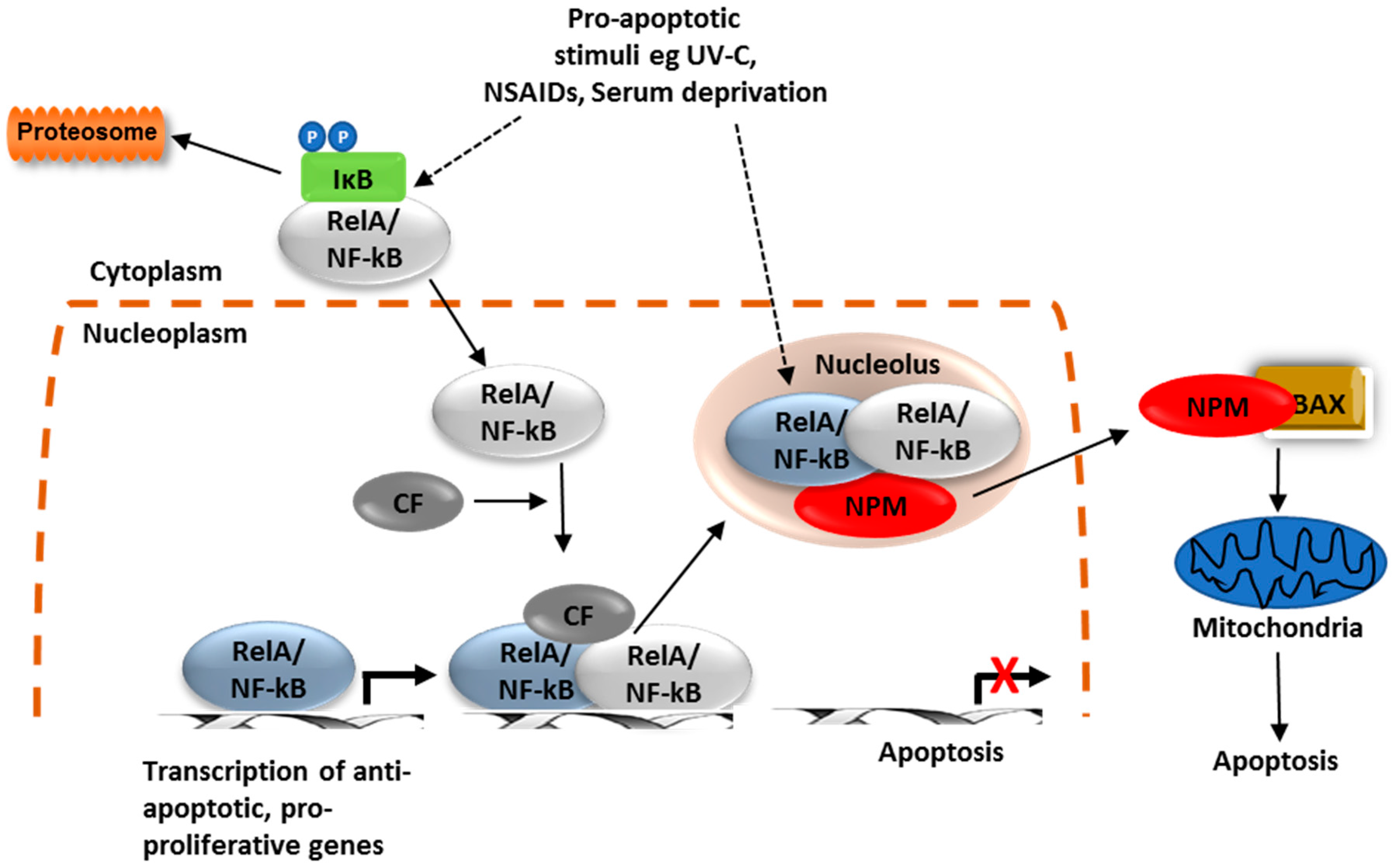{kind=link}
1. Aspirin and Cancer
2. NF-κB, Cancer and Aspirin
3. Crosstalk between the NF-κB Pathway and Nucleoli
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Tougeron, D.; Sha, D.; Manthravadi, S.; Sinicrope, F.A. Aspirin and colorectal cancer: Back to the future. Clin. Cancer Res. 2014, 20, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Patrono, C. Aspirin and Cancer. J. Am. Coll. Cardiol. 2016, 68, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Arber, N.; Burn, J.; Chia, W.K.; Elwood, P.; Hull, M.A.; Logan, R.F.; Rothwell, P.M.; Schrör, K.; Baron, J.A. Aspirin in the chemoprevention of colorectal neoplasia: An overview. Cancer Prev. Res. 2012, 5, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.J.; Hague, A.; Hicks, D.J.; Paraskeva, C. Differential growth inhibition by the aspirin metabolite salicylate in human colorectal tumor cell lines: Enhanced apoptosis in carcinoma and in vitro-transformed adenoma relative to adenoma relative to adenoma cell lines. Cancer Res. 1996, 56, 2273–2276. [Google Scholar] [PubMed]
- Williams, C.S.; Smalley, W.; DuBois, R.N. Aspirin use and potential mechanisms for colorectal cancer prevention. J. Clin. Invest. 1997, 100, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.; Dunlop, M.G.; Stark, L.A. Evidence for colorectal cancer cell specificity of aspirin effects on NF-κB signalling and apoptosis. Br. J. Cancer 2004, 91, 381–388. [Google Scholar] [PubMed]
- Stark, L.A.; Din, F.V.N.; Zwacka, R.M.; Dunlop, M.G. Aspirin-induced activation of the NF-κB signalling pathway: A novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001, 15, 1273–1275. [Google Scholar] [PubMed]
- Piazza, G.A.; Alberts, D.S.; Hixson, L.J.; Paranka, N.S.; Li, H.; Finn, T.; Bogert, C.; Guillen, J.M.; Brendel, K.; Gross, P.H.; et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997, 57, 2909–2915. [Google Scholar] [PubMed]
- Qiu, W.; Wang, X.; Leibowitz, B.; Liu, H.; Barker, N.; Okada, H.; Oue, N.; Yasui, W.; Clevers, H.; Scheen, R.E.; et al. Chemoprevention by nonsteroidal anti-inflammatory drugs eliminates oncogenic intestinal stem cells via SMAC-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2010, 107, 20027–20032. [Google Scholar] [CrossRef] [PubMed]
- Beazer-Barclay, Y.; Levy, D.B.; Moser, A.R.; Dove, W.F.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.M. Sulindac suppresses tumorigenesis in the Min mouse. Carcinogenesis 1996, 17, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Corpet, D.E.; Pierre, F. Point: From animal models to prevention of colon cancer. Systematic review of chemoprevention in min mice and choice of the model system. Cancer Epidemiol. Biomark. Prev. 2003, 12, 391–400. [Google Scholar]
