Abstract
Stem cell gene therapy approaches for Human Immunodeficiency Virus (HIV) infection have been explored in clinical trials and several anti-HIV genes delivered by retroviral vectors were shown to block HIV replication. However, gammaretroviral and lentiviral based retroviral vectors have limitations for delivery of anti-HIV genes into hematopoietic stem cells (HSC). Foamy virus vectors have several advantages including efficient delivery of transgenes into HSC in large animal models, and a potentially safer integration profile. This review focuses on novel anti-HIV transgenes and the potential of foamy virus vectors for HSC gene therapy of HIV.
1. Introduction
The latest statistics show that nearly 36.9 million people are living with Human Immunodeficiency Virus (HIV) and should be receiving treatment against HIV infection [1]. Highly active antiretroviral therapy (HAART) is the current standard of care which is a combination of drugs that effectively suppress HIV infection and increase the life expectancy of patients [2,3]. A strict adherence to drugs is required to suppress HIV infection and any interruption to a HAART regimen leads to re-emergence of the viral infection. Lifelong adherence to the drugs is also expensive. The annual cost of HAART drugs alone was estimated to be around $10,000–$12,000/patient in the United States, which might even be higher with more aggressive treatment options in patients with treatment failures, due to the development of resistant HIV variants. Importantly, continual usage of these drugs increases the risk of developing metabolic and cardiovascular disorders and non-HIV infection related morbidity [4,5].
The limitations of HAART have led to attempts to provide a permanent one time gene therapy approach using hematopoietic stem cells (HSCs) or T cells as the cell target. The “Berlin Patient” has provided a proof of concept for eradicating HIV infection by transplanting HSCs from an allogeneic donor who is naturally resistant to HIV [6]. This resistance is due to a mutation in the CCR5 allele, a HIV entry coreceptor that occurs in a small minority of the population [7,8,9]. The HSC transplantation approach used to treat the Berlin patient resulted in an effective cure without the need for antiretroviral drugs [7,10]. The Berlin patient has demonstrated the promise of HSC transplantation, but there are major limitations to this approach. The natural occurrence of CCR5 mutation is rare, with only around 1% of the Caucasian population estimated to harbor this mutation in homozygous form [8,11]. Thus, identifying an allogeneic matching donor with a homozygous CCR5 mutation is very rare [11,12], which is supported by the fact that only three additional transplants have been performed since the Berlin patient, but to date only the Berlin patient has had a functional cure against HIV [13]. Another limitation to the Berlin patient approach is that allogeneic transplantation comes with the risk of graft versus host disease. Alternatively, autologous HSC could be genetically modified ex vivo to express anti-HIV transgene(s) and infused into patients. HSCs have the ability to self-renew and differentiate into all hematopoietic lineages enabling long-term persistence of the therapeutic anti-HIV transgene(s) in the target cells for HIV infection, primarily CD4+ T cells and macrophages. Unlike life-long adherence to HAART, anti-HIV gene therapy could provide permanent protection against HIV with a single treatment. The promise of eradicating HIV by gene therapy is thus of great interest, and efforts to improve the efficacy and safety of HIV gene therapy are being intensively studied.
HSC gene therapy has been used to successfully treat monogenic diseases including adenosine deaminase deficient-severe combined immunodeficiency (ADA-SCID), X-linked SCID, X-linked adrenoleukodystrophy and hemophilia B [14]. In these HSC gene therapy clinical studies retroviral-based vector systems, gammaretroviral (GRV) or lentiviral (LV) vectors, were used to mediate permanent delivery of the therapeutic transgene [15]. Retroviral vectors have been used for HSC gene therapy due to their ability to stably integrate into the host genome, thereby resulting in stable genetic modification of all daughter cells by cell division [16]. This is critically important for HSC gene therapy where there is massive expansion and cell division during hematopoietic differentiation to all blood cell lineages. In the HSC gene therapy setting non-integrating vectors have not yet been effective in large animal models or in patients [15].
Unfortunately, this advantage of retroviral vectors also creates a problem. In the otherwise successful SCID-X1 gene therapy clinical trials, five out of twenty treated patients developed leukemia after treatment. The leukemia was caused due to the GRV provirus integration near proto-oncogenes including LIM Domain Only 2 (Rhombotin-Like 1). This adverse event caused by the integrated vector provirus is commonly referred to as vector mediated genotoxicity. This genotoxicity has raised serious safety concerns for using GRV vectors in HSC gene therapy. However, the clinical benefits achieved for the SCID-X1 trials and other gene therapy trials are compelling, and retroviral HSC gene therapy has moved forward for several diseases [17,18]. There are some challenges that need to be addressed before HSC gene therapy becomes a routine procedure for a wider range of diseases. Safer retroviral vectors need to be developed and thoroughly tested in animal models. To minimize vector mediated genotoxicity, self-inactivating (SIN)-LV vectors have gained favor [19]. Insulators and means to target integration away from proto-oncogenes are also being actively studied to further improve safety. Foamy viral (FV) vectors are particularly promising from a safety standpoint as they have a potentially safer integration profile [20] and are less likely to activate nearby genes than LV or GRV vectors [21].
HIV gene therapy will likely require long-term expression of potent anti-HIV transgenes in target cells. Several anti-HIV transgenes targeting various stages of the HIV life cycle have been identified and evaluated in tissue culture and in animal models. Some have progressed to clinical trials. Expressing multiple anti-HIV transgenes in a combinatorial approach has been more effective to suppress HIV replication than single transgene approaches [22,23,24]. This approach is analogous to the use of drug cocktails in HAART. However the efficient delivery of anti-HIV transgenes in clinical trials has been a major roadblock in part due to the fact that HIV-based LV vector titers are reduced by anti-HIV transgenes [25,26,27]. In this regard FV vectors are also advantageous as they do not share significant homology to HIV, and are not efficiently targeted by anti-HIV transgenes. This review focuses on the potency of anti-HIV transgenes and the potential of FV vectors for delivering anti-HIV transgenes.
2. Anti-HIV Transgenes
Anti-HIV transgenes can be either RNA or protein and can interfere either directly with viral components or indirectly with host cellular factors required for viral replication. The anti-HIV transgenes have been grouped into three classes based on where they target the viral life cycle, and mathematical modeling shows each class has very different effects on the HIV life cycle [28]. Class I transgenes inhibit the early stages of the HIV life cycle including viral entry, reverse transcription and integration of the provirus. Class II genes target the post-integration stages and inhibit viral genes that are critical for the translation and production of infectious HIV progeny. Class III transgenes target the late stages of the HIV life cycle, i.e., virion assembly, budding and release. Experimental evidence supports the modeling data that transgenes that block HIV entry are more potent inhibitors than the ones that target post-entry or late stages of HIV replication [28,29]. In the Berlin patient the success of providing a functional cure for HIV was attributed to the lack of a functional CCR5 coreceptor in allogeneic donor cells. Thus the CCR5 mutation which inhibits entry, acts like a class I anti-HIV gene [30]. A gene therapy approach of inhibiting CCR5 expression does have the limitation that it would only block R5-tropic HIV strains that specially use the CCR5 as an entry coreceptor, but not strains that use alternative coreceptors including CXCR4 (X4-tropic). Therefore individual and combinatorial anti-HIV transgenes that target multiple steps in HIV replication and can combat diverse strains of HIV are of great interest. To date, the efficacy of several anti-HIV transgenes have been tested under in vitro and in vivo settings (Table 1).

Table 1.
Anti-HIV transgenes and strategies used to block the expression.
2.1. Targeting Viral Genes to Inhibit HIV Infection
The HIV genome encodes multiple genes, including structural, regulatory and accessory genes that play diverse roles at various stages of HIV life cycle. Strategies to suppress the expression or function of these viral genes, including env, gag, pol, tat, rev, vpu and nef effectively inhibit HIV infection [29,70,71,72,73] (Figure 1). However, the relative potency of anti-HIV genes varies, and not all anti-HIV transgenes are promising for anti-HIV gene therapy [74,80]. HIV tat and rev regulate the transcription and translocation of HIV RNA from the nucleus to the cytoplasm, respectively, and have been commonly targeted viral genes for anti-HIV gene therapy. Strategies that include the expression of dominant-negative mutants, ribozymes, RNA decoys and siRNAs targeting tat and rev successfully inhibited HIV infection in both in vitro and in vivo models. The trans-dominant negative mutant, RevM10 competes with wild type rev for binding to the Rev responsive element (RRE) and inhibits cytoplasmic transport of the HIV transcript. RevM10 was shown to successfully block HIV replication when expressed in T cells and HSCs [61,62]. Ribozymes, small catalytic RNA molecules engineered to cleave specific RNA sequences, targeting tat and rev were shown to suppress viral transcript levels and inhibit HIV replication in CD34+-derived primary monocytes by 1000-fold [63]. Another successful strategy to block tat and rev activity is the use of small non-coding RNAs as decoys. The HIV Rev protein binds to the RRE of the viral transcript and promotes the translocation of viral RNA across the nuclear membrane into the cytoplasm. Tat, the trans-activator of transcription, is a regulatory protein that efficiently enhances production of viral transcripts by binding to transcription responsive elements (TAR) at the 5′ end of HIV RNA. RNA decoys that mimic RRE and TAR elements significantly interfered with these regulatory proteins and potently inhibited HIV replication up to five-fold [64,65,66,67]. RNAi mediated silencing of the tat and rev genes efficiently inhibited the expression of the respective proteins, and potently reduced HIV replication in gene modified human T cell lines, primary lymphocytes and CD4+ T cells by nearly 1000-fold as determined by p24 levels [68,70]. The success of targeting of tat and rev in blocking HIV replication allowed this approach to move forward to clinical studies [81,82]. In addition to tat and rev genes, structural and accessory genes were also targeted to block HIV replication. An approach utilizing siRNA-mediated suppression of viral structural genes, gag and pol reduced HIV replication by 4–5 logs in SupT1 and ~2–7-fold in PBMC, respectively [75]. Targeting accessory genes such as nef also conferred protection against HIV. However, the resistance did not last long due to the emergence of an escape mutant [77]. Recently, published work using CRISPR/Cas mediated targeting of various non-conserved HIV sequences also led to the emergence of escape mutants [69]. Both siRNA- and CRISPR/Cas mediated approaches to inhibit HIV replication confirmed that targeting conserved protein coding viral gene sequences could not only provide long lasting resistance against HIV infection but also delayed the emergence of escape mutants [75,76].
