Abstract
Nucleic acid aptamers are short RNA- or DNA-based affinity reagents typically selected from combinatorial libraries to bind to a specific target such as a protein, a small molecule, whole cells or even animals. Aptamers have utility in the development of diagnostic, imaging and therapeutic applications due to their size, physico-chemical nature and ease of synthesis and modification to suit the application. A variety of oligonucleotide modifications have been used to enhance the stability of aptamers from nuclease degradation in vivo. The non-bridging oxygen atoms of the phosphodiester backbones of RNA and DNA aptamers can be substituted with one or two sulfur atoms, resulting in thioaptamers with phosphorothioate or phosphorodithioate linkages, respectively. Such thioaptamers are known to have increased binding affinity towards their target, as well as enhanced resistance to nuclease degradation. In this review, we discuss the development of phosphorothioate chemistry and thioaptamers, with a brief review of selection methods.
1. Introduction
Nucleic acid aptamers are short segments of single-stranded DNA or RNA that mimic antibodies in their ability to specifically recognize and bind cognate target molecules with high affinities [1,2,3]. The aptamer field began in 1990 with the work of three groups. First, Tuerk and Gold [1] described an in vitro iterative process of RNA-T4 DNA polymerase binding experiments, with reverse transcription polymerase chain reaction (RT-PCR) amplification between each round, to progress from a starting RNA library with an eight-nucleotide random region down to two highly enriched RNA sequences. They named this process Systematic Evolution of Ligands through EXponential Enrichment (SELEX). Robertson and Joyce used a similar technique for the evolution of an RNA enzyme that cleaves ssDNA [2]. Ellington and Szostak [3] used a random RNA library containing 1013 sequences to develop the first aptamers targeting small molecules, specifically organic dyes. Many new aptamer selection and separation techniques have been developed since that time, and they have been reviewed in detail [4,5,6,7,8].
Aptamer selection methods have now been described using whole cells [7,9,10,11,12], excised tissues [13,14] and even live animals [15,16], because aptamers selected in vitro do not always work well in vivo [17,18]. Whereas typical SELEX is designed to produce aptamers targeting a known protein, the Cell-SELEX method targets the cells of interest directly, without the need for a predefined and purified target protein. It can be used to find aptamers that bind to the cell surface or become internalized. The first Cell-SELEX experiments targeted Trypanosoma brucei (T. brucei) [19,20] and Trypanosoma cruzi (T. cruzi) [21,22], although it has also been suggested [7] that earlier work targeting membranes from red blood cells [23] was the first Cell-SELEX experiment. The technique was subsequently used to derive aptamers targeting mammalian embryonic stem cells [24,25], infected cells [26] and cancerous cells [27]. Although Cell-SELEX has the advantage of not requiring a predefined target protein, it has numerous disadvantages. Cell-SELEX targets a cell surface decorated with a complex mixture of proteins and their post-translational variants, common to many cell types. Thus, it requires additional negative selection experiments against multiple common cell types. Secondly, lab-grown cells are a mixture of live and dead cells, so separation of these two cell types is warranted. Cell-SELEX is an excellent method for finding new biomarkers for specific cell types, and this is particularly true in cancer, as recently reviewed in 2016 by Chen et al. [7] and previously in 2014 by Sun et al. [28] and by Sun and Zu [29] in 2015. Live-animal-based-SELEX (also called in vivo-SELEX) has also been used to overcome the problems associated with in vitro protein-SELEX [17,18]. Mi et al. first used tumor-bearing mice as the selection medium to derive RNA aptamers targeting intrahepatic colorectal cancer and found an aptamer that targets p68, an RNA helicase [15]. Subsequently, Cheng et al. used this method to derive brain-penetrating aptamers [16], and just last year an aptamer targeting RNA helicase DHX9 [30]. Mai et al. used in vivo SELEX to derive DNA aptamers targeting lymphoma [31]. Tissue slide-based-SELEX is an intermediate method between selecting against live cells and live animals. In 2009, Li et al. first used the tissue slide-based-SELEX [13] method to develop an aptamer targeting hnRNP A1. Subsequently, Wang et al., in a method they call Morph-X-Select, incorporated the use of laser-microdissection to enable more precise morphology-based selection of aptamers or thioaptamers directly from pathological tissue slices [14]. They were able to simultaneously select aptamers (to unknown targets) and discover new biomarkers from tissue-bearing ovarian cancer, and do so for both the tumor vessels and the tumor cells.
A variety of techniques has been developed to improve the separation of aptamer:target complexes from unbound aptamers and target. Capillary electrophoresis separation methods (CE-SELEX [32,33,34,35] and nonequilibrium capillary electrophoresis of equilibrium mixtures (NECEEM) [36]) can remove the need for polymerase chain reaction (PCR) amplification and provide aptamers in as little as one round of selection. Likewise, other non-SELEX methods with separations based on chromatography (MonoLEX) [37,38], bead libraries [39], microfluidics (mSELEX) [40,41,42], atomic force microscopy (AFM) [43,44], graphene oxide (GO-SELEX) [45,46,47], surface plasmon resonance (SPR) [48,49,50] or fluorescence-activated cell sorting (FACS) [51,52] can reduce the time requirements and number of selection rounds required to obtain aptamers.
In order to create aptamers with enhanced stability to make them more relevant in the clinic, a variety of chemical modifications has been utilized to improve their in vivo life-times. Macugen (pegaptanib), a pegylated RNA aptamer that acts against vascular endothelial factor, was the first nucleic acid aptamer therapeutic approved by the United States Food and Drug Administration (FDA) for the treatment of age-related macular degeneration [53,54,55,56], and it contains 2′-fluoropyrimidines nucleotides and 2′-methoxy purines [56]. The use of 2′-fluoropyrimidines is fairly common [56,57,58]. Other 2′-ribose substitutions include 2′-amino, 2′-azido, 2′-hydroxymethyl and 2′-thio [59,60,61,62]. Other sugar modifications include bicyclic ribose derivatives in which the 2′- and 4′-positions are linked together forming blocked/locked nucleic acids [63,64,65], threose nucleic acids (TNA) [66,67], cyclohexenyl nucleic acids (CeNA) [68], glycerol nucleic acids (GNA) [69] and peptide nucleic acids (PNA) [70]. The phosphate backbones of aptamers have also been stabilized with the introduction of boranophosphates [71] and thio-/dithio-phosphates [72,73,74,75], which are the focus of this review.
2. Chemistry of Phosphorothioate and Phosphorodithioate DNA Backbones
The first phosphorothioate dinucleotide linkage was reported by Eckstein in 1967 [76], and he subsequently showed that RNA with phosphorothioate linkages is more resistant to ribonucleases (RNAses) compared to regular RNA [77]. This finding led to increased interest in sulfurization of oligonucleotides. A variety of sulfurizing reagents (Figure 1) has been used to synthesize phosphorothioate linkages from phosphite triesters, including elemental sulfur [78], tetraethylthiuram disulfide (TETD) [79], 3H-1,2-benzodithiol-3-one 1,1-dioxide also called Beaucage reagent [80], dibenzoyl tetrasulfide [81], bis(O,O-diisopropoxy phosphinothioyl)disulfide [82], 3-ethoxy-1,2,4-dithiazolidin-5-one (EDITH) [83], 1,2,4,-dithiazolidine-3,5-dione (DTSNH) [83], 3-methyl-1,2,4-dithiazolin-5-one (MEDITH) [84], 3-amino-1,2,4-dithiazole-5-thione (ADTT) [85] and, more recently, 3-((dimethylamino-methylidene)amino)-3H-1,2,4-dithiazole-3-thione (DDTT) [86]. 3-phenoxy 1,2,4-dithiazole-5-one and 3-phenoxy-1,2,4-dithiazole-5-one are also highly reactive oxidative sulfurizing agents, similar to EDITH [87]. Many of them contained a common five-membered ring system with a disulfide bond that undergoes a ring-opening reaction that starts with nucleophilic attack of the sulfur atom near the carbonyl carbon (Scheme 1).
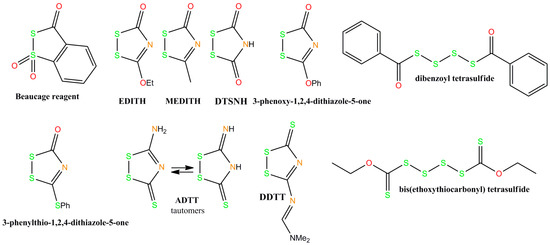
Figure 1.
Chemical structures of selected oxidative sulfurization reagents. EDITH: 3-ethoxy-1,2,4-dithiazolidin-5-one; MEDITH: 3-methyl-1,2,4-dithiazolin-5-one; DTSNH: 1,2,4,-dithiazolidine-3,5-dione; ADTT: 3-amino-1,2,4-dithiazole-5-thione; DDTT: 3-((dimethylamino-methylidene)amino)-3H-1,2,4-dithiazole-3-thione.
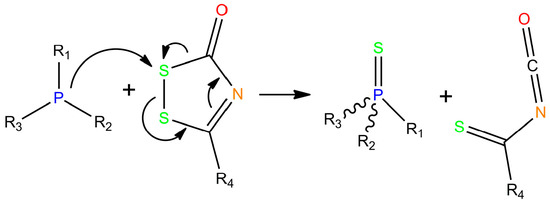
Scheme 1.
Oxidative sulfurization of a phosphotriester with a 1,2,4-dithiazole-5-one.
For a number of years, the Beaucage reagent [80] was widely used because it was inexpensive and stable when dry, but it suffered from stability problems once it was solvated and placed on a synthesizer. DTSNH and its precursor EDITH [83] gained popularity because they are very effective oxidative sulfurizing agents when using a short 2-min oxidation step, even at low (0.05 M) concentration. Ma et al. [88] found that when EDITH was used with fast deprotection methods (t-butylphenoxy acetyl protecting groups) and a capping step before the thio oxidation step, it provides oligonucleotides with fewer undesirable G substitutions and that mild deprotection can be used to synthesis oligonucleotides, such as thioated RNA, that are unstable to deprotection in concentrated ammonium hydroxide at elevated temperatures. An equally effective, but less expensive reagent, ADTT [85] was introduced in 2000 that offered similar benefits, but at a lower cost [85]. At present, DDTT [86] is often the preferred reagent for sulfurization due to its combination of low cost, reaction kinetics, efficacy, stability in a solvated form and its excellent oxidative sulfurization of RNA [89], but EDITH is also still used. DDTT can be shipped to the end user as a pre-made 0.1 M solution. As noted by Ponomarov et al., the 1,2,4,-dithiazole-5-ones are at least two orders of magnitude more reactive than the 1,2,4-dithiazoles [87]. Therapeutic phosphorothioates were recently reviewed by Eckstein [90]. At present, synthesizing oligonucleotides with phosphoromonothioate substitutions is easily performed on standard DNA and RNA synthesizers using solid-phase phosphoramidite chemistry [91], developed by Caruthers, and oxidative sulfurization in place of the standard oxidation step using iodine.