- Perkins, S.; Clarke, A.R.; Steward, W.; Gescher, A. Age-related difference in susceptibility of ApcMin/+ mice towards the chemopreventive efficacy of dietary aspirin and curcumin. Br. J. Cancer 2003, 88, 1480–1483. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Stark, L.A.; Dunlop, M.G.; Clarke, A.R. Suppression of intestinal and mammary neoplasia by lifetime administration of aspirin in ApcMin/+ and ApcMin/+, Msh2−/− mice. Cancer Res. 2001, 61, 7060–7064. [Google Scholar] [PubMed]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-Year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.F. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Elwood, P.C.; Morgan, G.; Galante, J.; Chia, J.W.; Dolwani, S.; Graziano, J.M.; Kelson, M.; Lanas, A.; Longley, M.; Phillips, C.J.; et al. Systematic Review and Meta-Analysis of Randomised Trials to Ascertain Fatal Gastrointestinal Bleeding Events Attributable to Preventive Low-Dose Aspirin: No Evidence of Increased Risk. PLoS ONE 2016, 11, e0166166. [Google Scholar] [CrossRef] [PubMed]
- Sandler, R.S.; Galanko, J.C.; Murray, S.C.; Helm, J.F.; Woosley, J.T. Aspirin and nonsteroidal anti-inflammatory agents and risk for colorectal adenomas. Gastroenterology 1998, 114, 441–447. [Google Scholar] [CrossRef]
- Sandler, R.S.; Halabi, S.; Baron, J.A.; Budinger, S.; Paskett, E.; Keresztes, R.; Petrelli, N.; Pipas, J.M.; Karp, D.D.; Loprinzi, C.L.; et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N. Engl. J. Med. 2003, 348, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Cole, B.F.; Logan, R.F.; Halabi, S.; Benamouzig, R.; Sandler, R.S.; Grainge, M.J.; Chaussade, S.; Baron, J.A. Aspirin for the chemoprevention of colorectal adenomas: Meta-analysis of the randomized trials. J. Natl. Cancer Inst. 2009, 101, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Ecclec, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef]
- Cook, N.R.; Lee, I.M.; Zhang, S.M.; Moorthy, M.V.; Buring, J.E. Alternate-day, low-dose aspirin and cancer risk: Long-term observational follow-up of a randomized trial. Ann. Intern. Med. 2013, 159, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.; Theodoratou, E.; Farrington, S.M.; Tenesa, A.; Barnetson, R.A.; Cetnarskyj, R.; Stark, L.; Porteous, M.; Campbell, H.; Dunlop, M.G. Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut 2010, 59, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Thorat, M.A.; Bosetti, C.; Brown, P.H.; Burn, J.; Cook, N.R.; Ford, L.G.; Jacobs, E.J.; Jankowski, J.A.; Vecchia, C.L.; et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann. Oncol. 2015, 26, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Nishihara, R.; Wu, K.; Wang, M.; Ogino, S.; Willett, W.C.; Spigelman, D.; Fuchs, C.S.; Giovannucci, E.L.; Chan, A.T. Population-wide Impact of Long-term Use of Aspirin and the Risk for Cancer. JAMA Oncol. 