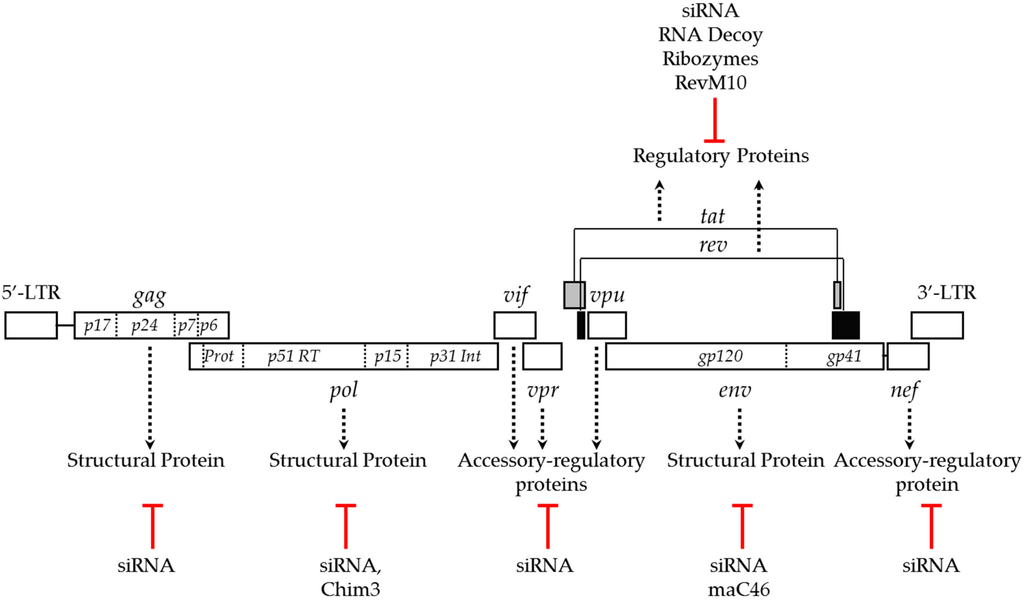
Figure 1.
HIV genome and gene therapy approaches targeting viral genes to block replication. Dotted arrows indicate the function of the viral genes and red lines indicate the gene-targeting approaches used to block HIV replication.
Viral infectivity factor (vif) is an auxiliary viral gene that supports HIV infectivity by countering the antiviral activity of the host restriction factor, apolipoprotein B mRNA-editing enzyme catalytic polypeptide (APOBEC)-3G [83]. Vif is also a part of HIV reverse transcription complex machinery and promotes reverse transcription and infectivity of HIV [84]. Approaches to disable the vif gene have been explored to block HIV replication. Expression of a naturally-occurring vif mutant, F12-Vif in CD4+ T cells was shown to inhibit HIV infection [85]. Subsequently, a more potent derivative of F-12 Vif, Chim3 was developed. Chim3 acts as dominant negative factor and inhibited the replication of R5- and X4-tropic HIV strains up to six-fold in macrophages and up to 25-fold in CD4+ T cells [78,79]. While all of the above anti-HIV transgenes inhibited post entry or late stages of the HIV life cycle, the membrane anchored fusion inhibitor, maC46 is a class I anti-HIV transgene derived from the HIV env gene that blocks HIV entry [59]. The maC46 is derived from the C-terminal heptad repeat of HIV gp41 fused to a membrane receptor to express as a membrane-anchored peptide. The maC46 is a 46 amino acid long membrane-bound version of the first clinical approved protein-fusion inhibitor, a 36 amino acid peptide, T20 (Enfuvirtide). T20 potently inhibits HIV replication of various strains, including multiple drug resistant mutants. However, prolonged treatment with T20 results in the emergence of HIV escape mutants that are resistant to T20 based treatment [86,87]. Several derivatives of T20 were subsequently generated that could suppress the replication of T20-resistant mutants [88,89,90]. Like the parental form, maC46 also confers resistance against a broad range of HIV strains [91] when expressed in human cell lines and CD4+ T cells [23,27,29,60]. Expression of maC46 was shown to block simian HIV infection up to 4 logs [23]. A direct comparison showed that cells expressing maC46 are highly resistant to HIV infection than cells expressing either anti-tat/rev siRNA or antisense RNA against env [29]. Though potent, emergence of escape mutants against T20 raises concerns for using maC46 based treatment. Strategies targeting viral genes, especially monotherapies targeting a single gene, are prone to the emergence of escape mutants due to the low fidelity of HIV reverse transcriptase. In order to prevent viral escape, strategies targeting either a host gene that facilitates HIV replication or that target multiple genes have been employed.
2.2. Using Host Cellular Proteins as Targets to Block HIV Infection
HIV depends on host factors to facilitate entry and integration of the viral genome, as well as transcription and translation of viral proteins in target cells (Figure 2). Disrupting the function or expression of these host genes can also block HIV infection. Targeting viral entry provides an effective way to interfere with HIV infection and CD4 is the primary receptor used by HIV to invade target cells. HIV uses additional coreceptors, primarily CCR5 or CXCR4 that determine the tropism, R5- and X4-tropic HIV strains for T cells and macrophages, respectively. Therapeutic approaches targeting these receptors have been developed to block HIV infection. Targeting CD4 is not as attractive as targeting CCR5 because of its essential role in immunological functions. However, strategies to block the interaction between the viral gp120 envelope protein and cellular CD4 receptor by expressing soluble CD4 [92], anti-CD4 monoclonal antibodies [93] and CD4-based bi-specific chimeric antigen receptors [94] all inhibited HIV entry. Strategies targeting HIV entry coreceptors have been extensively studied, with CCR5 being the most commonly targeted coreceptor [31,32,33]. Several strategies to disrupt CCR5 expression including intrabodies, ribozymes, siRNAs and gene editing approaches using zinc finger nucleases (ZFNs) and CRISPR/Cas have been evaluated. Blocking CCR5 expression by retaining CCR5 receptors in the endoplasmic reticulum with CCR5-specific single chain intrabodies protected lymphocyte cell lines, CD4+ T cells, and thymocytes against HIV infection in vitro [34]. Ribozymes targeting CCR5 efficiently suppressed CCR5 expression and inhibited HIV replication [35,36]. siRNA and miRNA based targeting of CCR5 mRNA effectively blocked HIV infection in vitro and ex vivo in xenotransplant mouse models [37,38,39]. Permanent disruption of the CCR5 allele using gene editing tools like ZFNs in T cells [40] and CD34+ cells [41] conferred resistance against R5-tropic HIV infection. A gene editing approach using the most recently discovered CRISPR/Cas system was also used to permanently disrupt the CCR5 allele [42,43]. Following allelic disruption of CCR5 via CRISPR/Cas and a CCR5 specific small guide RNA in induced-pluripotent stem cells, the cells were allowed to differentiate into monocytes and macrophages. The differentiated CCR5 mutant cells were resistant to HIV infection [42]. Wang and coworkers showed that efficient disruption of CCR5 by CRISPR/Cas in human CD4+ T cells confers resistance against R5-tropic HIV infection [44]. Some of these approaches targeting CCR5 have been evaluated in clinical trials [95,96]. Targeting CXCR4, an entry coreceptor used by X4-tropic strains, by siRNA- and CRISPR/Cas-mediated approaches was shown to confer resistance against X4-tropic HIV infection [45,46,47]. Unlike CCR5, CXCR4 is widely expressed in a variety of cells and is known to play a significant role in several physiological processes including stem cell homing [97,98]. CXCR4-null mice display severe hematopoietic and nervous disorders including reduced B-cell lymphopoiesis, myelopoiesis, and cerebellum development which often results in death at perinatal stage [99]. Conversely, CCR5-negative humans seems to have no deleterious effects [11]. Thus, the approach of targeting CCR5 is more attractive than targeting CXCR4.