As phosphoromonothioate oligonucleotides became more common, the search for methods to create phosphorodithioate oligonucleotides began. In 1993, two groups led by Gorenstein [92] and Caruthers [93] were awarded patents for the production of oligonucleotides containing dithiophosphate substitutions using thiophosphoramidites (Figure 2) in place of phosphoramidites, and subsequent oxidative sulfurization. A handful of improvements in the method, mostly in regards to the phosphoramidite leaving group, were patented in the following few years by Caruthers’ group [94,95,96,97,98] and are described in multiple publications [99,100,101,102]. Solid-phase synthesis of dithioate oligonucleotides has recently been reviewed [103]. As the chemistry and understanding of both monothioate and dithioate DNA/RNA improved and the field of aptamer selection matured, the two fields began to merge.
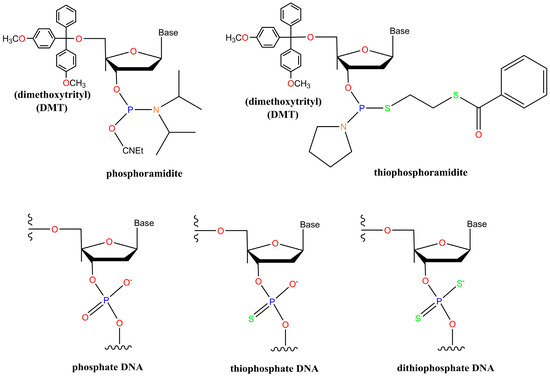
Figure 2.
Structures of DNA synthesis reagents and DNA containing normal DNA backbones or thioated DNA backbones.
3. Thioaptamer Development
3.1. The Beginning
David King et al. conducted the first combinatorial DNA thioaptamer selection experiments, using α-S-dATP in place of the usual dATP nucleotide during PCR, targeting the nuclear factor for interleukin 6 (NF-IL6), a basic leucine zipper involved in the inflammation cascade [104]. In this work using a 22-nucleotide random region flanked by two primers, they found that the thioaptamer out-competed the native binding sequence for NF-IL6 and bound in a two-to-one ratio, unlike the native binding 10-mer, which binds NF-IL6 at a one-to-one ratio.
Around this same time, Yang et al. investigated short decoy thioated DNA in the form of the monothioated and dithioated CK14 aptamer decoy binding to nuclear factor kappa B (NF-κB). In this work, it was shown that the binding characteristics of phosphorodithioates [105] were superior to both phosphoromonothioates and phosphate-only versions of CK14 and that structural changes occur in the DNA with the duplex taking on a more A-DNA-like structure [106]. Later ab initio calculations suggested that decreased binding of sodium atoms to the thioated phosphates may account for the increased binding properties of phosphorothioates and phosphorodithioates [107]. Marshall and Caruthers had also demonstrated previously that phosphorodithioate DNA oligomers strongly inhibit human immunodeficiency virus type-1 reverse transcriptase (HIV-1 RT), relative to non-thioated oligonucleotides [108]. This enhanced binding of dithioated DNA leads to such a strong binding interaction between heavily-dithioated DNA and hydrophobic columns typically used to purify them, that purification of the full-length oligonucleotide with trimethylamine acetate buffer at room temperature is often poor, even with the dimethoxytrityl group still attached. Separation of full-length oligonucleotides from shorter synthetic failures missing a few non-dithioated bases on the 5′-side is inadequate. To overcome this, Xin Bin Yang et al. devised a better purification scheme using anion-exchange chromatography with sodium thiocyanate salt [109]. The unusual binding strength of dithioates is exemplified by a report from last year, when Abeydeera et al. demonstrated that a single dithioate substitution in an α-thrombin RNA aptamer produced a 1000-fold improvement in binding [110]. As shown recently by Wu et al., the combination of dithioate DNA linkages and 2′-O-methyl substitutions enhanced the anti-tumor activity of a 25-base pair siRNA molecule by forming a more stable RISC complex [75].
Following their NF-IL6 thioaptamer selection experiments, King et al. used the same thioaptamer selection procedure to target the NF-κB proteins p65 (also called RelA)) and p50 [111]. With binding constants of 1–5 nM, the thioaptamers could effectively compete with the native binding element, immunoglobulin kappa B (IgκB), which has a weaker binding constant of ~82 nM, and the inflammation cascade could be altered by treatment with the NF-κB thioaptamer.
In exploring ways to improve the standard in vitro combinatorial aptamer selection process used at the time, which requires as many as 10–15 rounds of selection and PCR amplification, Yang et al. constructed the first bead-based thioaptamer/dithioaptamer selection process [112]. Using fluorescently-tagged proteins, the beads containing the best binding aptamers could be easily, albeit slowly, selected by hand under fluorescent lighting or they could be sorted in a high-speed manner using flow cytometry via fluorescence-assisted cell sorting (FACS) methods [39], as is also done with cell-based FACS-SELEX [51,52]. Although the bead sequences were read out by PCR of the sequences attached on the selected beads, an important feature is that amplification of the library was not required as part of the selection process.
3.2. Thioaptamers Targeting Infectious Diseases
In 2005, Anoma Somasunderam et al. developed double-stranded DNA thioaptamers targeting the ribonuclease H (RNase H) domain of human immunodeficiency virus-1 reverse transcriptase (HIV-RT) and demonstrated inhibition of the HIV-RT and anti-viral activity in vitro [113], in a dose-dependent manner. Although the top thioaptamer binds to the isolated RNase domain of HIV-RT, it did not bind to the RNase H domain from Escherichia coli, indicated that it is a sequence-specific aptamer. Subsequently, Ferguson et al. tested several delivery routes of the double-stranded aptamer and reported viral inhibition as high as 83% [114].
In 2006, Vaillant et al. tested the anti-inhibitory effects of three classes of random sequences (randomers) [115]. Randomer 1 sequences contained phosphorothioate DNA bases; Randomer 2 contained phosphorothioate, 2-O-methyl RNA bases; and Randomer 3 contained 2-O-methyl RNA bases only. They found that the phosphorothioate sequences bound to the heptad repeats on gp41, presumably on the hydrophobic face of this fusion peptide, leading to anti-viral activity. In contrast, the 2-O-methylated RNA did not lead to anti-viral activity, despite also binding to gp41, presumably on the hydrophilic face of the fusion peptides.
In 2007, Prusiner’s group investigated the effects of phosphorothioate DNA on the infectious prion protein PrPSc [116,117]. Karpuj et al. found that phosphorothioated 22-mers decreased levels of PrPSc and PrPC in prion-infected (scrapie-infected, Sc) neuroblastoma (N2a) cells (ScN2a) regardless of the DNA sequence [116]. The thioated DNA EC50 for half-maximal decrease of PrPSc was 50-times lower than the EC50 for PrPC, illustrating preferred action against the disease-causing form of the prion protein. In another paper from Prusiner’s group, published in the same month, King et al. [117] describe a thioaptamer family selected against Syrian hamster (Sha) recombinant prion protein (recSHaPrP). This family of aptamers contained a consensus sequence of twelve bases containing five critical monothioated deoxyadenosine (dA) bases. Their best aptamer bound to recSHaPrP with a 0.6 nM Kd, but with less affinity to PrP from bovine or human sources, thus showing protein sequence specificity.
Encouraged by earlier work targeting T. brucei and T. cruzi [19,20,21,22], Fennewald et al. demonstrated the in vivo utility of thioaptamer decoys for controlling the effects of viral hemorrhagic fever viruses in the arenavirus group [118]. In particular, studying the Lassa virus and Pichinde Virus (PICV), they demonstrated that their decoy thioaptamer called XBY-S2 bound to Fos-related antigen 2 (Fra-2) and Jun B, thereby enhancing cytokines levels (interleukin-6 (IL-6), interleukin-8 (IL-8), tumor necrosis factor alpha (TNF-α)) and greatly reducing mortality in guinea pigs infected with legal doses of Pichinde virus. When treated prophylactically with the XBY-S2 thioaptamer and again on Day 2 post-infection, 61% of the animals survived the PICV infection while only 21% of the untreated animals survived. Treatment with thioaptamer XBY-S1, which contains the same number of dithioate bases as XBY-S2, provided no significant change in survival.
In a more recent related study, Liu et al. described 20-mer dsDNA thioaptamers targeting the AP-1 transcription factor protein [119]. The aptamers contained either 3, 4, or 5 phosphorothioate end cap (EC) substitutions at each end of the thioaptamers and fluorescein isothiocyanate (FITC), gold or superparamagnetic iron oxide nanoparticles (SPIONs) for detection by optical imaging, transmission electron microscopy (TEM) or magnetic resonance imaging (MRI). In this study using C57black6 mice and a mutant with an altered A-Fos protein with enhanced binding affinity to Jun, they demonstrated multimodal imaging of transcription factor trafficking in the brain.
Kang et al. developed an RNA thioaptamer that targeted the capsid protein of the Venezuelan equine encephalitis (VEE) virus with a binding constant of about 70 nM in 2007 [120]. More recently, Gandham et al. developed a thioaptamer targeting the purified envelope protein domain III (ED3) of the Dengue 2 virus [121], but it has failed to reduce virus titers when tested against live cells up to this time.
3.3. Cancer-Related Thioaptamers
In 2008, Kang et al. developed a thioaptamer targeting tumor growth factor β-1 (TGF-β1) [122]. Surprisingly, despite the use of both thio-dA and thio-dC in the thioaptamer selection process, thus creating a heavily-monothioated aptamer, the TGF-β1 thioaptamer’s binding constant was only 90 nM. In 2015, Mathura et al. incorporated this thioaptamer into an aptasensor, which could detect TGF-β1 over a linear range of 1–250 ng/mL [123]. As part of a microfluidics device, the sensor could monitor the TGF-β1 release from activated hepatic stellate cells for nearly a day. Zhou et al. [124] had shown a similar lower level of detection for thrombin using two aptamers, named aptamer 15 (Apt15) [125] and aptamer 29 (Apt29) [126], targeting different epitopes of thrombin.