2016, 1, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Rahm, A.K.; Finn, T.S.; Fryer, B.H.; Li, H.; Stoumen, A.L.; Pamukcu, R.; Ahnen, D.J. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction. Cancer Res. 1997, 57, 2452–2459. [Google Scholar] [PubMed]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Jacobs, E.J.; Patrono, C. The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol. 2012, 9, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New. Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells during Therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; DuBois, R.N. Prostaglandins in cancer cell adhesion, migration, and invasion. Int. J. Cell Biol. 2012, 2012, 723419. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.S.; Mann, M.; DuBois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E. Cyclooxygenase-2 (cox-2) and the inflammogenesis of cancer. Subcell. Biochem. 2007, 42, 93–126. [Google Scholar] [PubMed]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med. 2007, 356, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Chulada, P.C.; Thompson, M.B.; Mahler, J.F.; Doyle, C.M.; Gaul, B.W.; Lee, C.; Tiano, H.F.; Morham, S.G.; Smithies, O.; Langenbach, R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000, 60, 4705–4708. [Google Scholar] [PubMed]
- Ruegg, C.; Zaric, J.; Stupp, R. Non steroidal anti-inflammatory drugs and COX-2 inhibitors as anti-cancer therapeutics: Hypes, hopes and reality. Ann. Med. 2003, 35, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Dinchuk, J.E.; Kargman, S.L.; Oshima, H.; Hancock, B.; Kwong, E.; Trzaskos, J.M.; Evans, J.F.; Taketo, M.M. Suppression of intestinal polyposis in Apc Δ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996, 87, 803–809. [Google Scholar] [CrossRef]
- Gupta, R.A.; DuBois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Guillem-Llobat, P.; Dovizio, M.; Bruno, A.; Ricciotti, E.; Cufino, V.; Sacco, A.; Grande, R.; Alberti, S.; Arena, V.; Cirillo, M.; et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 2016, 7, 32462–32477. [Google Scholar] [CrossRef] [PubMed]
- Guillem-Llobat, P.; Dovizio, M.; Alberti, S.; Bruno, A.; Patrignani, P. Platelets, cyclooxygenases, and colon cancer. Semin. Oncol. 2014, 41, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1996, 52, 237–245. [Google Scholar] [CrossRef]
- Rigas, B.; Shiff, S.J. Is inhibition of cyclooxygenase required for the chemopreventive effect of NSAIDs in colon cancer? A model reconciling the current contradiction. Med. Hypotheses 2000, 54, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Morham, S.G.; Langenbach, R.; Young, D.A. Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J. Exp. Med. 1999, 190, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, N.N.; Boolbol, S.K.; Dannenberg, A.J.; Mestre, J.R.; Bilinski, R.T.; Martucci, C.; Newmark, H.L.; Chadburn, A.; Bertagnolli, M.M. The sulfide metabolite of sulindac prevents tumors and restores enterocyte apoptosis in a murine model of familial adenomatous polyposis. Carcinogenesis 1998, 19, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Kawamori, T.; Lubet, R.A.; Steele, V.E.; Kelloff, G.J.; Rao, C.V. Chemopreventive efficacy of sulindac sulfone against colon cancer depends on time of administration during carcinogenic process. Cancer Res. 1999, 59, 3387–3391. [Google Scholar] [PubMed]
- Tegeder, I.; Pfeilschifter, J.; Geisslinger, G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001, 15, 2057–2072. [Google Scholar] [CrossRef] [PubMed]
- Bos, C.L.; Kodach, L.L.; van den Brink, G.R.; Diks, S.H.; van Santen, M.M.; Richel, D.J.; Peppelenbosch, M.P.; Hardwick, J.C.H. Effect of aspirin on the Wnt/β-catenin pathway is mediated via protein phosphatase 2A. Oncogene 2006, 25, 6447–6456. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Baldwin, A.S. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. NF-κB: Tumor promoter or suppressor? Trends Cell Biol. 2004, 14, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The IKK complex: An integrator of all signals that activate NF-κB? Trends Cell Biol. 2000, 10, 129–133. [Google Scholar] [CrossRef]
- Vlahopoulos, S.A.; Cen, O.; Hengen, N.; Agan, J.; Moschovi, M.; Critselis, E.; Adamaki, M.; Bacopoulou, F.; Copland, J.A.; Boldogh, I.; et al. Dynamic aberrant NF-κB spurs tumorigenesis: A new model encompassing the microenvironment. Cytokine Growth Factor Rev. 2015, 26, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wu, P.; Zhao, L.; Huang, L.; Zhang, Z.; Zhao, S.; Huang, J. NF-κB Expression and outcomes in solid tumors: A systematic review and meta-analysis. Medicine 2015, 94, e1687. [Google Scholar] [CrossRef] [PubMed]
- Shaked, H.; Hofseth, L.J.; Chumanevich, A.; Chumanevich, A.A.; Wang, J.; Wang, Y.; Taniguchi, K.; Guma, M.; Shenouda, S.; Clevers, H.; et al. Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 14007–14012. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.-L.; Athineos, O.; et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Kopp, E.; Ghosh, S. Inhibition of NF-κB by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-β. Nature 1998, 396, 77–80. [Google Scholar] [PubMed]
- Stark, L.A.; Dunlop, M.G. Nucleolar sequestration of RelA (p65) regulates NF-κB-driven transcription and apoptosis. Mol. Cell Biol. 2005, 25, 5985–6004. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Bhardwaj, A.; Potdar, P.; Aggarwal, B.B. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-κB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 2004, 23, 9247–9258. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yin, M.J.; Lin, K.M.; Gaynor, R.B. Sulindac inhibits activation of the NF-κB pathway. J. Biol. Chem. 1999, 274, 27307–27314. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Zhong, L.; Duan, T.; Zhang, R.H.; Wang, X.; Wang, G.; Hu, K.; Lv, X.; Kang, T. Aspirin suppresses the growth and metastasis of osteosarcoma through the NF-κB pathway. Clin. Cancer Res. 2015, 21, 5349–5359. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Song, S.H.; Kim, S.G.; Chun, K.S.; Lim, S.Y.; Na, H.K.; Kim, J.W.; Surh, Y.-J.; Bang, Y.-J.; Song, Y.-S. Celecoxib induces apoptosis in cervical cancer cells independent of cyclooxygenase using NF-κB as a possible target. J. Cancer Res. Clin. Oncol. 2004, 130, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Gwak, J.; Park, S.; Won, J.; Kim, D.E.; Yea, S.S.; Cha, I.-J.; Kim, T.K.; Shin, J.-G.; Oh, S. Diclofenac attenuates Wnt/β-catenin signaling in colon cancer cells by activation of NF-κB. FEBS Lett. 2005, 579, 4213–4218. [Google Scholar] [CrossRef] [PubMed]
- Loveridge, C.J.; Macdonald, A.D.; Thoms, H.C.; Dunlop, M.G.; Stark, L.A. The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the RelA(p65) subunit of NF-κB. Oncogene 2008, 27, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Mladenova, D.; Pangon, L.; Currey, N.; Ng, I.; Musgrove, E.A.; Grey, S.T.; Kohonen-Corish, M.R.J. Sulindac activates NF-κB signaling in colon cancer cells. Cell Commun. Signal. 2013, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.B.; Yang, X.; Clark, R.; Choi, J.; Baek, S.J.; Lee, S.H. A mechanistic study of the proapoptotic effect of tolfenamic acid: Involvement of NF-κB activation. Carcinogenesis 2013, 34, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Jo, J.R.; Hong, H.; Nam, K.Y.; Kim, J.B.; Hwang, S.H.; Choi, M.S.; Ryu, N.H.; Jang, H.J.; Lee, S.H.; et al. Aspirin induces apoptosis in YD-8 human oral squamous carcinoma cells through activation of caspases, down-regulation of Mcl-1, and inactivation of ERK-1/2 and AKT. Toxicol. In Vitro 2010, 24, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, E.J.; Madigan, J.P.; Boardman, L.A.; Rosenberg, D.W. Ibuprofen inhibits activation of nuclear β-catenin in human colon adenomas and induces the phosphorylation of GSK-3β. Cancer Prev. Res. 2011, 4, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Reid, K.; Sansom, O.J.; Din, F.V.; Guichard, S.; Mayer, I.; Jodrell, D.I.; Clarke, A.R.; Dunlop, M.G. Aspirin activates the NF-κB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis 2007, 28, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lobb, I.; Pierre, M.; Sonia, M.N.; James, S.; Kathrin, K.; Kathrin, K.; Kathrin, K.; Oakley, F.; Stark, L.A. Disruption of the PolI complex links inhibition of CDK4 activity to activation of the NF-κB pathway. Available online: http://biorxiv.org/content/biorxiv/early/2017/01/13/100255.full.pdf (accessed on 13 January 2017).