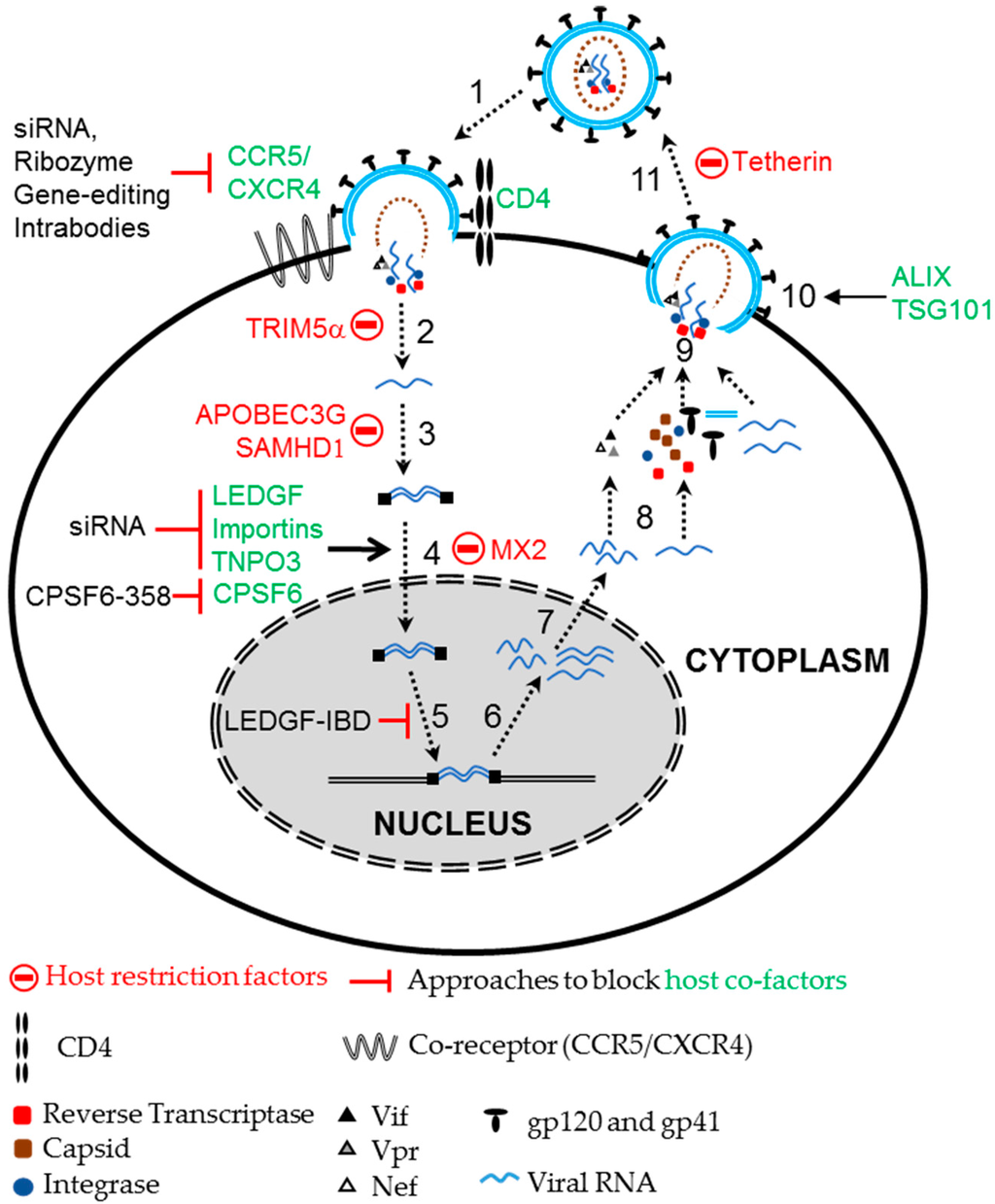
Figure 2.
HIV-1 life cycle and host co-factors. HIV exploits several host co-factors for its infection and replication. For entry, HIV uses the host receptor CD4 with the coreceptors CCR5 or CXCR4. Nuclear import and integration of the viral genome is supported by several host co-factors including LEDGF, Importin and TNPO3. Host cell cycle regulators support viral transcription, while proteins like ALIX and Tsg101 aid in budding of HIV virions. In contrast, HIV infection is restricted by several host restriction factors like TRIM5α, SAMHD1, APOBEC3G, Tetherin and MX2. Altering these co-factors and restriction factors by various strategies like siRNA, gene editing, ribozymes, expression of dominant negative variants and host restriction factors can block HIV replication. Numbers represent the steps in HIV life cycle, 1: Entry; 2: Uncoating; 3: Reverse Transcription; 4: Nuclear import; 5: Integration; 6: Transcription; 7: Nuclear export; 8: Translation; 9: Assembly; 10: Budding; and 11: Release and maturation.
Following entry, the virion uncoats, allowing the viral RNA to be reverse transcribed, imported into the nucleus to integrate into the host genome and continue its life cycle. HIV depends on various host factors for nuclear import and integration in the host cell genome. Lens epithelium derived growth factor (LEDGF) is a co-factor of HIV integrase that aids nuclear import of the viral pre-integration complex and supports chromosomal tethering of HIV [100,101,102]. Depleting LEDGF or blocking the interaction between LEDGF and the HIV integrase, either by expressing the integrase binding domain (IBD) of LEDGF or a small molecular inhibitor, LEDGIN, reduced chromosomal integration of HIV [50,51,103]. Expression of the IBD was shown to compete with endogenous LEDGF, interfere with HIV integration, and block HIV replication by more than 100-fold [103]. Stable expression of both the LEDGF-IBD, a peptide that lacks the chromatin binding domain, and a siRNA targeting LEDGF inhibited HIV infection in human cell lines and CD4+ T cells by 40-fold [52]. Mice engrafted with human CD4+ T cells expressing LEDGF/IBD showed significantly reduced levels of HIV load compared to mice engrafted with control CD4+ T cells upon HIV challenge [52]. HIV also depends on various other host factors such as importins and transportin 3 (TNPO3) for nuclear import [104,105]. Knockdown of importin-7 inhibited nuclear import of the HIV genome in macrophages [106], but had no effect on HIV replication [107]. A study by Zielske and Stevenson demonstrated that Importin-7 plays a critical role in accelerating the nuclear import of HIV [107]. Similarly, TNPO3 was also shown to be a key component of nuclear entry by various reports [108], although this function is debated [109]. Depletion of TNPO3 blocks nuclear entry and inhibits HIV replication, and this is dependent on another host protein, cleavage and polyadenylation specificity factor 6 (CPSF6) [109,110]. CPSF6, a nuclear protein, interacts with the HIV capsid protein and is implicated in regulating nuclear entry of HIV. However, suppression of endogenous CPSF6 did not affect HIV replication in vitro. The expression of a C-terminal truncated version of CPSF6 (CPSF6-358) was enriched in the cytoplasm, targeted the viral capsid protein and restricted nuclear import of HIV, inhibiting replication [55,56]. The anti-HIV efficacy of CPSF6-358 was shown to be equivalent to that of TRIM5α. Expression of CPSF6 blocked the replication of various HIV strains, but did not block the replication of variants with a N74D mutation in the HIV capsid protein [55].
Other cellular factors like autophagy related 16-like (Atg-16), autophagy related 5-like (Atg-5), Heat Shock 60 kDa protein (Chaperonin), TSG101, and ALIX which supports various stages of the HIV life cycle, were also shown to block HIV replication. A study to determine the effects of 30 known cellular factors on HIV replication demonstrated that cell lines expressing siRNA against ALIX, Atg-16 and TRBP genes were resistant to HIV replication for up to two months [53]. A subsequent study demonstrated that knockdown of various autophagy factors including Beclin-1, WIPI-1, PIK3R4, Atg-4, Atg-5, and Atg-16, inhibited HIV replication by blocking the production of viral particles. Further, the authors showed that simultaneous knockdown of two autophagy related proteins, Atg-5 and Atg-16 could delay HIV replication in SupT1 cells up to 100-fold compared to control cells [54].
In addition to host factors that support HIV replication, several host factors are known to restrict viral replication [111]. These include tripartite motif-containing protein 5 alpha (TRIM5α), APOBEC3G, tetherin and SAMHD1 (SAM domain and HD domain-containing protein 1) [112,113,114,115,116,117,118]. Strategies targeting these host restriction factors to block HIV replication have also been explored. In turn HIV expresses several accessory proteins to counter the antiviral activities of host restriction factors [111]. The viral accessory genes, vif, vpx and vpu were shown to counter the anti-viral restriction activities of APOBEC3G, SAMHD1 and Tetherin, respectively. Nonhuman primate TRIM5α confers potent resistance against HIV infection in old world monkeys [112]. Although the inhibitor mechanism is not fully understood, nonhuman primate TRIM5α binds to the viral capsid and blocks uncoating of HIV virions in old world monkeys. Human TRIM5α only modestly restricts HIV replication but ectopic expression of Rhesus macaque TRIM5α in human cells efficiently restricted HIV infection [112]. Similarly, a chimeric TRIM5α-CyclophilinA (AoT5Cyp) fusion found in the Aeotus genus of the new world owl monkey inhibited HIV infection when expressed in human cells [119]. Expressing a non-human transgene in humans has the potential to generate an immune response to the foreign transgene, which has hindered the use of nonhuman primate TRIM5α as an anti-HIV transgene. However, altering a small number of amino acids in the active or the capsid binding domains of human TRIM5α resulted in effective restriction of HIV infection in human cells [48,120,121,122,123]. Neagu and coworkers engineered a humanized version of TRIM5α-CyclophilinA fusion (hT5Cyp) that was able to restrict HIV infection in human cells at a comparable efficiency to the potent Rhesus/human chimeric AoT5CypA [49]. When hT5CypA is expressed in primary CD4+ T cells and macrophages it confers a survival advantage upon HIV infection in vitro. Transplantation of hT5CypA expressing CD4+ T in a mouse xenotransplant model resulted in effective engraftment and protection against HIV infection. Subsequently, another fusion protein of TRIM21 and CyclophilinA was also shown to possess potent anti-HIV inhibition similar to hT5CypA [124]. It remains to be determined if these proteins will be immunogenic in the setting of HSC transplantation in patients. Given the fact that allogeneic cells can be used for HSC transplantation, it is likely that immune responses to specific transgenes could be managed.
Host factors stimulated by type-1 interferon (IFN) upon viral infection also inhibit HIV replication [125,126]. The IFN stimulated gene, human myxovirus resistance 2 (MX2) has been shown to inhibit HIV replication [57,58]. MX2 was demonstrated to be a late post-entry suppressor of HIV, which blocks nuclear import of sub-viral complexes. Ectopic expression of MX2 was shown to potently inhibit the replication of several laboratory HIV isolates [57]. In summary, there are many anti-HIV transgenes available for an HSC gene therapy approach. Designing safe and effective vectors that express multiple transgenes in a combinatorial gene therapy approach is an important goal to bring HIV gene therapy to the clinic.