Somasunderam et al. developed a thioaptamer targeting the CD44 hyaluronic acid binding domain, or CD44 HABD [127]. The CD44 protein plays a large role in tumor growth and metastasis and is the primary binding site of hyaluronic acid. Although CD44 has many splice variants, its HABD is invariant and thus served as an excellent target for thioaptamer binding. In this work, they developed thioaptamers, with binding constants in the 200–300 nM range, which were able to block the binding of hyaluronic acid because hyaluronic acid only binds to CD44 HABD with a micromolar binding constant. The thioaptamers exhibited excellent binding to three human ovarian cancer cell lines—SKOV3 (Sloan-Kettering ovarian cancer-3), IGROV-1 (Institut Gustave Roussy Ovarian cancer-1) and A2780 (ovarian adenocarcinoma), but did not bind to CD44-negative cells, suggesting that they could be useful targeting agents against cancer. In 2016, Fan et al. demonstrated safe delivery of siRNA using a CD44 thioaptamer in a dendritic polyamidoamine system in vivo [128].
Another cancer-related DNA thioaptamer, called ESTA1 (E-Selection ThioAptamer 1), was selected against the soluble portion of the E-selectin protein [129]. The NF-κB and AP-1-inducible glycoprotein E-selectin (also called CD62 antigen-like family member E (CD62E), endothelial leukocyte adhesion molecule-1 (ELAM-1) or leukocyte-endothelial cell adhesion molecule-2 (LECAM-2)) is upregulated in the inflamed vasculature associated with many cancers, including, ovarian, breast and colon cancers, while not being constitutively expressed in endothelial cells [130]. The selectin proteins, including E-, L- and P-selectin, are surface glycoproteins that play a role in leukocyte tethering and rolling along endothelial walls [131,132]. Thus, they are attractive targets for localized delivery of cytotoxic chemotherapy drugs.
In 2010, Mann et al. showed that ESTA1 could be used to guide biodegradable mesoporous silicon microparticles to the bone marrow vasculature as part of a multifunctional drug delivery system for imaging or for therapeutic purposes [133]. In 2011, Mann et al. also showed that liposomes conjugated to ESTA1 could target inflamed vasculature associated with cancer [134]. Zong et al. demonstrated targeted delivery of ESTA-1-labeled multistage drug delivery vehicles to acute myelogenous leukemia (AML) blasts and leukemia stem cells [135] and reduced AML tumor burden by up to 60% while greatly reducing drug dosage and dosing frequency [136]. Later, Mai et al. demonstrated the therapeutic effects of the ESTA1-conjugated multistage vector (ESTA-MSV) drug carrier for the treatment of metastatic breast cancer in bones by targeting the bone marrow [31]. This directed delivery of siRNA targeting the STAT3 gene (ESTA-MSV/STAT3 siRNA) knocked down STAT3 protein production in nearly 50% of cancer cells in the bone and led to enhanced survival (25% extension of life) in mice bearing MDA-MB-231 bone metastasis. It was then shown that ESTA1 can be used to block the metastatic-niche and reduce the spread of breast cancer metastasis in the lungs [137] and that it does so in a safe manner [138]. This work implies that ESTA1 may work in a variety of other hematogenous metastasis. It was also shown that PEGylation of ESTA1 provides enhanced circulation time for this aptamer [139].
An RNA aptamer [140] stabilized with 2′-amino pyrimidine nucleotides and a DNA aptamer [141] targeting L-selectin have been reported. Although the RNA and DNA aptamers targeting L-selectin were both effective at blocking the interaction between L-selectin and sialyl Lewis X carbohydrate, only the DNA aptamer was also capable of blocking lymphocyte trafficking in vivo in severe combined immune deficiency (SCID) mice containing the scid mutation. Although the RNA aptamer affinity to L-selectin was high at lower temperatures, its weak interaction at 37 °C made it unsuitable for in vivo testing.
Gutsaeva et al. produced an anti-mouse P-selectin RNA aptamer (ARC5690) stabilized with 2′-fluoro pyrimidines and 2′-methoxy purines [142] and tested it both in vitro and in a sickle cell disease (SCD) transgenic mouse model. In vitro tests indicate that the core of the aptamer binds with an impressive Kd of ~15 pM with little to no binding with E-selectin. In SCD mice dosed at 20 mg/kg of body weight via intraperitoneal injection, the ARC5690 aptamer outperformed an anti-P-selectin antibody in anti-adhesion tests and performed comparably to the antibody in red blood cell (RBC) velocity measurements and the reduction of leukocyte rolling flux. In a hypoxia/normoxia stress experiment, SCD mice treated with aptamer ARC5690 had a higher survival rate (86%, 12/14) compared to saline-treated (67%, 14/21) or scrambled aptamer-treated mice (53%, 8/15).
In 2012, Watanabe et al. tested the effects of DNA thioaptamers targeting high mobility group A (HMGA) proteins HMGA1 and HMGA2 in pancreatic cancer cell lines [143]. These proteins are architectural transcription factors known to bind in the minor groove of adenosine:thymine (A:T) tracks [144], and high expression levels of HMGA1 are associated with chemotherapy resistance [145]; while HMGA2 is associated with Ras-induced invasion and metastasis, and its downregulation inhibits pancreatic cell lines [146]. Most notably, the AT-rich phosphorothioate aptamer (AT-sDNA) increased the sensitivity of Miapaca-2 and AsPC-1 pancreatic cancer cell lines to gemcitabine treatment, presumably by acting as a decoy to bind to excess HMGA1 [143].
In 2015, Hu et al. described selecting a thioaptamer containing thio-dA, named HY6 [147], against an extracellular domain (ECD) peptide of the human epithelial growth factor receptor 2 (HER2, ErbB2). HER2 overexpression is associated with many cancers, including breast cancer [148], lung cancer [149], ovarian cancer [150] and gastric and esophageal carcinomas [151], and treatment targeting HER2 with antibodies is known [152,153]. HY6 binds to the extracellular domain of HER2 with a binding constant of 178 nM, and they showed binding to HER2-positive SK-BR-3 and MDA-MD-453 breast cancer cells, but not to HER2-negative MDA-MB-231 cells. Liu et al. had previously developed a non-thioated aptamer (HB5) targeting a HER2 ECD peptide [154]. HB5 was found to bind the ECD of HER2 with a 316 nM Kd and bind more tightly to an epitope peptide with an 18.9 nM Kd [154]. In vitro testing of a HB5-doxorubicin complex showed preferential treatment to HER2-positive breast cancer cells. The HB5 and HY6 sequences have some similarity in that they both contain T stretches. While the thioaptamer HY6 was stable in serum and was able to bind its target after long exposure in serum, its analog without thiophosphate substitutions could not [147].
In 2016, Mangala et al. described the selection of thioaptamers targeting ovarian cancer-associated endothelial cells generated by using a thioaptamer version of the Cell-SELEX technique [155] against ovarian cancer associated-endothelial cells derived from ten different patients while using cells derived from normal ovaries as controls. They found that the Endo28 thioaptamer targets annexin A2, a protein upregulated in cancer [156,157]. When Endo28 was conjugated to the surface of Chitosan (CH) Nanoparticles (NP) containing siRNA targeting micro-RNAs miR106b-5p and miR30c-5p, the CH/Endo28-siRNA-NP provided enhanced targeting of the tumor relative to other organs. Most striking was the near elimination of particle accumulation in the liver, which absorbs the majority of untargeted particles. Targeted delivery of these particles into orthotopic ovarian cancer models led to enhanced maturation of the vasculature surrounding the tumor and led to anti-tumor effects, suggesting that this therapy could lead to better therapy targeting of angiogenesis. The Endo28 thioaptamer has been connected to one arm of a stable three-Way Junction (3WJ) RNA from phi29 packaging RNA to form stable DNA/RNA hybrid nanoparticles that could be loaded with doxorubicin for targeted delivery to ovarian cancer cells [158]. The Endo28-3WJ-Sph1/Dox particles were delivered to annexin-2-positive IGROV-1 (71%) and SKOV-3 (52%) ovarian cancer cell lines, with very little binding (17%) to annexin-A2-negative human embryonic kidney 293 (HEK293) cells. In vivo testing showed that 6 h post-injection of 10 µM particles, the fluorescently-labeled particles were only detected in the xenograft tumors, demonstrating their potential utility as a targeting agent for annexin-2-positive cancers [158].
In 2015, Wu et al. developed two DNA aptamers, XQ-2 and a truncated version called XQ-2d, targeting Pancreatic Ductal Adenocarcinoma (PDAC) [159], the most common pancreatic adenocarcinoma [160]. The longer original aptamer, XQ-2, was selected against the PDAC PL45 cell line using Cell-SELEX, and it was shown to bind tumor sites in athymic BALB/c nude mice. They created both phosphorothioate and 2′-OMe derivatives of XQ-2d to increase serum stability, but found that the thioaptamer version of XQ-2d exhibited reduced binding to PL45 cells, while the 2-OMe version did not.
AXL-tyrosine receptor kinase plays a role in many cancers, including breast [161], lung [162], ovarian [163], pancreatic [164] and prostate [165] cancers, as well as leukemia [166]. In 2012, Cerchia et al. developed GL21.T, an RNA aptamer targeting the AXL-tyrosine receptor kinase (AXL) protein [167]. They showed that GL21.T inhibits the formation of lung tumors in xenograft mouse models, presumably by binding to the extracellular domain of AXL and blocking downstream phosphorylation of Erk and Akt proteins. Because AXL downregulation has been shown to be an attractive therapeutic target for ovarian cancer [156,168,169,170,171], Kanlikilicer et al. created and tested a serum-stable DNA version of GL21.T, called AXL-DNA-APTAMER, containing both 2′-fluoropyrimidine nucleosides and thiophosphates [172], and tested its effects on ovarian cancer. In two models of intraperitoneal ovarian cancer, the AXL-DNA-APTAMER suppresses metastasis, migration/invasion and tumor growth on its own and enhances the effects of paclitaxel treatment [172], presumably by disrupting focal adhesion kinase (FAK) phosphorylation and matrix-metalloproteinases. These aptamers may serve as alternate future treatments for ovarian cancers, especially those that have become drug-resistant.
3.4. Other Thioaptamers
In 2012, Lui et al. described an aptamer they called Adipo8 that specifically targets mature white adipocytes with an 18 nM Kd [173]. Using the Cell-SELEX procedure, they first targeted differentiated 3T3-L1 adipocytes before doing counter selection with undifferentiated 3T3-L1 preadipocytes or HepG2 cells during aptamer selection rounds. They found that Adipo8 did not bind to six types of cancer cells, B cells or T cells, nor to hepatocytes or muscle cells known to display some of the same proteins displayed on adipocytes after differentiation. More recently [174], Chen et al. tested a 5′-PEG-modified version of Adipo8 with phosphorothio linkages located just after the first base and just before the last base. The addition of these two thioate linkages increased the serum stability by a small, yet statistically significant amount when tested on tissue culture and in vivo. Because this aptamer can selectively distinguish white adipocytes from brown adipocytes and pre-adipocytes, it may serve as a useful delivery agent for adipocyte-specific therapy.