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Marzbani, E.; Inatsuka, C.; Lu, H.; Disis, M.L. The invisible arm of immunity in common cancer chemoprevention agents. Cancer Prev. Res. 2013, 6, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Nishihara, R.; Qian, Z.R.; Song, M.; Mima, K.; Inamura, K.; Nowak, J.A.; Drew, D.A.; Lochhead, P.; Nosho, K.; et al. Regular aspirin use associates with lower risk of colorectal cancers with low numbers of tumor-infiltrating lymphocytes. Gastroenterology 2016, 151, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I. The nucleolus-guardian of cellular homeostasis and genome integrity. Chromosoma 2013, 122, 487–497. [Google Scholar] [CrossRef] [PubMed]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Russo, G. Ribosomal Proteins Control or Bypass p53 during Nucleolar Stress. Int. J. Mol. Sci. 2017, 18, 140. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait, B.H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.S.; Lam, Y.W.; Leung, A.K.; Ong, S.E.; Lyon, C.E.; Lamond, A.I.; Mann, M. Nucleolar proteome dynamics. Nature 2005, 433, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; Lam, Y.W.; Lamont, D.; Lamond, A.I. A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell. Proteom. 2010, 9, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Coccia, M.; Rossi, A.; Riccio, A.; Trotta, E.; Santoro, M.G. Human NF-κB repressing factor acts as a stress-regulated switch for ribosomal RNA processing and nucleolar homeostasis surveillance. Proc. Natl. Acad. Sci. USA 2017, 114, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Rubbi, C.P.; Milner, J. Non-activated p53 co-localizes with sites of transcription within both the nucleoplasm and the nucleolus. Oncogene 2000, 19, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Audas, T.E.; Jacob, M.D.; Lee, S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol. Cell 2012, 45, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L. Nucleolar aggresomes as counterparts of cytoplasmic aggresomes in proteotoxic stress. Bioessays 2011, 33, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L.; Moore, H.M.; Bai, B.; Jaamaa, S.; Laiho, M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 2011, 30, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Ehm, P.; Nalaskowski, M.M.; Wundenberg, T.; Jucker, M. The tumor suppressor SHIP1 colocalizes in nucleolar cavities with p53 and components of PML nuclear bodies. Nucleus 2015, 6, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Campbell, K.J.; Perkins, N.D. p53- and Mdm2-independent repression of NF-κB transactivation by the ARF tumor suppressor. Mol. Cell 2003, 12, 15–25. [Google Scholar] [CrossRef]
- Sweet, T.; Khalili, K.; Sawaya, B.E.; Amini, S. Identification of a novel protein from glial cells based on its ability to interact with NF-κB subunits. J. Cell. Biochem. 2003, 90, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; Lynn, B.C.; Daosukho, C.; St Clair, D.K. Identification of nucleophosmin as an NF-κB co-activator for the induction of the human SOD2 gene. J. Biol. Chem. 2004. [Google Scholar] [CrossRef] [PubMed]
- Birbach, A.; Bailey, S.T.; Ghosh, S.; Schmid, J.A. Cytosolic, nuclear and nucleolar localization signals determine subcellular distribution and activity of the NF-κB inducing kinase NIK. J. Cell Sci. 2004, 117, 3615–3624. [Google Scholar] [CrossRef] [PubMed]
- Niedick, I.; Froese, N.; Oumard, A.; Mueller, P.P.; Nourbakhsh, M.; Hauser, H.; Köster, M. Nucleolar localization and mobility analysis of the NF-κB repressing factor NRF. J. Cell Sci. 2004, 117, 3447–3458. [Google Scholar] [CrossRef] [PubMed]
- Bierhoff, H.; Dundr, M.; Michels, A.A.; Grummt, I. Phosphorylation by casein kinase 2 facilitates rRNA gene transcription by promoting dissociation of TIF-IA from elongating RNA polymerase I. Mol. Cell. Biol. 2008, 28, 4988–4998. [Google Scholar] [CrossRef] [PubMed]