2.3. Efficacy of Combinatorial Approaches to Combat HIV Infection
Anti-HIV gene therapy is expected to be more effective when a combinatorial approach targeting multiple stages of the HIV life cycle is used. This increases the potency due to expression of multiple anti-HIV transgenes in the same cell and also can prevent the emergence of escape mutants. Kimpel and colleagues evaluated the potency of three anti-HIV transgenes, an anti-tat/rev siRNA, RNA antisense against the viral envelope, and maC46. They found that cells expressing maC46 and env RNA antisense strongly inhibited HIV infection in in vitro conditions [29]. Cells expressing maC46 had a better survival advantage over the cells expressing env RNA antisense and anti-tat/rev siRNA in a humanized xenotransplant mouse model upon HIV infection. Although the anti-tat/rev siRNA was less potent, their combination with entry inhibitors such as anti-CCR5 and maC46 expression improved the overall potency of the combinatorial construct [23,127]. Further, the protection developed by single anti-HIV transgene is more likely to fail as the result of the development of an escape mutant or phenotypic switch [77,128]. Escape occurs due to the error prone reverse transcriptase of HIV. Phenotypic switching between R5- to X4-tropic HIV strain during late stages of infection, resulting in emergence of X4 tropic strains in the patients treated with anti-CCR5 drugs occurs frequently [129,130,131]. Targeting both CCR5 and CXCR4 could provide a broader protection against diverse strains but targeting CXCR4 is problematic, as described above. Simultaneous expression of multiple anti-HIV transgenes targeting various HIV genes showed broad resistance against diverse HIV isolates and can delay the emergence of escape mutants [75,128].
For HSC gene therapy a combinatorial anti-HIV transgene cassette must be efficiently and stably delivered to a HSC. To ensure each transduced HSC gets all the transgenes, a single vector with multiple transgenes has commonly been used. Several groups have designed a single vector comprising a gene cassette that express multiple anti-HIV genes targeting various stage of the HIV life cycle. Including at least one class I anti-HIV transgene that targets viral entry has proved to be highly effective [22,27,75,76]. A combinatorial vector expressing a TAR decoy and an anti-CCR5 ribozyme displayed improved resistance against HIV-infection relative to expressing a TAR decoy alone both in vitro and in vivo. Further, a preclinical study revealed that the transgenes from the above combinatorial vector did not interfere with thymopoiesis [22]. The addition of a third anti-HIV transgene, i.e., tat/rev siRNA, to the above combinatorial anti-HIV transgenes (anti-CCR5 ribozyme and TAR decoy) had better anti-HIV effect than each anti-HIV transgene alone [127]. Cells transduced with the triple combinatorial anti-HIV vector suppressed HIV replication by nearly 2 logs [127]. Subsequent studies showed that human CD34+ cells transduced with the same triple combinatorial anti-HIV transgenes were able to differentiate into T cells in mice. The differentiated transgene-expressing T cells were resistant to HIV infection by more than 10-fold when challenged ex vivo [132]. In yet another approach, CD34+ cells transduced with a vector expressing three highly potent anti-HIV transgenes, i.e., CCR5 siRNA, chimeric TRIM5α and TAR decoy were shown to efficiently inhibit HIV replication [133]. Importantly, the expression of the above combinatorial anti-HIV transgenes significantly delayed the emergence of escape mutants as evaluated by long-term culture of challenged cells. The emergence of escape mutants can also be avoided by using a combinatorial approach of targeting conserved protein coding viral sequences. Simultaneous targeting of viral genes including a combination of gag, pol, nef and tat/rev by siRNA-mediated suppression provided long lasting resistance against HIV infection and also delayed the emergence of escape mutants compared to targeting a single viral gene [75,76,134].
In yet another combinatorial approach, CD34+ derived macrophages transduced with a combinatorial vector expressing the maC46 and anti-tat/rev siRNA were highly resistant to HIV infection and had a survival advantage upon HIV challenge compared to cells transduced with vector expressing individual anti-HIV transgene. Cells transduced with a triple combinatorial vector expressing anti-tat/rev siRNA, anti-CCR5 siRNA and maC46 showed significantly better protection against HIV infection than cells expressing the maC46 alone [23]. A preclinical study demonstrated that human CD34+ cells transduced with a dual combination anti-HIV vector, expressing CCR5 siRNA and maC46, could efficiently support long term engraftment and differentiation into multilineage hematopoietic cells in a humanized mouse model and provide resistance against both R5- and X4-tropic HIV infection [135,136].
3. Anti-HIV Gene Therapy Clinical Trails
The success of gene therapy approaches in preclinical models led to clinical trials to test the efficacy of anti-HIV gene therapy approach in patients. These clinical trials evaluated therapeutic benefits and also examined the safety, transduction efficacy, engraftment, immunological tolerance and long-term persistence of the gene modified cells in the infused patients. Moloney murine leukemia viral based GRV or LV vectors have been used to deliver the anti-HIV transgene by ex vivo transduction of either human CD34+ cells or T cells. The anti-HIV transgenes evaluated in clinical trials include: RRE decoy [137], RevM10 [81,138], anti-tat/vpr ribozyme [82,139], maC46 [140], CCR5 gene editing by ZFN [96] and a triple combinatorial anti-HIV gene cassette expressing anti-CCR5 ribozyme, anti-tat/rev siRNA and TAR decoy [95].
The efficacy of RRE decoy was tested in pediatric HIV patients. Patients infused with GRV vector-mediated modified autologous bone marrow derived CD34+ cells expressing a RRE decoy showed no evidence of adverse effects on patients. However, gene marking in peripheral blood samples was negligible after one year of transplantation [137]. This study highlighted the importance of improving gene transfer and engraftment efficiency [137]. Similarly, an anti-tat ribozyme (RRz2) anti-HIV HSC gene therapy was evaluated in phase I clinical studies [82,139]. Results showed that the gene modified T cells expressing RRz2 were not eliminated from the patient and expression of RRz2 was detected in the gene modified cells up to 4 years, but at a very low frequency [82]. Gene marking in peripheral blood samples in the patients ranged from 0.1% to 0.005% at 24 week point after infusion. A phase II study with seventy-four HIV patients evaluated a class II OZ1 (tat/vpr ribozyme, referred previously as RRz2) transgene delivered by a replication-incompetent GRV vector into CD34+ cells. Similar to the phase I study, transplantation of OZ1 transduced HSC showed no adverse clinical implications. Though gene marking and OZ1 expression was detectable in peripheral blood cells, the levels never reached the quantifiable range. Moreover, OZ1 modified cells that were detectable in 94% of the participants after four weeks of infusion gradually decreased to 7% at 100 weeks. In spite of this low gene marking and expression, the CD4+ T cell counts were higher in OZ1 treatment patients than the placebo group throughout the 100 weeks [141]. However, these trials were limited by very low marking.
The efficacy of Class I anti-HIV transgenes have also been evaluated in human trials. A German clinical trial employed infusion of T cells transduced with a GRV vector expressing the potent maC46 anti-HIV transgene to treat ten HIV-patients with advanced disease [140]. Infusion of T cells expressing maC46 did not result in any detectable immune response to the foreign transgene but had low gene marking, below 0.01% in total leukocytes after seven days. Gene modified cells were detectable in peripheral blood, lymph node and bone marrow up to one year post infusion but gradually decreased over time and was very low at one year (ranged below 0.001%–0.0001%). However, due to a lack of any significant change in HIV load during the first 4 months post infusion, seven out of the ten patients returned to a HAART regimen. Interestingly, four out of seven patients showed low viral loads, however the reason for this outcome is not clear.
The safety and the tolerability of a conditionally replicating LV vector expressing an antisense gene against the HIV envelope has also been evaluated in a phase I clinical trial [142]. A conditionally replicating LV vector retained the complete 5′- and 3′-LTR and all the cis-regulatory elements required for retroviral replication that replicated only in presence of wild type HIV, i.e., by HIV infection in the transduced cells. Five patients with chronic HIV infection were infused with gene-modified autologous CD4+ T cells. Cells expressing the antisense env were well tolerated in all patients. The engraftment frequency ranged from 0.023% to 0.04% after two years of infusion. CD4+ counts in four out the five patients either remained stable or increased; and at least one patient showed sustained decrease in the viral load. In yet another clinical study, the safety of a combinatorial anti-HIV gene therapy approach was evaluated, in this case a SIN-LV vector was used [95]. Four AIDS/lymphoma patients were transplanted with HSCs transduced with SIN-LV vector with combinatorial anti-HIV gene cassette expressing: anti-CCR5 ribozyme, anti-tat/rev siRNA and a TAR decoy. Expression of the anti-HIV transgenes were detectable for up to two years, but the gene marking frequency in the peripheral blood mononuclear cells was low and ranged between from 0.02%–0.32%. A more recent anti-HIV gene therapy trial evaluated the efficacy of infusing autologous CD4+ T cells that were gene-edited to disrupt CCR5 by ZFNs [96]. CCR5 disrupted CD4+ T cells readily engrafted and persisted long-term without any adverse events. This study showed a survival advantage of CCR5 disrupted CD4+ T cells in the HIV infected patients.
These clinical trials have demonstrated that an HSC based anti-HIV gene transfer approach to combat HIV infection is potentially safe and feasible, but the very low gene marking observed highlights a need for more effective ways to deliver transgenes with improved engraftment levels [143]. Therefore, efforts to develop improved vectors and transplantation protocols are needed to advance HSC anti-HIV gene therapy [143].
5. Foamy Viral Vectors
FV vectors have shown a great promise for HSC gene therapy [159]. FV are member of Spumaretrovirinea, a subfamily of retroviruses. FV are endemic in various non-human primates, cats, cattle and horses, and have co-evolved with their natural hosts. Although, humans are not a natural host, FV has been detected in humans through zoonotic infection. FV infection has not resulted in any disease in either natural or accidental hosts, including humans [160]. Moreover, FV replication is unique in that reverse transcription occurs during the late stage of FV life cycle resulting in packaging of DNA in infectious virions, rather than RNA. However, the relative amount of RNA and DNA in FV virions is still debated. FV have a relatively large genome allowing FV vectors to efficiently deliver transgenes up to 9.2 kb [161]. The wide cell tropism mediated by the FV envelope allows FV vectors to transduce essentially any mammalian cells, including dog HSC and human CD34+ cells with high efficiency [162,163,164,165,166]. FV vector transduction requires mitosis but FV vectors can persist as a stable transduction intermediate in quiescent cells and can transduce quiescent G0 cells at similar efficiency to LV vectors [167]. FV vectors compare favorably to LV vectors for HSC transduction in the large animal dog model [168] and in human CD34+ cells, and capable of engrafting in immunodeficient mice [169]. FV vectors also appear safe and effective when tested in the large animal, dog model. FV vector mediated HSC gene therapy was able to provide a functional cure for pyruvate kinase deficiency [170] and leukocyte adhesion deficiencies in dogs [171,172]. The poor safety profile of GRV vectors and the close phylogenetic relationship between LV vector and HIV has led to the development of FV vectors for anti-HIV HSC gene therapy. FV vectors efficiently transduced HSCs and can efficiently deliver some anti-HIV transgenes that lower the titer of LV vectors.