In 2013, Higashimoto et al. derived the first thioaptamers, containing both thio-dA and thio-dT, targeting Advanced Glycation End-products (AGEs) [175], and found that three of them, #4s, #7s and #9s, bound to AGE-Human Serum Albumin (HSA) with subnanomolar binding. Elevated levels of AGEs and interaction with their Receptor (RAGE) increase vascular hyperpermeability and thrombosis by inducing Vasculature Endothelial Growth Factor (VEGF) and Plasminogen Activator Inhibitor-1 (PAI-1) [175,176]. AGEs are associated with complications of diabetes, such as retinopathy, and other vascular and degenerative diseases, and suppression of AGEs is a potential therapeutic strategy [176]. Higashimoto et al. found that when Human Umbilical Vein Endothelial Cells (HUVECs) were exposed to 20 mM thioaptamer #4s, induction of RAGE, VEGF and PAI-1 was significantly reduced and that the aptamer inhibited binding to AGE-HSA in a dose-dependent manner [175]. In subsequent work [177], Maeda et al. tested the effects of an AGE-aptamer in a Streptozotocin (STZ)-induced diabetic Wistar rats using continuous intraperitoneal infusion and found that it prevented elevation in AGEs and abnormalities in Electroretinograms (ERGs). In earlier work, Higashimoto et al. [178] showed that a group of non-thioated aptamers targeting AGEs could inhibit apoptotic cell death and decrease DNA synthesis in pericytes and that one of them, clone 9, blocked the toxic effects of AGEs despite its relatively weak binding interaction (1 µM).
Mai et al. recently develop thioaptamers using in vivo-SELEX [31] to develop robust targeting agents for lymphoma. In this work, they demonstrated that metastases in the bone of living animals could be selectively targeted for treatment. Han et al. have also shown that thioaptamers attached to PEGylated Single-Walled Nanotubes (SWNTs) also provide targeting to breast cancer cells in vivo and in vitro [179].
4. Recent Developments in Thioaptamer Methods
4.1. Conjugate-SELEX
Most thioaptamer particle targeting experiments start by conjugating a known thioaptamer to a particle. However, recently, Mu et al. developed what they call the conjugate-SELEX method [180]. In this method, liposomes were created under dilute aptamer library conditions so that each liposome contained only one, or possibly a few, random DNA strands exposed on the surface. In each selection round, they collected the cytosolic fluid from head and neck squamous cell carcinomas. This method ensured that they were selecting only those thioaptamers that were successfully delivered to the cancer while attached to the liposomes.
4.2. X-Aptamers
In 2012, He et al. [181] described X-aptamers (Figure 1) in which a DNA CD44 thioaptamer and a drug, which both bind to CD44 separately, were conjugated to create an improved binding agent. Combining the results of high-throughput computational drug screening, NMR binding verification, and a bead-based thioaptamer method [38] with a CD44 thioaptamer [121], a better binding reagent was created that was superior to either the drug or the thioaptamer alone. Specifically, a bead-based library of sequences with randomly-placed allylamino groups was created based on a CD44 thioaptamer [121], and these allylamino groups were conjugated to drug hits. Although other high-throughput analysis techniques, such as that reported by Thiel et al. [182], work well for random DNA/RNA libraries, Lu et al. developed Aptaligner [183] for the automated alignment and decoding of the pseudo-random X-Aptamer sequences. At present, over a dozen modified deoxyuridine reagents are available for use in aptamer synthesis and their relative merits have been reviewed recently [184]. A bead-based X-Aptamer Selection Kit (Figure 3), which contains a subset of these deoxyuracil derivatives (Figure 4) with protein-like side chains and others, is now available from AM Biotechnologies, LLC (Houston, TX, USA), and a detailed step-by-step protocol for its use will be available soon [185]. Despite misconceptions, the X-Aptamer process does not require the “SELEX” process, only DNA-protein binding and sequencing. The advantages include selection in one or two rounds and a diverse set of bases or drugs, but its major disadvantages are the pseudo-random nature of the designs and limited sequence diversity space (~108 sequences) relative to traditional random libraries. Although the X-Aptamer method has similarities to slow off-rate modified aptamers (SOMAmers) [186], which were developed by Larry Gold’s group and which also contain protein-like side chains, X-Aptamers allow more freedom in that all occurrences of a particular base (dT/dU) are not required to all be modified, or all not be unmodified.
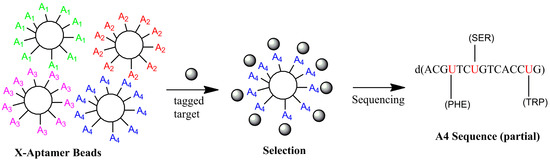
Figure 3.
X-aptamer selection process using tagged proteins. Each aptamer bead contains many copies of a single X-aptamer. Beads are selected by fluorescence or magnets, and a second optional solution-phase selection step is not shown.
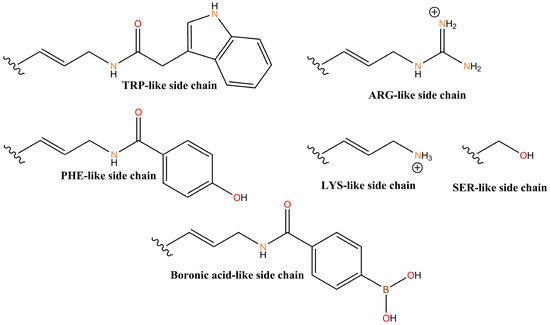
Figure 4.
Example side chains (X) in 5-X-dU containing X-aptamer beads.
5. Conclusions
A wide variety of oligonucleotide substitutions are now available, and these methods have been exhaustively reviewed in several recent articles [7,8,184,187] and older publications [188,189,190]. The review by Zhou and Rossi published a few months ago is particularly thorough. While thioaptamers are becoming more prevalent, they still lag behind the development of RNA aptamers. Preclinical testing of these thioaptamers in animals shows the promise of future therapies in a number of diseases.
Author Contributions
Both authors drafted the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; DeRosa, M.C. Challenges and opportunities for small molecule aptamer development. J. Nucleic Acids 2012, 2012, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.Y.; Byun, J. Nucleic acid aptamers: New methods for selection, stabilization, and application in biomedical science. Biomol. Ther. 2013, 21, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Szeitner, Z.; András, J.; Gyurcsányi, R.E.; Mészáros, T. Is less more? Lessons from aptamer selection strategies. J. Pharm. Biomed. Anal. 2014, 101, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yu, Y.; Jiang, F.; Zhou, J.; Li, Y.; Liang, C.; Dang, L.; Lu, A.; Zhang, G. Development of Cell-SELEX technology and its application in cancer diagnosis and therapy. Int. J. Mol. Sci. 2016, 17, 2079. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.-T.; Ziemer, G.; Paul, A.; Wendel, H.P. Cell-SELEX: Novel perspectives of aptamer-based therapeutics. Int. J. Mol. Sci. 2008, 9, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, L.; Giangrande, P.H.; McNamara, J.O.; Franciscis, V. Cell-Specific Aptamers for Targeted Therapies. In Nucleic Acid and Peptide Aptamers; Mayer, G., Ed.; Humana Press: Totowa, NJ, USA, 2009; Volume 535, pp. 59–78. ISBN 978-1-934115-89-3. [Google Scholar]
- Ohuchi, S. Cell-SELEX Technology. BioRes. Open Access 2012, 1, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Donovan, M.J.; Jiang, J. Aptamers from cell-based selection for bioanalytical applications. Chem. Rev. 2013, 113, 2842–2862. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, H.; Ding, H.; Huang, Y.; Cao, X.; Yang, G.; Li, J.; Xie, Z.; Meng, Y.; Li, X.; et al. Identification of an aptamer targeting hnRNP A1 by tissue slide-based SELEX. J. Pathol. 2009, 218, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, X.; Volk, D.E.; Lokesh, G.L.-R.; Elizondo-Riojas, M.-A.; Li, L.; Nick, A.M.; Sood, A.K.; Rosenblatt, K.P.; Gorenstein, D.G. Morph-X-Select: Morphology-based tissue aptamer selection for ovarian cancer biomarker discovery. BioTechniques 2016, 61. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Liu, Y.; Rabbani, Z.N.; Yang, Z.; Urban, J.H.; Sullenger, B.A.; Clary, B.M. In vivo selection of tumor-targeting RNA motifs. Nat. Chem. Biol. 2010, 6, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Chen, Y.H.; Lennox, K.A.; Behlke, M.A.; Davidson, B.L. In vivo SELEX for identification of brain-penetrating aptamers. Mol. Ther. Nucleic Acids 2013, 2, e67. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Healy, J.M.; Lewis, S.D.; Kurz, M.; Boomer, R.M.; Thompson, K.M.; Wilson, C.; McCauley, T.G. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm. Res. 2004, 21, 2234–2246. [Google Scholar] [CrossRef] [PubMed]
- Homann, M.; Göringer, H.U. Combinatorial selection of high affinity RNA ligands to live African trypanosomes. Nucleic Acids Res. 1999, 27, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Homann, M.; Göringer, H.U. Uptake and intracellular transport of RNA aptamers in African trypanosomes suggest therapeutic “piggy-back” approach. Bioorg. Med. Chem. 2001, 9, 2571–2580. [Google Scholar] [CrossRef]
- Lorger, M.; Engstler, M.; Homann, M.; Göringer, H.U. Targeting the variable surface of African trypanosomes with variant surface glycoprotein-specific, serum-stable RNA aptamers. Eukaryot. Cell 2003, 2, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.; Magdesian, M.H.; Alves, M.J.M.; Colli, W. In vitro selection of RNA aptamers that bind to cell adhesion receptors of Trypanosoma cruzi and inhibit cell invasion. J. Biol. Chem. 2002, 277, 20756–20762. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.N.; Jensen, K.B.; Julin, C.M.; Weil, M.; Gold, L. High affinity ligands from in vitro selection: Complex targets. Proc. Natl. Acad. Sci. USA 1998, 95, 2902–2907. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.-T.; Schäfer, R.; Paul, A.; Ziemer, G.; Wendel, H.P. Aptamer-based strategies for stem cell research. Mini Rev. Med. Chem. 2007, 7, 701–705. [Google Scholar] [PubMed]