- Kato, T., Jr.; Delhase, M.; Hoffmann, A.; Karin, M. CK2 is a C-terminal IκB kinase responsible for NF-κB activation during the UV response. Mol. Cell 2003, 12, 829–839. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the α subunit of eukaryotic initiation factor 2 is required for activation of NF-κB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.N.; Owen, C.R.; White, B.C.; Rafols, J.A. Ultrastructural localization of phosphorylated eIF2α [eIF2α(P)] in rat dorsal hippocampus during reperfusion. Acta Neuropathol. 1999, 98, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Thoms, H.C.; Loveridge, C.J.; Simpson, J.; Clipson, A.; Reinhardt, K.; Dunlop, M.G.; Stark, L.A. Nucleolar targeting of RelA(p65) is regulated by COMMD1-dependent ubiquitination. Cancer Res. 2010, 70, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Parrondo, R.; de las, P.A.; Reiner, T.; Rai, P.; Perez-Stable, C. NF-κB activation enhances cell death by antimitotic drugs in human prostate cancer cells. Mol. Cancer 2010, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Sniderhan, L.F.; Garcia-Bates, T.M.; Burgart, M.; Bernstein, S.H.; Phipps, R.P.; Maggirwar, S.B. Neurotrophin signaling through tropomyosin receptor kinases contributes to survival and proliferation of non-Hodgkin lymphoma. Exp. Hematol. 2009, 37, 1295–1309. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Forscher, C.; Di, V.D.; Koeffler, H.P. Induction of p53-independent apoptosis by ectopic expression of HOXA5 in human liposarcomas. Sci. Rep. 2015, 5, 12580. [Google Scholar] [CrossRef] [PubMed]
- Thoms, H.C.; Dunlop, M.G.; Stark, L.A. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007, 67, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Dadsetan, S.; Balzano, T.; Forteza, J.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Hernandez-Rabaza, V.; Gil-Perotin, S.; Cubas-Nunez, L.; Garcia-Verdugo, J.M.; Agusti, A.; et al. Reducing peripheral inflammation with infliximab reduces neuroinflammation and improves cognition in rats with hepatic encephalopathy. Front. Mol. Neurosci. 2016, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, N.; Simpson, J.; Taylor, G.; Rafique, S.; Whitehouse, A.; Hiscox, J.; Stark, L.A. Nucleolar NF-κB/RelA mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death Differ. 2011, 18, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Kerr, L.E.; Birse-Archbold, J.L.; Short, D.M.; McGregor, A.L.; Heron, I.; Macdonald, D.C.; Thompson, J.; Carlson, G.J.; Kelly, J.S.; McCulloch, J.; et al. Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene 2007, 26, 2554–2562. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gall, J.M.; Bonegio, R.; Havasi, A.; Illanes, K.; Schwartz, J.H.; Borkan, S.C. Nucleophosmin, a critical Bax cofactor in ischemia-induced cell death. Mol. Cell. Biol. 2013, 33, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, J.; Park, B.H.; Kinzler, K.W.; Vogelstein, B. Role of BAX in the apoptotic response to anticancer agents. Science 2000, 290, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Cole, C.; Scullion, P.; Wilkie, R.; Westwood, N.J.; Stark, L.A.; Hey, R.T. A proteomic approach to analyze the aspirin-mediated lysine acetylome. Mol. Cell. Proteom. 2017, 16, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, C.J.; Zhang, J.; He, Y.; Lee, Y.M.; Chen, S.; Lim, T.K.; Ng, S.; Shen, H.M.; Lin, Q. Mapping sites of aspirin-induced acetylations in live cells by quantitative acid-cleavable activity-based protein profiling (QA-ABPP). Sci. Rep. 2015, 5, 7896. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Charruyer, A.; Grazide, S.; Bezombes, C.; Muller, S.; Laurent, G.; Jaffrezou, J.P. UV-C light induces raft-associated acid sphingomyelinase and JNK activation and translocation independently on a nuclear signal. J. Biol. Chem. 2005, 280, 19196–19204. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Stark, L.A. Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus. Biomedicines 2017, 5, 43. https://doi.org/10.3390/biomedicines5030043
Chen J, Stark LA. Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus. Biomedicines. 2017; 5(3):43. https://doi.org/10.3390/biomedicines5030043
Chicago/Turabian StyleChen, Jingyu, and Lesley A. Stark. 2017. "Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus" Biomedicines 5, no. 3: 43. https://doi.org/10.3390/biomedicines5030043
APA StyleChen, J., & Stark, L. A. (2017). Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus. Biomedicines, 5(3), 43. https://doi.org/10.3390/biomedicines5030043