5.1. FV Vectors Have a Promising Safety Profile
Vector-mediated genotoxicity is a major concern of using retroviral vectors in gene therapy approach. LV vectors prefer to integrate within genes [20,173] and can generate chimeric transcripts by read-through transcription [21]. Proviral integration profile analysis and genotoxicity studies suggest FV vectors may be safer than other retroviral vectors. FV vectors integrate near transcription start sites less often than GRVs and integrate within genes less often than LV vectors [20,169,172,173]. A genotoxicity study comparing the ability of retroviral vectors to transactivate nearby genes revealed that FV vectors are less likely to transactivate nearby genes compared to GRV and LV vectors [21]. A high throughput integration site analysis of FV vector transduced long-term repopulating gene-modified cells in dog and mouse models showed no signs of clonal dominance [171,172,174]. Together, these studies support the use of FV vectors for anti-HIV gene therapy.
5.2. Anti-HIV Transgenes in FV Vectors
Another important feature that favors FV vectors to HIV-based LV vectors for anti-HIV gene therapy is the lack of homology to HIV. At least two studies revealed that anti-rev siRNA expression significantly drops LV viral vector titer up to 2 logs, whereas the same anti-HIV transgene had no significant effect on the titers of FV vectors [23,27]. Therefore, unlike LV vectors, anti-HIV transgenes are less likely to inhibit FV vector production and reduce vector titers. High titer vector is crucial for translation to the clinic.
5.3. FV Vectors for Anti-HIV Gene Therapy
The efficacy of various anti-HIV transgenes in a FV vector backbone and in stem cell based anti-HIV gene therapy have been explored [23,27,175]. The ability to block SIV infection, a close relative of HIV, by expressing anti-tat/rev siRNA encouraged the use of FV vectors in expressing anti-HIV transgenes and its utility in anti-HIV gene therapy [176]. Park and colleagues designed a FV vector expressing a combinatorial anti-HIV miRNA cassette targeting two regions of the HIV LTR and HIV-rev under the control of a Tat-inducible heat shock protein promoter [175]. Expression of anti-HIV miRNAs inhibited HIV replication in an in vitro HIV challenge assay by more than 98%. This study demonstrated that combinatorial anti-HIV transgene cassettes expressing TAR and anti-rev siRNA or TAR, anti-rev and anti-LTR siRNAs had showed an increased ability to block HIV replication than the cell expressing TAR alone [175]. Yet another study revealed the efficacy of FV vectors expressing combinatorial anti-HIV transgenes in inhibiting HIV replication [27]. CD34+ derived macrophages transduced with either maC46 or anti-tat/rev siRNA or the anti-HIV combinatorial cassette expressing RevM10, anti-tat/rev siRNA and maC46 showed ~4 logs reduction in HIV infection relative to untransduced and cells transduced with RevM10 alone. Benefits of the combinatorial anti-HIV transgene were further evaluated using an in vitro competitive survival advantage assay. The results revealed that cells expressing triple combinatorial anti-HIV transgenes had ~5.2 times more surviving cells after 16 days of HIV challenge, compared to cells expressing either anti-tat/rev siRNA or RevM10.
Similarly, Kiem and colleagues revealed that lymphocytes expressing multiple anti-HIV transgenes including maC46, siRNAs against tat/rev and CCR5, were highly resistant to HIV infection [23]. In a single-cycle infection assay, the authors observed that cells transduced with FV vector expressing maC46 or anti-HIV combinatorial FV vector expressing maC46, siRNAs against tat/rev and CCR5, showed ~20- and ~23-fold reduction in HIV infection compared to cells transduced with a control FV vector. This study was first to use an FV vector with a P140K mutant of methylguanine methyltransferase gene (MGMTP140K) in mediating in vivo selection of human repopulating cells with an anti-HIV transgene in a mouse model. Expression of MGMTP140K makes the cells resistant to O6-benzylguanine (O6BG), but functionally active to counter the effect of the bis-chloroethyl nitrosourea (BCNU), a chemotherapeutic drug. Administration of BCNU and O6BG selectively kills untransduced cells and allows selective enrichment of transgenic cells expressing MGMTP140K. This approach of in vivo selection was successful in increasing the percentage of gene marked cells and in expansion of gene modified repopulating cells [23,174]. In summary, the non-pathogenicity to humans, ability to transduce HSC and promising integration profile support FV vectors for anti-HIV gene therapy.
5.4. FV in HIV Vaccine Development
In addition to the gene therapy applications, FV vectors have also been explored for vaccine development including HIV vaccines [177]. Viral vaccines are derived from pathogenic viruses which are attenuated or inactivated and carry epitopes to elicit an immune response upon immunization. Replication-competent FV have also been used as vaccines. Though non-pathogenic, FV showed persistent infection in various hosts and induced strong cellular and humoral immune responses. The combination of being non-pathogenic, immunogenic, and the ability to integrate in the host and replicate in lymphoid cells has the potential to result in prolonged epitope expression eliciting a robust host immune response. Replication-competent FV vectors carrying various epitopes were designed to induce specific antibodies against their epitopes [178]. Gag, Env and Bet proteins of FVs were used as epitope carriers. The conserved domains in the above FV genes were replaced with the epitope sequence of the pathogen. Among the three, alterations in Bet protein had the least impact on FV capsid formation, virions release and viral titers [178]. For the successful development of a HIV vaccine, the epitope used should able to induce a broad array of neutralizing antibodies that prevent infection and the integration of the viral genome into target cells. Several neutralizing antibodies against surface and transmembrane (TM) and membrane proximal external region (MPER) of HIV gp41, were isolated from HIV infected patients. A Chimeric FV vector expressing TM and MPER HIV epitopes was designed that could successfully induce antibodies against HIV in a rat model [177]. In this study, the FV Bet protein was altered by replacing conserved regions with MPER of HIV gp41 proteins. The epitope expressed by the chimeric FV vector readily reacted with 2F5 and 4E10 HIV antibodies in vitro. Further, rats immunized with the chimeric FV vector induced antibodies specific to HIV gp41 that could bind to HIV particles. However, the antibodies induced by the chimeric FV vector showed no effect on HIV infection when tested in an indicator cell-line based neutralizing assay. While the lack of neutralizing effects by these antibodies requires further testing, this study opened up the feasibility of using a FV vector for HIV vaccines.
6. Conclusions and Future Directions
Gene therapy has the potential to offer a life-long therapeutic option for controlling HIV infection. However, there are several challenges that must be overcome before anti-HIV gene therapy can become a widely used therapy to treating HIV patients. First, anti-HIV transgenes that can inhibit diverse HIV strains and inhibit escape are needed. Expressing multiple anti-HIV transgenes, especially targeting viral entry, in the same cells using a single combinatorial anti-HIV vector, is a feasible approach to increase the efficacy of blocking HIV that can even reduce the chance of the emergence of escape mutants. The success of HIV gene therapy, depends on the efficient engraftment and long-term persistence of gene modified cells in a human system. For this, safe and efficient delivering systems are needed for ex vivo transduction of HSC. FV vectors have several advantages for HIV HSC gene therapy, including efficient transduction of HSCs, efficient delivery of anti-HIV transgenes and a promising safety profile.
Acknowledgments
This research was supported by NIH grants AI097100 and AI102672 (Grant D. Trobridge).