- Iwagawa, T.; Ohuchi, S.P.; Watanabe, S.; Nakamura, Y. Selection of RNA aptamers against mouse embryonic stem cells. Biochimie 2012, 94, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Parekh, P.; Turner, P.; Moyer, R.W.; Tan, W. Generating aptamers for recognition of virus-infected cells. Clin. Chem. 2009, 55, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Shangguan, D.; Wang, K.; Shi, H.; Sefah, K.; Mallikratchy, P.; Chen, H.W.; Li, Y.; Tan, W. Selection of aptamers for molecular recognition and characterization of cancer cells. Anal. Chem. 2007, 79, 4900–4907. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zu, Y. A highlight of recent advances in aptamer technology and its application. Molecules 2015, 20, 11959–11980. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Ray, P.; Liu, J.; Kuan, C.-T.; Xu, J.; Hsu, D.; Sullenger, B.A.; White, R.R.; Clary, B.M. In vivo selection against human colorectal cancer xenografts identifies an aptamer that targets RNA helicase protein DHX9. Mol. Ther. Nucleic Acids 2016, 5, e315. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.; Li, X.; Zhang, G.; Huang, Y.; Xu, R.; Shen, Q.; Lokesh, G.L.; Thiviyanathan, V.; Chen, L.; Liu, H.; et al. A DNA thioaptamer with homing specificity to lymphoma bone marrow involvement. Nucleic Acids Res. 2017. under review. [Google Scholar]
- Mosing, R.K.; Mendonsa, S.D.; Bowser, M.T. Capillary electrophoresis-SELEX selection of aptamers with affinity for HIV-1 reverse transcriptase. Anal. Chem. 2005, 77, 6107–6112. [Google Scholar] [CrossRef] [PubMed]
- Drabovich, A.P.; Berezovski, M.; Okhonin, V.; Krylov, S.N. Selection of smart aptamers by methods of kinetic capillary electrophoresis. Anal. Chem. 2006, 78, 3171–3178. [Google Scholar] [CrossRef] [PubMed]
- Yufa, R.; Krylova, S.M.; Bruce, C.; Bagg, E.A.; Schofield, C.J.; Krylov, S.N. Emulsion PCR significantly improves nonequilibrium capillary electrophoresis of equilibrium mixtures-based aptamer selection: Allowing for efficient and rapid selection of aptamer to unmodified ABH2 protein. Anal. Chem. 2015, 87, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Berezovski, M.V.; Musheev, M.U.; Drabovich, A.P.; Jitkova, J.V.; Krylov, S.N. Non-SELEX: Selection of aptamers without intermediate amplification of candidate oligonucleotides. Nat. Protoc. 2006, 1, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Berezovski, M.; Drabovich, A.; Krylova, S.M.; Musheev, M.; Okhonin, V.; Petrov, A.; Krylov, S.N. Nonequilibrium capillary electrophoresis of equilibrium mixtures: A universal tool for development of aptamers. J. Am. Chem. Soc. 2005, 127, 3165–3171. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, A.; Kurth, A.; Dunkhorst, A.; Pänke, O.; Sielaff, H.; Junge, W.; Muth, D.; Scheller, F.; Stöcklein, W.; Dahmen, C.; et al. One-step selection of vaccinia virus-binding DNA aptamers by MonoLEX. BMC Biotechnol. 2007, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Dickman, M.; Hornby, D.P. Isolation of single-stranded DNA using denaturing DNA chromatography. Anal. Biochem. 2000, 284, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, X.; Prow, T.W.; Reece, L.M.; Bassett, S.E.; Luxon, B.A.; Herzog, N.K.; Aronson, J.; Shope, R.E.; Leary, J.F.; et al. Immunofluorescence assay and flow-cytometry selection of bead-bound aptamers. Nucleic Acids Res. 2003, 31, e54. [Google Scholar] [CrossRef]
- Hybarger, G.; Bynum, J.; Williams, R.F.; Valdes, J.J.; Chambers, J.P. A microfluidic SELEX prototype. Anal. Bioanal. Chem. 2006, 384, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.; Qian, J.; Xiao, Y.; Viel, L.; Gerdon, A.E.; Lagally, E.T.; Atzberger, P.; Tarasow, T.M.; Heeger, A.J.; Soh, H.T. Micromagnetic selection of aptamers in microfluidic channels. Proc. Natl. Acad. Sci. USA 2009, 106, 2989–2994. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ahn, J.-Y.; Jo, M.; Lee, D.; Lis, J.T.; Craighead, H.G.; Kim, S. Selection and elution of aptamers using nanoporous sol-gel arrays with integrated microheaters. Lab Chip 2009, 9, 1206. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Stephens, B.J.; Bonin, K.; Cubicciotti, R.; Guthold, M. A combined atomic force/fluorescence microscopy technique to select aptamers in a single cycle from a small pool of random oligonucleotides. Microsc. Res. Tech. 2007, 70, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Miyachi, Y.; Shimizu, N.; Ogino, C.; Kondo, A. Selection of DNA aptamers using atomic force microscopy. Nucleic Acids Res. 2010, 38, e21. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-W.; Tatavarty, R.; Kim, D.W.; Jung, H.-T.; Gu, M.B. Immobilization-free screening of aptamers assisted by graphene oxide. Chem. Commun. 2012, 48, 2071–2073. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.-T.; Kwon, Y.S.; Kim, J.H.; Gu, M.B. Multiple GO-SELEX for efficient screening of flexible aptamers. Chem. Commun. 2014, 50, 10513–10516. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Duan, N.; Wu, S.; Hao, L.; Xia, Y.; Ma, X.; Wang, Z. Graphene oxide-assisted non-immobilized SELEX of okdaic acid aptamer and the analytical application of aptasensor. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sayer, N.; Ibrahim, J.; Turner, K.; Tahiri-Alaoui, A.; James, W. Structural characterization of a 2′F-RNA aptamer that binds a HIV-1 SU glycoprotein, gp120. Biochem. Biophys. Res. Commun. 2002, 293, 924–931. [Google Scholar] [CrossRef]
- Spiga, F.M.; Maietta, P.; Guiducci, C. More DNA-aptamers for small drugs: A capture-SELEX coupled with surface plasmon resonance and high-throughput sequencing. ACS Comb. Sci. 2015, 17, 326–333. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Guo, L.; He, J.; Xu, H.; Xie, J. Stepping library-based post-SELEX strategy approaching to the minimized aptamer in SPR. Anal. Chem. 2017, 89, 6559–6566. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, M.L.; Dolf, A.; Endl, E.; Knolle, P.; Famulok, M.; Mayer, G. Enrichment of cell-targeting and population-specific aptamers by fluorescence-activated cell sorting. Angew. Chem. Int. Ed. 2008, 47, 5190–5193. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G.; Ahmed, M.-S.L.; Dolf, A.; Endl, E.; Knolle, P.A.; Famulok, M. Fluorescence-activated cell sorting for aptamer SELEX with cell mixtures. Nat. Protoc. 2010, 5, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.A.; Keating, G.M. Pegaptanib: In exudative age-related macular degeneration. Drugs 2005, 65, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- VEGF Inhibition Study in Ocular Neovascularization (V.I.S.I.O.N.) Clinical Trial Group. Year 2 efficacy results of 2 randomized controlled clinical trials of pegaptanib for neovascular age-related macular degeneration. Ophthalmology 2006, 113, 1508.e1–1508.e25. [Google Scholar] [CrossRef]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165) inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998, 273, 20556–20567. [Google Scholar] [CrossRef] [PubMed]
- Khati, M.; Schüman, M.; Ibrahim, J.; Sattentau, Q.; Gordon, S.; James, W. Neutralization of infectivity of diverse R5 clinical isolates of human immunodeficiency virus type 1 by gp120-binding 2’F-RNA aptamers. J. Virol. 2003, 77, 12692–12698. [Google Scholar] [CrossRef] [PubMed]
- Layzer, M.; McCaffrey, A.P.; Tanner, A.K.; Huang, Z.; Kay, M.A.; Sullenger, B.A. In vivo activity of nuclease-resistant siRNAs. RNA 2004, 10, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Jellinek, D.; Green, L.S.; Bell, C.; Lynott, C.K.; Gill, N.; Vargeese, C.; Kirschenheuter, G.; McGee, D.P.C.; Abesinghe, P. Potent 2’-amino-2’-deoxypyrimidine RNA inhibitors of basic fibroblast growth factor. Biochemistry 1995, 34, 11363–11372. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Gao, X.; Zhang, Z. Isolation and characterization of 2′-amino-modified RNA aptamers for human TNFα. Genom. Proteom. Bioinform. 2004, 2, 32–42. [Google Scholar] [CrossRef]
- Padilla, R.; Sousa, R. A Y639F/H784A T7 RNA polymerase double mutant displays superior properties for synthesizing RNAs with non-canonical NTPs. Nucleic Acids Res. 2002, 30, e138. [Google Scholar] [CrossRef] [PubMed]
- Dellafiore, M.A.; Montserrat, J.M.; Iribarren, A.M. Modified nucleoside triphosphates for in vitro selection techniques. Front. Chem. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Obika, S.; Nagashima, J.; Ohta, Y.; Suto, Y.; Ozaki, H.; Sawai, H.; Imanishi, T. Systematic analysis of enzymatic DNA polymerization using oligo-DNA templates and triphosphate analogs involving 2′,4′-bridged nucleosides. Nucleic Acids Res. 2008, 36, 4257–4265. [Google Scholar] [CrossRef] [PubMed]
- Barciszewski, J.; Medgaard, M.; Koch, T.; Kurreck, J.; Erdmann, V.A. Locked Nucleic Acid Aptamers. In Nucleic Acid and Peptide Aptamers; Mayer, G., Ed.; Humana Press: Totowa, NJ, USA, 2009; Volume 535, pp. 165–186. ISBN 978-1-934115-89-3. [Google Scholar]
- Kasahara, Y.; Irisawa, Y.; Ozaki, H.; Obika, S.; Kuwahara, M. 2′,4′-BNA/LNA aptamers: CE-SELEX using a DNA-based library of full-length 2′-O,4′-C-methylene-bridged/linked bicyclic ribonucleotides. Bioorg. Med. Chem. Lett. 2013, 23, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Horhota, A.; Zou, K.; Ichida, J.K.; Yu, B.; McLaughlin, L.W.; Szostak, J.W.; Chaput, J.C. Kinetic analysis of an efficient DNA-dependent TNA polymerase. J. Am. Chem. Soc. 2005, 127, 7427–7434. [Google Scholar] [CrossRef] [PubMed]