Author Contributions
Arun K. Nalla and Grant D. Trobridge authored and drafted the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization (WHO). Unaids Report: How AIDS Changed Everything; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Shen, L.; Siliciano, R.F. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J. Allergy Clin. Immunol. 2008, 122, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.G.; Lima, V.; Gouws, E. Modelling the impact of antiretroviral therapy on the epidemic of HIV. Curr. HIV Res. 2011, 9, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Camacho, R.; Teofilo, E. Antiretroviral therapy in treatment-naive patients with HIV infection. Curr. Opin. HIV AIDS 2011, 6 (Suppl. 1), S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Olszko, M.E.; Trobridge, G.D. Foamy virus vectors for HIV gene therapy. Viruses 2013, 5, 2585–2600. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 D32/D32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5 D32/D32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia growth and development study, multicenter aids cohort study, multicenter hemophilia cohort study, san francisco city cohort, ALIVE study. Science 1996, 273, 1856–1862. [Google Scholar] [PubMed]
- Hutter, G.; Thiel, E. Allogeneic transplantation of CCR5-deficient progenitor cells in a patient with HIV infection: An update after 3 years and the search for patient No. 2. AIDS 2011, 25, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Martinson, J.J.; Chapman, N.H.; Rees, D.C.; Liu, Y.T.; Clegg, J.B. Global distribution of the CCR5 gene 32-basepair deletion. Nat. Genet. 1997, 16, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Dhanda, R.; Bamshad, M.; Mummidi, S.; Geevarghese, R.; Catano, G.; Anderson, S.A.; Walter, E.A.; Stephan, K.T.; Hammer, M.F.; et al. Global survey of genetic variation in CCR5, RANTES, and MIP-1a: Impact on the epidemiology of the HIV-1 pandemic. Proc. Natl. Acad. Sci. USA 2001, 98, 5199–5204. [Google Scholar] [CrossRef] [PubMed]
- Cannon, P.M.; Kohn, D.B.; Kiem, H.P. HIV eradication—From Berlin to Boston. Nat. Biotechnol. 2014, 32, 315–316. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO-2 associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Hendrie, P.C.; Huo, Y.; Stolitenko, R.B.; Russell, D.W. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol. Ther. 2008, 16, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Banerjea, A.; Li, M.J.; Remling, L.; Rossi, J.; Akkina, R. Lentiviral transduction of TAR decoy and CCR5 ribozyme into CD34+ progenitor cells and derivation of HIV-1 resistant T cells and macrophages. AIDS Res. Ther. 2004, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von Laer, D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.W.; Younan, P.; Jerome, K.R.; Kiem, H.P. Combinatorial anti-HIV gene therapy: Using a multipronged approach to reach beyond HAART. Gene Ther. 2013, 20, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Mautino, M.R.; Morgan, R.A. Potent inhibition of human immunodeficiency virus type 1 replication by conditionally replicating human immunodeficiency virus-based lentiviral vectors expressing envelope antisense mRNA. Hum. Gene Ther. 2000, 11, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Bahner, I.; Sumiyoshi, T.; Kagoda, M.; Swartout, R.; Peterson, D.; Pepper, K.; Dorey, F.; Reiser, J.; Kohn, D.B. Lentiviral vector transduction of a dominant-negative Rev gene into human CD34+ hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Mol. Ther. 2007, 15, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.A.; Vojtech, L.; Bahner, I.; Kohn, D.B.; Laer, D.V.; Russell, D.W.; Richard, R.E. Foamy virus vectors expressing anti-HIV transgenes efficiently block HIV-1 replication. Mol. Ther. 2008, 16, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Von Laer, D.; Hasselmann, S.; Hasselmann, K. Impact of gene-modified T cells on HIV infection dynamics. J. Theor. Biol. 2006, 238, 60–77. [Google Scholar] [CrossRef] [PubMed]
- Kimpel, J.; Braun, S.E.; Qiu, G.; Wong, F.E.; Conolle, M.; Schmitz, J.E.; Brendel, C.; Humeau, L.M.; Dropulic, B.; Rossi, J.J.; et al. Survival of the fittest: Positive selection of CD4+ T cells expressing a membrane-bound fusion inhibitor following HIV-1 infection. PLoS ONE 2010, 5, e12357. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Allers, K.; Schneider, T. The additional use of viral entry inhibitors during autologous hematopoietic stem cell transplantation in patients with non-hodgkin lymphoma and HIV-1 infection. Biol. Blood Marrow Transpl. 2011, 17, 586–587. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Butticaz, C.; Ciuffi, A.; Munoz, M.; Thomas, J.; Bridge, A.; Pebernard, S.; Iggo, R.; Meylan, P.; Telenti, A. Protection from HIV-1 infection of primary CD4 T cells by CCR5 silencing is effective for the full spectrum of CCR5 expression. Antivir. Ther. 2003, 8, 373–377. [Google Scholar] [PubMed]
- Martinez, M.A.; Gutierrez, A.; Armand-Ugon, M.; Blanco, J.; Parera, M.; Gomez, J.; Clotet, B.; Este, J.A. Suppression of chemokine receptor expression by RNA interference allows for inhibition of HIV-1 replication. AIDS 2002, 16, 2385–2390. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Banerjea, A.; Akkina, R. Bispecific short hairpin siRNA constructs targeted to CD4, CXCR4, and CCR5 confer HIV-1 resistance. Oligonucleotides 2003, 13, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Swan, C.H.; Buhler, B.; Steinberger, P.; Tschan, M.P.; Barbas, C.F., 3rd; Torbett, B.E. T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther. 2006, 13, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Leavitt, M.; Tritz, R.; Duarte, E.; Kang, D.; Mamounas, M.; Gilles, P.; Wong-Staal, F.; Kennedy, S.; Merson, J.; et al. Inhibition of CCR5-dependent HIV-1 infection by hairpin ribozyme gene therapy against CC-chemokine receptor 5. Virology 2000, 276, 271–278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cagnon, L.; Rossi, J.J. Downregulation of the CCR5 beta-chemokine receptor and inhibition of HIV-1 infection by stable VA1-ribozyme chimeric transcripts. Antisense Nucleic Acid Drug Dev. 2000, 10, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Hong, P.; Arumugam, B.; Pokomo, L.; Boyer, J.; Koizumi, N.; Kittipongdaja, P.; Chen, A.; Bristol, G.; Galic, Z.; et al. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood 2010, 115, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Kamata, M.; Chen, K.N.; Pariente, N.; An, D.S.; Chen, I.S. Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J. Gene Med. 2010, 12, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Myburgh, R.; Ivic, S.; Pepper, M.S.; Gers-Huber, G.; Li, D.; Audige, A.; Rochat, M.A.; Jaquet, V.; Regenass, S.; Manz, M.G.; et al. Lentivector knockdown of CCR5 in hematopoietic stem and progenitor cells confers functional and persistent HIV-1 resistance in humanized mice. J. Virol. 2015, 89, 6761–6772. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5D32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Ferreira, L.M.; Collins, R.; Meissner, T.B.; Boutwell, C.L.; Friesen, M.; Vrbanac, V.; Garrison, B.S.; Stortchevoi, A.; Bryder, D.; et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 2014, 15, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ye, C.; Liu, J.; Zhang, D.; Kimata, J.T.; Zhou, P. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS ONE 2014, 9, e115987. [Google Scholar] [CrossRef] [PubMed]
- Steen, A.; Schwartz, T.W.; Rosenkilde, M.M. Targeting CXCR4 in HIV cell-entry inhibition. Mini Rev. Med. Chem. 2009, 9, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Banerjea, A.; Planelles, V.; Akkina, R. Potent suppression of HIV type 1 infection by a short hairpin anti-CXCR4 siRNA. AIDS Res. Hum. Retroviruses 2003, 19, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Chen, S.; Wang, S.; Yu, X.; Chen, Y.; Jiang, M.; Zhuang, K.; Ho, W.; Hou, W.; Huang, J.; et al. Genome editing of CXCR4 by CRISPR/Cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015, 5, 15577. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.; Urak, K.; Veillette, M.; Nepveu-Traversy, M.E.; Pham, Q.T.; Hamel, S.; Rossi, J.J.; Berthoux, L. Preclinical assessment of mutant human TRIM5a a as an anti-HIV-1 transgene. Hum. Gene Ther. 2015, 26, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.R.; Ziegler, P.; Pertel, T.; Strambio-De-Castillia, C.; Grutter, C.; Martinetti, G.; Mazzucchelli, L.; Grutter, M.; Manz, M.G.; Luban, J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J. Clin. Investig. 2009, 119, 3035–3047. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Vets, S.; Kimpel, J.; Volk, A.; De Rijck, J.; Schrijvers, R.; Verbinnen, B.; Maes, W.; Von Laer, D.; Debyser, Z.; Gijsbers, R. Lens epithelium-derived growth factor/p75 qualifies as a target for HIV gene therapy in the NSG mouse model. Mol. Ther. 2012, 20, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Geerts, D.; Jeeninga, R.E.; Berkhout, B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antivir. Res. 2011, 89, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Sagnier, S.; Geerts, D.; Jeeninga, R.E.; Biard-Piechaczyk, M.; Berkhout, B. Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol. J. 2012, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Mulky, A.; Yuen, W.; Martin, T.D.; Meyerson, N.R.; Choi, L.; Yu, H.; Sawyer, S.L.; Kewalramani, V.N. HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. J. Virol. 2012, 86, 3851–3860. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Egelhofer, M.; Brandenburg, G.; Martinius, H.; Schult-Dietrich, P.; Melikyan, G.; Kunert, R.; Baum, C.; Choi, I.; Alexandrov, A.; von Laer, D. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J. Virol. 2004, 78, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Riley, J.L.; Carroll, R.G.; von Laer, D.; June, C.H. Suppression of HIV-1 infection in primary CD4 T cells transduced with a self-inactivating lentiviral vector encoding a membrane expressed gp41-derived fusion inhibitor. Clin. Immunol. 2005, 115, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bonyhadi, M.L.; Moss, K.; Voytovich, A.; Auten, J.; Kalfoglou, C.; Plavec, I.; Forestell, S.; Su, L.; Bohnlein, E.; Kaneshima, H. RevM10-expressing T cells derived in vivo from transduced human hematopoietic stem-progenitor cells inhibit human immunodeficiency virus replication. J. Virol. 1997, 71, 4707–4716. [Google Scholar] [PubMed]
- Su, L.; Lee, R.; Bonyhadi, M.; Matsuzaki, H.; Forestell, S.; Escaich, S.; Bohnlein, E.; Kaneshima, H. Hematopoietic stem cell-based gene therapy for acquired immunodeficiency syndrome: Efficient transduction and expression of RevM10 in myeloid cells in vivo and in vitro. Blood 1997, 89, 2283–2290. [Google Scholar] [PubMed]
- Bauer, G.; Valdez, P.; Kearns, K.; Bahner, I.; Wen, S.F.; Zaia, J.A.; Kohn, D.B. Inhibition of human immunodeficiency virus-1 (HIV-1) replication after transduction of granulocyte colony-stimulating factor-mobilized CD34+ cells from HIV-1-infected donors using retroviral vectors containing anti-HIV-1 genes. Blood 1997, 89, 2259–2267. [Google Scholar] [PubMed]
- Lee, S.W.; Gallardo, H.F.; Gilboa, E.; Smith, C. Inhibition of human immunodeficiency virus type 1 in human T cells by a potent rev response element decoy consisting of the 13-nucleotide minimal rev-binding domain. J. Virol. 1994, 68, 8254–8264. [Google Scholar] [PubMed]
- Smith, C.; Lee, S.W.; Wong, E.; Gallardo, H.; Page, K.; Gaspar, O.; Lebkowski, J.; Gilboa, E. Transient protection of human T-cells from human immunodeficiency virus type 1 infection by transduction with adeno-associated viral vectors which express RNA decoys. Antivir. Res. 1996, 32, 99–115. [Google Scholar] [CrossRef]
- Michienzi, A.; Li, S.; Zaia, J.A.; Rossi, J.J. A nucleolar TAR decoy inhibitor of HIV-1 replication. Proc. Natl. Acad. Sci. USA 2002, 99, 14047–14052. [Google Scholar] [CrossRef] [PubMed]
- Michienzi, A.; De Angelis, F.G.; Bozzoni, I.; Rossi, J.J. A nucleolar localizing rev binding element inhibits HIV replication. AIDS Res. Ther. 2006, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Bauer, G.; Michienzi, A.; Yee, J.K.; Lee, N.S.; Kim, J.; Li, S.; Castanotto, D.; Zaia, J.; Rossi, J.J. Inhibition of HIV-1 infection by lentiviral vectors expressing pol III-promoted anti-HIV RNAs. Mol. Ther. 2003, 8, 196–206. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Coburn, G.A.; Cullen, B.R. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J. Virol. 2002, 76, 9225–9231. [Google Scholar] [CrossRef] [PubMed]
- Jacque, J.M.; Triques, K.; Stevenson, M. Modulation of HIV-1 replication by RNA interference. Nature 2002, 418, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.J.; Liu, X.; He, J. Lentiviral siRNAs targeting multiple highly conserved RNA sequences of human immunodeficiency virus type 1. Gene Ther. 2005, 12, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.S.; Li, Y.; Kameoka, M.; Ng, T.B.; Wan, D.C. Suppression of HIV replication using RNA interference against HIV-1 integrase. FEBS Lett. 2007, 581, 3253–3259. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Berkhout, B. Lentiviral vectors that carry anti-HIV shRNAs: Problems and solutions. J. Gene Med. 2007, 9, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; ’t Hooft, K.; Liu, Y.P.; Centlivre, M.; von Eije, K.J.; Berkhout, B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol. Ther. 2008, 16, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, S.; Alberici, L.; Gubinelli, F.; Lupo, R.; Olgiati, C.; Rizzardi, G.P.; Bovolenta, C. The F12-Vif derivative Chim3 inhibits HIV-1 replication in CD4+ T lymphocytes and CD34+-derived macrophages by blocking HIV-1 DNA integration. Blood 2009, 113, 3443–3452. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, S.; Gubinelli, F.; Alberici, L.; Piovani, B.M.; Rizzardi, G.P.; Bovolenta, C. Chim3 confers survival advantage to CD4+ T cells upon HIV-1 infection by preventing HIV-1 DNA integration and HIV-1-induced G2 cell-cycle delay. Blood 2010, 115, 4021–4029. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Vink, M.A.; Westerink, J.T.; Ramirez de Arellano, E.; Konstantinova, P.; Ter Brake, O.; Berkhout, B. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 2010, 16, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Podsakoff, G.M.; Engel, B.C.; Carbonaro, D.A.; Choi, C.; Smogorzewska, E.M.; Bauer, G.; Selander, D.; Csik, S.; Wilson, K.; Betts, M.R.; et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34+ cells. Mol. Ther. 2005, 12, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, J.L.; Boyd, M.P.; Arndt, A.J.; Todd, A.V.; Fanning, G.C.; Ely, J.A.; Elliott, F.; Knop, A.; Raponi, M.; Murray, J.; et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J. Gene Med. 2005, 7, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Harris, R.S.; Neuberger, M.S. The vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 2003, 13, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.M.; Coolen, C.; Davis, A.J.; Burrell, C.J.; Li, P. Human immunodeficiency virus 1 (HIV-1) virion infectivity factor (Vif) is part of reverse transcription complexes and acts as an accessory factor for reverse transcription. Virology 2008, 372, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Vallanti, G.; Lupo, R.; Federico, M.; Mavilio, F.; Bovolenta, C. T lymphocytes transduced with a lentiviral vector expressing F12-vif are protected from HIV-1 infection in an APOBEC3G-independent manner. Mol. Ther. 2005, 12, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.E.; Sanders, R.W.; Deng, Y.; Jurriaans, S.; Lange, J.M.; Lu, M.; Berkhout, B. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J. Virol. 2004, 78, 12428–12437. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.; Berkhout, B. Mechanistic studies of a T20-dependent human immunodeficiency virus type 1 variant. J. Virol. 2008, 82, 7735–7740. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Cai, L.; Lu, H.; Qi, Z.; Jiang, S. Combinations of the first and next generations of human immunodeficiency virus (HIV) fusion inhibitors exhibit a highly potent synergistic effect against enfuvirtide-sensitive and -resistant HIV type 1 strains. J. Virol. 2009, 83, 7862–7872. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.; Kawaji, K.; Miyamoto, F.; Shimane, K.; Shimura, K.; Sakagami, Y.; Hattori, T.; Watanabe, K.; Oishi, S.; Fujii, N.; et al. Mechanism of resistance to S138A substituted enfuvirtide and its application to peptide design. Int. J. Biochem. Cell Biol. 2013, 45, 908–915. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cheng, J.; Lu, H.; Li, J.; Hu, J.; Qi, Z.; Liu, Z.; Jiang, S.; Dai, Q. Potent HIV fusion inhibitors against enfuvirtide-resistant HIV-1 strains. Proc. Natl. Acad. Sci. USA 2008, 105, 16332–16337. [Google Scholar] [CrossRef] [PubMed]
- Lohrengel, S.; Hermann, F.; Hagmann, I.; Oberwinkler, H.; Scrivano, L.; Hoffmann, C.; von Laer, D.; Dittmar, M.T. Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J. Virol. 2005, 79, 10237–10246. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Baler-Bitterlich, G.; Ragheb, J.A.; Wong-Staal, F.; Gallo, R.C.; Anderson, W.F. Further evaluation of soluble CD4 as an anti-HIV type 1 gene therapy: Demonstration of protection of primary human peripheral blood lymphocytes from infection by HIV type 1. AIDS Res. Hum. Retroviruses 1994, 10, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Fessel, W.J.; Anderson, B.; Follansbee, S.E.; Winters, M.A.; Lewis, S.T.; Weinheimer, S.P.; Petropoulos, C.J.; Shafer, R.W. The efficacy of an anti-CD4 monoclonal antibody for HIV-1 treatment. Antivir. Res. 2011, 92, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Patel, B.; Ghanem, M.H.; Bundoc, V.; Zheng, Z.; Morgan, R.A.; Rosenberg, S.A.; Dey, B.; Berger, E.A. Novel CD4-based bispecific chimeric antigen receptor designed for enhanced anti-HIV potency and absence of HIV entry receptor activity. J. Virol. 2015, 89, 6685–6694. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34+ cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36ra43. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Waite, J.; Brewer, F.; Sunshine, M.J.; Littman, D.R.; Zou, Y.R. The role of CXCR4 in maintaining peripheral B-cell compartments and humoral immunity. J. Exp. Med. 2004, 200, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An essential role for LEDGF/p75 in HIV integration. Science 2006, 314, 461–464. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J.; Vandekerckhove, L.; Gijsbers, R.; Hombrouck, A.; Hendrix, J.; Vercammen, J.; Engelborghs, Y.; Christ, F.; Debyser, Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J. Virol. 2006, 80, 11498–11509. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Fassati, A.; Görlich, D.; Harrison, I.; Zaytseva, L.; Mingot, J.M. Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. EMBO J. 2003, 22, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
- Zielske, S.P.; Stevenson, M. Importin 7 may be dispensable for human immunodeficiency virus type 1 and simian immunodeficiency virus infection of primary macrophages. J. Virol. 2005, 79, 11541–11546. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Thys, W.; De Rijck, J.; Gijsbers, R.; Albanese, A.; Arosio, D.; Emiliani, S.; Rain, J.C.; Benarous, R.; Cereseto, A.; et al. Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 2008, 18, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- De Iaco, A.; Luban, J. Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 2011, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.; Valle-Casuso, J.C.; White, T.E.; Brandariz-Nuñez, A.; Bosche, W.J.; Reszka, N.; Gorelick, R.; Diaz-Griffero, F. The ability of TNPO3-depleted cells to inhibit HIV-1 infection requires CPSF6. Retrovirology 2013, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Bloch, N.; Landau, N.R. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 2015, 16, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5a restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Lynch, C.; Stoye, J.P.; Yap, M.W. A TRIM5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. USA 2004, 101, 13324–13328. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Perron, M.; Welikala, S.; Sodroski, J. Species-specific variation in the B30.2(SPRY) domain of TRIM5a determines the potency of human immunodeficiency virus restriction. J. Virol. 2005, 79, 3139–3145. [Google Scholar] [CrossRef] [PubMed]