- Ichida, J.K.; Zou, K.; Horhota, A.; Yu, B.; McLaughlin, L.W.; Szostak, J.W. An in vitro selection system for TNA. J. Am. Chem. Soc. 2005, 127, 2802–2803. [Google Scholar] [CrossRef] [PubMed]
- Kempeneers, V. Investigation of the DNA-dependent cyclohexenyl nucleic acid polymerization and the cyclohexenyl nucleic acid-dependent DNA polymerization. Nucleic Acids Res. 2005, 33, 3828–3836. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-H.; Chen, J.; Szostak, J.W. Enzymatic synthesis of DNA on glycerol nucleic acid templates without stable duplex formation between product and template. Proc. Natl. Acad. Sci. USA 2007, 104, 14598–14603. [Google Scholar] [CrossRef] [PubMed]
- Wittung, P.; Nielsen, P.E.; Buchardt, O.; Egholm, M.; Norde, B. DNA-like double helix formed by peptide nucleic acid. Nature 1994, 368, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Lato, S.M.; Ozerova, N.D.S.; He, K.; Sergueeva, Z.; Shaw, B.R.; Burke, D.H. Boron-containing aptamers to ATP. Nucleic Acids Res. 2002, 30, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Andreola, M.-L.; Calmels, C.; Michel, J.; Toulmé, J.-J.; Litvak, S. Towards the selection of phosphorothioate aptamers. FEBS J. 2000, 267, 5032–5040. [Google Scholar] [CrossRef]
- Yang, X.; Gorenstein, D. Progress in thioaptamer development. Curr. Drug Targets 2004, 5, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Thiviyanathan, V.; Gorenstein, D.G. Aptamers and the next generation of diagnostic reagents. Proteom. Clin. Appl. 2012, 6, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Yang, X.; Gharpure, K.M.; Hatakeyama, H.; Egli, M.; McGuire, M.H.; Nagaraja, A.S.; Miyake, T.M.; Rupaimoole, R.; Pecot, C.V.; et al. 2′-OMe-phosphorodithioate-modified siRNAs show increased loading into the RISC complex and enhanced anti-tumour activity. Nat. Commun. 2014, 5, 3459. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F. A dinucleoside phosphorothioate. Tetrahedron Lett. 1967, 8, 1157–1160. [Google Scholar] [CrossRef]
- De Clerq, E.; Eckstein, E.; Merigan, T.C. Interferon induction increased through chemical modification of a synthetic polyribonucleotide. Science 1969, 165, 1137–1139. [Google Scholar] [CrossRef]
- Burgers, P.M.; Eckstein, F. Synthesis of dinucleoside monophosphorothioates via addition of sulphur to phosphite triesters. Tetrahedron Lett. 1978, 19, 3835–3838. [Google Scholar] [CrossRef]
- Vu, H.; Hirschbein, B.L. Internucleotide phosphite sulfurization with tetraethylthiuram disulfide. Phosphorothioate oligonucleotide synthesis via phosphoramidite chemistry. Tetrahedron Lett. 1991, 32, 3005–3008. [Google Scholar] [CrossRef]
- Iyer, R.P.; Egan, W.; Regan, J.B.; Beaucage, S.L. 3H-1,2-Benzodithiole-3-one 1,1-dioxide as an improved sulfurizing reagent in the solid-phase synthesis of oligodeoxyribonucleoside phosphorothioates. J. Am. Chem. Soc. 1990, 112, 1253–1254. [Google Scholar] [CrossRef]
- Rao, M.V.; Reese, C.B.; Zhao, Z.Y. Dibenzoyl tetrasulfide: A rapid sulfur-transfer agent in the synthesis of phosphorothioate analogues of oligonucleotides. Tetrahderon Lett. 1992, 33, 4839–4842. [Google Scholar] [CrossRef]
- Stec, W.J.; Uznanski, B.; Wilk, A. Bis(O,O-diisopropoxy phosphinothioyl)disulfide: A highly efficient sulfurizing reagent for cost-effective synthesis of oligo(nucleoside phosphorothioate)s. Tetrahedron Lett. 1993, 34, 5317–5320. [Google Scholar] [CrossRef]
- Xu, Q.H.; Musierforsyth, K.; Hammer, R.P.; Barany, G. Use of 1,2,4-dithiazolidine-3,5-dione (DTSNH) and 3-ethoxy-1,2,4-dithiazoline-5-one (EDITH) for synthesis of phosphorothioate-containing oligodeoxyribonucleotides. Nucleic Acids Res. 1996, 24, 1602–1607. [Google Scholar]
- Zhang, Z.; Nichols, A.; Tang, J.X.; Han, Y.; Tang, J.Y. Solid phase synthesis of oligonucleotide phosphorothioate analogues using 3-methyl-1,2,4-dithiazolin-5-one (MEDITH) as a new sulfur-transfer reagent. Tetrahedron Lett. 1999, 40, 2095–2098. [Google Scholar] [CrossRef]
- Tang, J.-Y.; Han, Y.; Tang, J.X.; Zhang, Z. Large-scale synthesis of oligonucleotide phosphorothioates using 3-amino-1,2,4-dithiazole-5-thione as an efficient sulfur-transfer reagent. Org. Process Res. Dev. 2000, 4, 194–198. [Google Scholar] [CrossRef]
- Lemaître, M.M.; Murphy, A.S.; Somers, R.L. Sulfurizing reagent II and its use in synthesizing oligonucleotide phosphoramidites. Glen Rep. 2006, 18, 4–5. [Google Scholar]
- Ponomarov, O.; Laws, A.P.; Hanusek, J. 1,2,4-Dithiazole-5-ones and 5-thiones as efficient sulfurizing agents of phosphorus (III) compounds—A kinetic comparative study. Org. Biomol. Chem. 2012, 10, 8868. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.Y.-X.; Dignam, J.C.; Fong, G.W.; Li, L.; Gray, S.H.; Jacob-Samuel, B.; George, S.T. Evaluation of 3-ethoxy-1, 2, 4-dithiazoline-5-one (EDITH) as a new sulfurizing reagent in combination with labile exocyclic amino protecting groups for solid-phase oligonucleotide synthesis. Nucleic Acids Res. 1997, 25, 3590–3593. [Google Scholar] [CrossRef] [PubMed]
- Bergot, B.J.; Egan, W. Separation of synthetic phosphorothioate oligodeoxynucleotides from their oxygenated (phosphodiester) defect species by strong-anion-exchange high-performance liquid chromatography. J. Chromatogr. A 1992, 599, 35–42. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Beaucage, S.L.; Caruthers, M.H. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981, 22, 1859–1862. [Google Scholar] [CrossRef]
- Gorenstein, D.; Farschtschi, N. Process for Preparing Dithiophosphate Oligonucleotide Analogs via Nucleoside Thiophosphoramidite Intermediates. Patent US5,218,088 A, 8 June 1993. [Google Scholar]
- Caruthers, M.H.; Ma, Y.-X.; Yau, E.K.; Nielsen, J.; Brill, W. Nucleoside Thiophosphoramidites. Patent US5,218,103 A, 8 June 1993. [Google Scholar]
- Caruthers, M.H.; Marshall, W.S. Polynucleotide Phosphorodithioates. Patent US5,278,302 A, 11 January 1994. [Google Scholar]
- Caruthers, M.H.; Marshall, W.S.; Brill, W.; Nielsen, J. Polynucleotide Phosphorodithioate. Patent US5,453,496 A, 26 September 1995. [Google Scholar]
- Caruthers, M.H.; Brill, W.K.D.; Yau, E.; Ma, M.; Nielsen, J. Nucleoside Thiophosphoramidites. Patent US5,684,148 A, 4 November 1997. [Google Scholar]
- Caruthers, M.H.; Brill, W.K.-D.; Yau, E.; Ma, M.; Nielsen, J. Polynucleotide Phosphorodithioate Compounds. Patent US5,602,244 A, 11 February 1997. [Google Scholar]
- Caruthers, M.H.; Brill, W.K.-D.; Yau, E.; Ma, M.; Nielsen, J. Polynucleotide Phosphorodithioate Compounds. Patent US5,750,666 A, 12 May 1998. [Google Scholar]
- Fischer, R.W.; Caruthers, M.H. Synthesis of a dinucleotide phosphoramidimidate. Tetrahedron Lett. 1995, 36, 6807–6810. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Caruthers, M.H. Oxidative formation of phosphorodithiaotes via H-phosphonodithioates. Tetrahedron Lett. 1995, 36, 695–698. [Google Scholar] [CrossRef]
- Bjergrde, K.; Dahl, O.; Caruthers, M.H. Synthesis of dinucleoside phosphoramidimidates. Tetrahedron Lett. 1994, 35, 2941–2944. [Google Scholar] [CrossRef]
- Greef, C.H.; Seeberger, P.H.; Caruthers, M.H.; Beaton, G.; Bankaitis-Davis, D. Synthesis of phosphorodithioate RNA by the H-phosphonothioate method. Tetrahedron Lett. 1996, 37, 4451–4454. [Google Scholar] [CrossRef]
- Yang, X. Solid-phase synthesis of Oligodeoxynucleotide Analogs Containing Phosphorodithioate Linkages: Synthesis of Oligodeoxynucleotide Analogs Containing PS2 Linkages. In Current Protocols in Nucleic Acid Chemistry; Egli, M., Herdewijn, P., Matusda, A., Sanghvi, Y.S., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 4.71.1–4.71.14. ISBN 978-0-471-14270-6. [Google Scholar]
- King, D.J.; Ventura, D.A.; Brasier, A.R.; Gorenstein, D.G. Novel combinatorial selection of phosphorothioate oligonucleotide aptamers. Biochemistry 1998, 37, 16489–16493. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Fennewald, S.; Luxon, B.A.; Aronson, J.; Herzog, N.K.; Gorenstein, D.G. Aptamers containing thymidine 3′-O-phosphorodithioates: Synthesis and binding to nuclear factor-κB. Bioorg. Med. Chem. Lett. 1999, 9, 3357–3362. [Google Scholar] [CrossRef]
- Volk, D.E.; Yang, X.; Fennewald, S.M.; King, D.J.; Bassett, S.; Venkitachalam, S.; Herzog, N.; Luxon, B.A.; Gorenstein, D.G. Solution structure and design of dithiophosphate backbone aptamers targeting transcription factor NF-κB. Bioorg. Chem. 2002, 30, 396–419. [Google Scholar] [CrossRef]
- Volk, D.E.; Power, T.D.; Gorenstein, D.G.; Luxon, B.A. An ab initio study of phosphorothioate and phosphorodithioate interactions with sodium cation. Tetrahedron Lett. 2002, 43, 4443–4447. [Google Scholar] [CrossRef]
- Marshall, W.S.; Caruthers, M.H. Phosphorodithioate DNA as a potential therapeutic drug. Science 1993, 259, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hodge, R.P.; Luxon, B.A.; Shope, R.; Gorenstein, D.G. Separation of synthetic oligonucleotide dithioates from monothiophosphate impurities by anion-exchange chromatography on a mono-Q column. Anal. Biochem. 2002, 306, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Abeydeera, N.D.; Egli, M.; Cox, N.; Mercier, K.; Conde, J.N.; Pallan, P.S.; Mizurini, D.M.; Sierant, M.; Hibti, F.-E.; Hassell, T.; et al. Evoking picomolar binding in RNA by a single phosphorodithioate linkage. Nucleic Acids Res. 2016, 44, 8052–8064. [Google Scholar] [CrossRef] [PubMed]
- King, D.J.; Bassett, S.E.; Li, X.; Fennewald, S.A.; Herzog, N.K.; Luxon, B.A.; Shope, R.; Gorenstein, D.G. Combinatorial selection and binding of phosphorothioate aptamers targeting human NF-κB RelA(p65) and p50. Biochemistry 2002, 41, 9696–9706. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Bassett, S.E.; Li, X.; Luxon, B.A.; Herzog, N.K.; Shope, R.E.; Aronson, J.; Prow, T.W.; Leary, J.F.; Kirby, R.; et al. Construction and selection of bead-bound combinatorial oligonucleotide phosphorothioate and phosphorodithioate aptamer libraries designed for rapid PCR-based sequencing. Nucleic Acids Res. 2002, 30, e132. [Google Scholar] [CrossRef] [PubMed]
- Somasunderam, A.; Ferguson, M.R.; Rojo, D.R.; Thiviyanathan, V.; Li, X.; O’Brien, W.A.; Gorenstein, D.G. Combinatorial selection, inhibition and antiviral activity of DNA thioaptamers targeting the RNase H domain of HIV-1 reverse transcriptase. Biochemistry 2005, 44, 10388–10395. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.R.; Rojo, D.R.; Somasunderam, A.; Thiviyanathan, V.; Ridley, B.; Yang, X.; Gorenstein, D.G. Delivery of double-stranded DNA thioaptamers into HIV-1 infected cells for antiviral activity. Biochem. Biophys. Res. Commun. 2006, 344, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, A.; Juteau, J.-M.; Lu, H.; Liu, S.; Lackman-Smith, C.; Ptak, R.; Jiang, S. Phosphorothioate oligonucleotides inhibit human immunodeficiency virus type 1 fusion by blocking gp41 core formation. Antimicrob. Agents Chemother. 2006, 50, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Karpuj, M.V.; Giles, K.; Gelibter, S.; Scott, M.R.; Lingappa, V.R.; Szoka, F.C.; Peretz, D.; Denetclaw, W.; Prusiner, S.B. Phosphorothioate oligonucleotides reduce PrPsc levels and prion infectivity in cultured cells. Mol. Med. 2007, 13, 190. [Google Scholar] [CrossRef] [PubMed]
- King, D.J.; Safar, J.G.; Legname, G.; Prusiner, S.B. Thioaptamer interactions with prion proteins: Sequence-specific and non-specific binding sites. J. Mol. Biol. 2007, 369, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Fennewald, S.M.; Scott, E.P.; Zhang, L.H.; Aronson, J.F.; Gorenstein, D.G.; Luxon, B.A.; Shope, R.E.; Beasley, D.W.C.; Herzog, N.K. Thioaptamer decoy targeting AP-1 proteins influences cytokine expression and the outcome of arenavirus infections. J. Gen. Virol. 2007, 88, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Ren, J.; Liu, C.-M.; Liu, P.K. Intracellular gene transcription factor protein-guided MRI by DNA aptamers in vivo. FASEB J. 2014, 28, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lee, M.S.; Watowich, S.J.; Gorenstein, D.G. Combinatorial selection of a RNA thioaptamer that binds to Venezuelan equine encephalitis virus capsid protein. FEBS Lett. 2007, 581, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Gandham, S.H.A.; Volk, D.E.; Rao, L.G.L.; Neerathilingam, N.; Gorenstein, D.G. Thioaptamers targeting Dengue virus type-2 envelope protein domain III. Biochem. Biophys. Res. Commun. 2014, 453, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lee, M.S.; Copland, J.A.; Luxon, B.A.; Gorenstein, D.G. Combinatorial selection of a single stranded DNA thioaptamer targeting TGF-β-1 protein. Bioorg. Med. Chem. Lett. 2008, 18, 1835–1839. [Google Scholar] [CrossRef] [PubMed]
- Matharu, Z.; Patel, D.; Gao, Y.; Haque, A.; Zhou, Q.; Revzin, A. Detecting transforming growth factor-β release from liver cells using an aptasensor integrated with microfluidics. Anal. Chem. 2014, 86, 8865–8872. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Tang, Y.; Xing, D. One-step homogeneous protein detection based on aptamer probe and fluorescence cross-correlation spectroscopy. Anal. Chem. 2011, 83, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Tasset, D.M.; Kubik, M.F.; Steiner, W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes. J. Mol. Biol. 1997, 272, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Somasunderam, A.; Thiviyanathan, V.; Tanaka, T.; Li, X.; Neerathilingam, M.; Lokesh, G.L.; Mann, A.; Peng, Y.; Ferrari, M.; Klostergaard, J.; et al. Combinatorial selection of DNA thioaptamers targeted to the HA binding domain of CD44. Biochemistry 2010, 49, 9106–9112. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Wang, X.; Ding, B.; Cai, H.; Wang, X.; Fan, Y.; Li, Y.; Liu, S.; Nie, S.; Lu, Q. Thioaptamer-conjugated CD44-targeted delivery system for the treatment of breast cancer in vitro and in vivo. J. Drug Target. 2016, 24, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.P.; Somasunderam, A.; Nieves-Alicea, R.; Li, X.; Hu, A.; Sood, A.K.; Ferrari, M.; Gorenstein, D.G.; Tanaka, T. Identification of thioaptamer ligand against E-selectin: Potential application for inflamed vasculature imaging. PLoS ONE 2010, 5, e13050. [Google Scholar] [CrossRef] [PubMed]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef] [PubMed]
- Berg, E.L.; Robinson, M.K.; Mansson, O.; Butcher, E.C.; Magnani, J.L. A carbohydrate domain common to both sialyl Le (a) and sialyl Le (X) is recognized by the endothelial cell leukocyte adhesion molecule ELAM-1. J. Biol. Chem. 1991, 266, 14869–14872. [Google Scholar] [PubMed]
- Ehrhardt, C.; Kneuer, C.; Bakowsky, U. Selectins-an emerging target for drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.P.; Somasunderam, A.; Liu, X.; Gorenstein, D.G.; Ferrari, M. E-selectin-targeted porous silicon particle for nanoparticle delivery to the bone marrow. Adv. Mater. 2011, 23, H278–H282. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.P.; Bhavane, R.C.; Somasunderam, A.; Montalvo-Ortiz, B.L.; Ghaghada, K.B.; Volk, D.; Nieves-Alicea, R.; Suh, K.S.; Ferrari, M.; Annapragada, A.; et al. Thioaptamer conjugated liposomes for tumor vasculature targeting. Oncotarget 2011, 2, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Sen, S.; Zhang, G.; Gorenstein, D.G.; Liu, X.; Ferrari, M.; Roboz, G.J.; Shen, H.; Guzman, M.L. Novel multistage nanoparticle drug delivery to ablate leukemia stem cells in their niche. Blood 2012, 120, 2631. [Google Scholar]
- Zong, H.; Sen, S.; Zhang, G.; Mu, C.; Albayati, Z.F.; Gorenstein, D.G.; Liu, X.; Ferrari, M.; Crooks, P.A.; Roboz, G.J.; et al. In vivo targeting of leukemia stem cells by directing parthenolide-loaded nanoparticles to the bone marrow niche. Leukemia 2016, 30, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-A.; Hasan, N.; Mann, A.P.; Zheng, W.; Zhao, L.; Morris, L.; Zhu, W.; Zhao, Y.D.; Suh, K.S.; Dooley, W.C.; et al. Blocking the adhesion cascade at the premetastatic niche for prevention of breast cancer metastasis. Mol. Ther. 2015, 23, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tsolmon, B.; Zheng, W.; Zhao, L.; Volk, D.E.; Lokesh, G.L.-R.; Hagensick, C.; Gupta, V.; Morris, L.; Zhao, Y.D.; et al. Safety evaluation of intravenously administered mono-thioated aptamer against E-selectin in mice. Toxicol. Appl. Pharmacol. 2015, 287, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Kamal, M.; Kang, S.-A.; Zhang, R.; Lokesh, G.L.R.; Thiviyanathan, V.; Hasan, N.; Woo, S.; Zhao, Y.; Leslie, M.; et al. E-selectin targeting PEGylated-thioaptamer prevents breast cancer metastases. Mol. Ther. Nucleic Acids 2016, 5, e399. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, D.; Koenig, A.; Jennings, S.; Hicke, B.; Han, H.-L.; Fitzwater, T.; Chang, Y.-F.; Varki, N.; Parma, D.; Varki, A. Calcium-dependent oligonucleotide antagonists specific for L-selectin. Proc. Natl. Acad. Sci. USA 1996, 93, 5883–5887. [Google Scholar] [CrossRef] [PubMed]
- Hicke, B.J.; Watson, S.R.; Koenig, A.; Lynott, C.K.; Bargatze, R.F.; Chang, Y.F.; Ringquist, S.; Moon-McDermott, L.; Jennings, S.; Fitzwater, T.; et al. DNA aptamers block L-selectin function in vivo. Inhibition of human lymphocyte trafficking in SCID mice. J. Clin. Investig. 1996, 98, 2688–2692. [Google Scholar] [CrossRef] [PubMed]
- Gutsaeva, D.R.; Parkerson, J.B.; Yerigenahally, S.D.; Kurz, J.C.; Schaub, R.G.; Ikuta, T.; Head, C.A. Inhibition of cell adhesion by anti-P-selectin aptamer: A new potential therapeutic agent for sickle cell disease. Blood 2011, 117, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Sheriff, S.; Lewis, K.B.; Tinch, S.L.; Cho, J.; Balasubramaniam, A.; Kennedy, M.A. HMGA-targeted phosphorothioate DNA aptamers increase sensitivity to gemcitabine chemotherapy in human pancreatic cancer cell lines. Cancer Lett. 2012, 315, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.; Nissen, M.S. The AT-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J. Biol. Chem. 1990, 265, 8573–8582. [Google Scholar] [PubMed]
- Liau, S.-S.; Whang, E. HMGA1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin. Cancer Res. 2008, 14, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ueda, Y.; Akaboshi, S.; Hino, Y.; Sekita, Y.; Nakao, M. HMGA2 maintains oncogenic Ras-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am. J. Pathol. 2009, 174, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Duan, J.; Cao, B.; Zhang, L.; Lu, X.; Wang, F.; Yao, F.; Zhu, Z.; Yuan, W.; Wang, C.; et al. Selection of a novel DNA thioaptamer against HER2 structure. Clin. Transl. Oncol. 2015, 17, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, F.; Fontanella, C.; Amoroso, V.; Bianchi, G.V.; Bisagni, G.; Falci, C.; Fontana, A.; Generali, D.; Gianni, L.; Grassadonia, A.; et al. Current challenges in HER2-positive breast cancer. Crit. Rev. Oncol./Hematol. 2016, 98, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Ross, D.S.; Aisner, D.L.; Chaft, J.E.; Hsu, M.; Kako, S.L.; Kris, M.G.; Varella-Garcia, M.; Arcila, M.E. HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J. Thorac. Oncol. 2016, 11, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-L.; Lee, M.-Y.; Chao, W.-R.; Han, C.-P. The status of HER2 amplification and Kras mutations in mucinous ovarian carcinoma. Hum. Genom. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Abrahao-Machado, L.F.; Scapulatempo-Neto, C. HER2 testing in gastric cancer: An update. World J. Gastroenterol. 2016, 22, 4619. [Google Scholar] [CrossRef] [PubMed]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Liu, Z.; Duan, J.-H.; Song, Y.-M.; Ma, J.; Wang, F.-D.; Lu, X.; Yang, X.-D. Novel HER2 aptamer selectively delivers cytotoxic drug to HER2-positive breast cancer cells in vitro. J. Transl Med. 2012, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Mangala, L.S.; Wang, H.; Jian, D.; Wu, S.Y.; Somasunderam, A.; Volk, D.E.; Lokesh, G.L.R.; Li, X.; Pradeep, S.; Yang, X.; et al. Improving vascular maturation using non-coding RNAs increases anti-tumor effect of chemotherapy. JCI Insight 2016, 1, e87754. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, P.M.; Hare, R.J.; Gryczynski, I.; Vishwanatha, J.K. Exosomal Annexin II promotes angiogenesis and breast cancer metastasis. Mol. Cancer Res. 2017, 15, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Bystricky, B.; Cierna, Z.; Sieberova, G.; Janega, P.; Karaba, M.; Minarik, G.; Benca, J.; Sedlackova, T.; Jurisova, S.; Gronesova, P.; et al. Relationship between circulating tumor cells and annexin A2 in early breast cancer patients. Anticancer Res. 2017, 37, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Pi, F.; Zhang, H.; Li, H.; Thiviyanathan, V.; Gorenstein, D.G.; Sood, A.K.; Guo, P. RNA nanoparticles harboring annexin A2 aptamer can target ovarian cancer for tumor-specific doxorubicin delivery. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, Z.; Bai, H.; Fu, T.; Yang, C.; Hu, X.; Liu, Q.; Champanhac, C.; Teng, I.-T.; Ye, M.; et al. DNA Aptamer selected against pancreatic ductal adenocarcinoma for in vivo imaging and clinical tissue recognition. Theranostics 2015, 5, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; DePinho, R.A. Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2002, 2, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Orfi, L.; Szabadkai, I.; Daub, H.; Keri, G.; Ullrich, A. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Shinh, Y.-S.; Lai, C.-Y.; Kao, Y.-R.; Shiah, S.-G.; Chu, Y.-W.; Lee, H.-S.; Wu, C.-W. Expression of AXL in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [CrossRef]
- Sun, W.; Fujimoto, J.; Tamaya, T. Coexpression of Gas6/AXL in human ovarian cancers. Oncology 2004, 66, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Koorstra, J.-B.M.; Karikari, C.A.; Feldmann, G.; Bisht, S.; Rojas, P.L.; Offerhaus, G.J.A.; Alvarez, H.; Maitra, A. The AXL receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Boil. Ther. 2009, 8, 618–626. [Google Scholar] [CrossRef]
- Sainaghi, P.P.; Castello, L.; Bergamasco, L.; Galletti, M.; Bellosta, P.; Avanzi, G.C. Gas6 induces proliferation in prostate carcinoma cell lines expressing the AXL receptor. J. Cell. Physiol. 2005, 204, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Boysen, J.; Nelson, M.; Secreto, C.; Warner, S.L.; Bearss, D.J.; Lesnick, C.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. Targeted AXL inhibition primes chronic lymphocytic leukemia b cells to apoptosis and shows synergistic/additive effects in combination with BTK inhibitors. Clin. Cancer Res. 2015, 21, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, L.; Esposito, C.L.; Camorani, S.; Rienzo, A.; Stasio, L.; Insabato, L.; Affuso, A.; de Franciscis, V. Targeting AXL with an high-affinity inhibitory aptamer. Mol. Ther. 2012, 20, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Tan, T.Z.; Kelly, Z.; Low, J.; Choolani, M.; Recchi, C.; Gabra, H.; Thiery, J.P.; Huang, R.Y.-J. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci. Signal. 2016, 9, ra97. [Google Scholar] [CrossRef] [PubMed]
- Halmos, B.; Haura, E.B. New twists in the AXL(e) of tumor progression. Sci. Signal. 2016, 9, fs14. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.; Villalobos, V.M.; Gevaert, O.; Abramovitz, M.; Williams, C.; Sikic, B.I.; Leyland-Jones, B. Single gene prognostic biomarkers in ovarian cancer: A meta-analysis. PLoS ONE 2016, 11, e0149183. [Google Scholar] [CrossRef] [PubMed]
- Rea, K.; Pinciroli, P.; Sensi, M.; Alciato, F.; Bisaro, B.; Lozneanu, L.; Raspagliesi, F.; Centritto, F.; Cabodi, S.; Defilippi, P.; et al. Novel AXL-driven signaling pathway and molecular signature characterize high-grade ovarian cancer patients with poor clinical outcome. Oncotarget 2015, 6, 30859. [Google Scholar] [PubMed]
- Kanlikilicer, P.; Ozpolat, B.; Aslan, B.; Bayraktar, R.; Gurbuz, N.; Rodriguez-Aguayo, C.; Bayraktar, E.; Denizli, M.; Gonzalez-Villasana, V.; Ivan, C.; et al. Hampering AXL tyrosine kinase mediated ovarian cancer metastasis via serum stable DNA-aptamer therapeutics. Mol. Ther. Nucleic Acids 2017, in press. [Google Scholar] [CrossRef]
- Liu, J.; Liu, H.; Sefah, K.; Liu, B.; Pu, Y.; van Simaeys, D.; Tan, W. Selection of Aptamers specific for adipose tissue. PLoS ONE 2012, 7, e37789. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, J.; Tong, G.; Liu, B.; Wang, G.; Liu, H. Adipo8, a high-affinity DNA aptamer, can differentiate among adipocytes and inhibit intracellular lipid accumulation in vitro. Sci. China Chem. 2015, 58, 1612–1620. [Google Scholar] [CrossRef]
- Higashimoto, Y.; Matsui, T.; Nishino, Y.; Taira, J.; Inoue, H.; Takeuchi, M.; Yamagishi, S. Blockade by phosphorothioate aptamers of advanced glycation end products-induced damage in cultured pericytes and endothelial cells. Microvasc. Res. 2013, 90, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.-B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Matsui, T.; Ojima, A.; Suematsu, M.; Kaseda, K.; Higashimoto, Y.; Yamakawa, R.; Yamagishi, S. DNA aptamer raised against advanced glycation end products prevents abnormalities in electroretinograms of experimental diabetic retinopathy. Ophthalmic Res. 2015, 54, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Higashimoto, Y.; Yamagishi, S.; Nakamura, K.; Matsui, T.; Takeuchi, M.; Noguchi, M.; Inoue, H. In vitro selection of DNA aptamers that block toxic effects of AGE on cultured retinal pericytes. Microvasc. Res. 2007, 74, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Han, X.; Wang, Z.; Wu, S.; Zheng, R. Thioaptamer conjugated single-wall carbon nanotubes in human breast cancer targeted photothermal therapy in vivo in vivo and in vitro in vitro. Int. J. Clin. Exp. Med. 2016, 9, 58–64. [Google Scholar]
- Mu, Q.; Annapragada, A.; Srivastava, M.; Li, X.; Wu, J.; Thiviyanathan, V.; Wang, H.; Williams, A.; Gorenstein, D.; Annapragada, A.; et al. Conjugate-SELEX: A high-throughput screening of thioaptamer-liposomal nanoparticle conjugates for targeted intracellular delivery of anticancer drugs. Mol. Ther. Nucleic Acids 2016, 5, e382. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Elizondo-Riojas, M.-A.; Li, X.; Lokesh, G.L.R.; Somasunderam, A.; Thiviyanathan, V.; Volk, D.E.; Durland, R.H.; Englehardt, J.; Cavasotto, C.N.; et al. X-Aptamers: A bead-based selection method for random incorporation of drug-like moieties onto next-generation aptamers for enhanced binding. Biochem. 2012, 51, 8321–8323. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Bair, T.; Peek, A.S.; Liu, X.; Dassie, J.; Stockdale, K.R.; Behlke, M.A.; Miller, F.J., Jr.; Giangrande, P.H. Rapid identification of cell-specific, internalizing RNA aptamers with bioinformatics analyses of a cell-based aptamer selection. PLoS ONE 2012, 7, e43836. [Google Scholar] [CrossRef] [PubMed]
- Lu, E.; Elizondo-Riojas, M.-A.; Chang, J.T.; Volk, D.E. Aptaligner: Automated software for aligning pseudo-random DNA X-aptamers from next-generation sequencing data. Biochemistry 2014, 53, 3523–3525. [Google Scholar] [CrossRef] [PubMed]
- Lipi, F.; Chen, S.; Chakravarthy, M.; Rakesh, S.; Veedu, R.N. In vitro evolution of chemically-modified nucleic acid aptamers: Pros and cons, and comprehensive selection strategies. RNA Biol. 2016, 13, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- Lokesh, G.L.; Wang, H.; Lam, C.H.; Thiviyanathan, V.; Ward, N.; Gorenstein, D.G.; Volk, D.E. X-Aptamer Selection and Validation. In RNA Nanostructures: Methods and Protocols; Bindewald, E., Shapiro, B., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1632, in press. [Google Scholar]
- Kraemer, S.; Vaught, J.D.; Bock, C.; Gold, L.; Katilius, E.; Keeney, T.R.; Kim, N.; Saccomano, N.A.; Wilcox, S.K.; Zichi, D.; et al. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS ONE 2011, 6, e26332. [Google Scholar] [CrossRef] [PubMed]
- Mercier, M.-C.; Dontenwill, M.; Choulier, L. Selection of nucleic acid aptamers targeting tumor cell-surface protein biomarkers. Cancers 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.E.; Wu, H.; Niu, Y.; Cai, J. Improving the stability of aptamers by chemical modification. Curr. Med. Chem. 2011, 18, 4126–4138. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ren, X.; Schluesener, H.J.; Zhang, Z. Aptamers: Selection, modification and application to nervous system diseases. Curr. Med. Chem. 2011, 18, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hahn, U.; Rentmeister, A. Cell-specific aptamers as emerging therapeutics. J. Nucleic Acids 2011, 2011, 1–18. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).