- Pham, Q.T.; Bouchard, A.; Grutter, M.G.; Berthoux, L. Generation of human TRIM5a mutants with high HIV-1 restriction activity. Gene Ther. 2010, 17, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Stremlau, M.; Lee, M.; Sodroski, J. Removal of Arginine 332 allows human TRIM5a to bind human immunodeficiency virus capsids and to restrict infection. J. Virol. 2006, 80, 6738–6744. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.W.; Nisole, S.; Stoye, J.P. A single amino acid change in the SPRY domain of human TRIM5a leads to HIV-1 restriction. Curr. Biol. 2005, 15, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Schaller, T.; Eddaoudi, A.; Zhan, H.; Tan, C.P.; Jacobsen, M.; Thrasher, A.J.; Towers, G.J.; Qasim, W. Lentiviral gene therapy against human immunodeficiency virus type 1, using a novel human TRIM21-cyclophilin a restriction factor. Hum. Gene Ther. 2012, 23, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Bitzegeio, J.; Sampias, M.; Bieniasz, P.D.; Hatziioannou, T. Adaptation to the interferon-induced antiviral state by human and simian immunodeficiency viruses. J. Virol. 2013, 87, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Malim, M.H. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 2010, 84, 9254–9266. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Delobel, P.; Sandres-Saune, K.; Cazabat, M.; Pasquier, C.; Marchou, B.; Massip, P.; Izopet, J. R5 to X4 switch of the predominant HIV-1 population in cellular reservoirs during effective highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2005, 38, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Hermann, F.G.; Egerer, L.; Brauer, F.; Gerum, C.; Schwalbe, H.; Dietrich, U.; von Laer, D. Mutations in gp120 contribute to the resistance of human immunodeficiency virus type 1 to membrane-anchored C-peptide maC46. J. Virol. 2009, 83, 4844–4853. [Google Scholar] [CrossRef] [PubMed]
- Desmezieres, E.; Gupta, N.; Vassell, R.; He, Y.; Peden, K.; Sirota, L.; Yang, Z.; Wingfield, P.; Weiss, C.D. Human immunodeficiency virus (HIV) gp41 escape mutants: Cross-resistance to peptide inhibitors of HIV fusion and altered receptor activation of gp120. J. Virol. 2005, 79, 4774–4781. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Li, M.J.; Palmer, B.; Remling, L.; Li, S.; Yam, P.; Yee, J.K.; Rossi, J.; Zaia, J.; Akkina, R. Safety and efficacy of a lentiviral vector containing three anti-HIV genes—CCR5 ribozyme, tat-rev siRNA, and TAR decoy in SCID—Hu mouse-derived T cells. Mol. Ther. 2007, 15, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Javien, J.; Nolta, J.A.; Bauer, G. Preintegration HIV-1 inhibition by a combination lentiviral vector containing a chimeric TRIMa protein, a CCR5 shRNA, and a TAR decoy. Mol. Ther. 2009, 17, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; von Eije, K.J.; Schopman, N.C.; Westerink, J.T.; ter Brake, O.; Haasnoot, J.; Berkhout, B. Combinatorial RNAi against HIV-1 using extended short hairpin RNAs. Mol. Ther. 2009, 17, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Wolstein, O.; Boyd, M.; Millington, M.; Impey, H.; Boyer, J.; Howe, A.; Delebecque, F.; Cornetta, K.; Rothe, M.; Baum, C.; et al. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol. Ther. Methods Clin. Dev. 2014, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.P.; Levin, B.R.; Zhang, J.; Sahakyan, A.; Boyer, J.; Carroll, M.V.; Colon, J.C.; Keech, N.; Rezek, V.; Bristol, G.; et al. Engineering cellular resistance to HIV-1 infection in vivo using a dual therapeutic lentiviral vector. Mol. Ther. Nucleic Acids 2015, 4, e236. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Bauer, G.; Rice, C.R.; Rothschild, J.C.; Carbonaro, D.A.; Valdez, P.; Hao, Q.; Zhou, C.; Bahner, I.; Kearns, K.; et al. A clinical trial of retroviral-mediated transfer of a REV-responsive element decoy gene into CD34+ cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood 1999, 94, 368–371. [Google Scholar] [PubMed]
- Kang, E.M.; De Witte, M.; Malech, H.; Morgan, R.A.; Carter, C.; Leitman, S.F.; Childs, R.; Barrett, A.J.; Little, R.; Tisdale, J.F. Gene therapy-based treatment for HIV-positive patients with malignancies. J. Hematother. Stem Cell Res. 2002, 11, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Mitsuyasu, R.T.; Rosenblatt, J.D.; Ngok, F.K.; Bakker, A.; Cole, S.; Chorn, N.; Lin, L.S.; Bristol, G.; Boyd, M.P.; et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: Myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum. Gene Ther. 2004, 15, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Van Lunzen, J.; Glaunsinger, T.; Stahmer, I.; von Baehr, V.; Baum, C.; Schilz, A.; Kuehlcke, K.; Naundorf, S.; Martinius, H.; Hermann, F.; et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol. Ther. 2007, 15, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.L.; Humeau, L.M.; Boyer, J.; MacGregor, R.R.; Rebello, T.; Lu, X.; Binder, G.K.; Slepushkin, V.; Lemiale, F.; Mascola, J.R.; et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17372–17377. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Stan, R.; Krishnan, A.; Li, H.; Rossi, J.J.; Zaia, J.A. Development of hematopoietic stem cell based gene therapy for HIV-1 infection: Considerations for proof of concept studies and translation to standard medical practice. Viruses 2013, 5, 2898–2919. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Thrasher, A.J. Gene therapy for PIDS: Progress, pitfalls and prospects. Gene 2013, 525, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. Integration of murine leukemia virus dna depends on mitosis. EMBO J. 1993, 12, 2099–2108. [Google Scholar] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, S.I.; Schambach, A.; Howe, S.J.; Ulaganathan, M.; Grassman, E.; Williams, D.; Schiedlmeier, B.; Sebire, N.J.; Gaspar, H.B.; Kinnon, C.; et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. 2008, 16, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Avedillo Diez, I.; Zychlinski, D.; Coci, E.G.; Galla, M.; Modlich, U.; Dewey, R.A.; Schwarzer, A.; Maetzig, T.; Mpofu, N.; Jaeckel, E.; et al. Development of novel efficient sin vectors with improved safety features for Wiskott-Aldrich syndrome stem cell based gene therapy. Mol. Pharm. 2011, 8, 1525–1537. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human b-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.T.; Garcia, J.V. Lentivirus vector mobilization and spread by human immunodeficiency virus. Hum. Gene Ther. 2000, 11, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Klimatcheva, E.; Planelles, V.; Day, S.L.; Fulreader, F.; Renda, M.J.; Rosenblatt, J. Defective lentiviral vectors are efficiently trafficked by HIV-1 and inhibit its replication. Mol. Ther. 2001, 3, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.D.; Hu, W.S. RNAs from genetically distinct retroviruses can copackage and exchange genetic information in vivo. J. Virol. 1997, 71, 6237–6242. [Google Scholar] [PubMed]
- Rizvi, T.A.; Panganiban, A.T. Simian immunodeficiency virus RNA is efficiently encapsidated by human immunodeficiency virus type 1 particles. J. Virol. 1993, 67, 2681–2688. [Google Scholar] [PubMed]
- White, S.M.; Renda, M.; Nam, N.Y.; Klimatcheva, E.; Zhu, Y.; Fisk, J.; Halterman, M.; Rimel, B.J.; Federoff, H.; Pandya, S.; et al. Lentivirus vectors using human and simian immunodeficiency virus elements. J. Virol. 1999, 73, 2832–2840. [Google Scholar] [PubMed]
- Browning, M.T.; Schmidt, R.D.; Lew, K.A.; Rizvi, T.A. Primate and feline lentivirus vector RNA packaging and propagation by heterologous lentivirus virions. J. Virol. 2001, 75, 5129–5140. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D. Foamy virus vectors for gene transfer. Expert Opin. Biol. Ther. 2009, 9, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar] [PubMed]
- Trobridge, G.; Josephson, N.; Vassilopoulos, G.; Mac, J.; Russell, D.W. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 2002, 6, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, G.; Trobridge, G.; Josephson, N.C.; Russell, D.W. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood 2001, 98, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.P.; Allen, J.; Trobridge, G.; Olson, E.; Keyser, K.; Peterson, L.; Russell, D.W. Foamy-virus-mediated gene transfer to canine repopulating cells. Blood 2007, 109, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Zucali, J.R.; Ciccarone, T.; Kelley, V.; Park, J.; Johnson, C.M.; Mergia, A. Transduction of umbilical cord blood CD34+ NOD/SCID-repopulating cells by simian foamy virus type 1 (SFV-1) vector. Virology 2002, 302, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Josephson, N.C.; Trobridge, G.; Russell, D.W. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Hum. Gene Ther. 2004, 15, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Mergia, A.; Chari, S.; Kolson, D.L.; Goodenow, M.M.; Ciccarone, T. The efficiency of simian foamy virus vector type-1 (SFV-1) in nondividing cells and in human PBLS. Virology 2001, 280, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.; Russell, D.W. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J. Virol. 2004, 78, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Allen, J.; Peterson, L.; Ironside, C.; Russell, D.W.; Kiem, H.P. Foamy and lentiviral vectors transduce canine long-term repopulating cells at similar efficiency. Hum. Gene Ther. 2009, 20, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Nasimuzzaman, M.; Kim, Y.S.; Wang, Y.D.; Persons, D.A. High-titer foamy virus vector transduction and integration sites of human CD34+ cell-derived SCID-repopulating cells. Mol. Ther. Methods Clin. Dev. 2014, 1, 14020. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Beard, B.C.; Wu, R.A.; Ironside, C.; Malik, P.; Kiem, H.P. Stem cell selection in vivo using foamy vectors cures canine pyruvate kinase deficiency. PLoS ONE 2012, 7, e45173. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.R., Jr.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.W.; et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.R., Jr.; Tuschong, L.M.; Calvo, K.R.; Shive, H.R.; Burkholder, T.H.; Karlsson, E.K.; West, R.R.; Russell, D.W.; Hickstein, D.D. Long-term follow-up of foamy viral vector-mediated gene therapy for canine leukocyte adhesion deficiency. Mol. Ther. 2013, 21, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Nowrouzi, A.; Dittrich, M.; Klanke, C.; Heinkelein, M.; Rammling, M.; Dandekar, T.; von Kalle, C.; Rethwilm, A. Genome-wide mapping of foamy virus vector integrations into a human cell line. J. Gen. Virol. 2006, 87, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Olszko, M.E.; Adair, J.E.; Linde, I.; Rae, D.T.; Trobridge, P.; Hocum, J.D.; Rawlings, D.J.; Kiem, H.P.; Trobridge, G.D. Foamy viral vector integration sites in SCID-repopulating cells after MGMTP140K-mediated in vivo selection. Gene Ther. 2015, 22, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Nadeau, P.E.; Mergia, A. Activity of TAR in inducible inhibition of HIV replication by foamy virus vector expressing siRNAs under the control of HIV LTR. Virus Res. 2009, 140, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Nadeau, P.; Zucali, J.R.; Johnson, C.M.; Mergia, A. Inhibition of simian immunodeficiency virus by foamy virus vectors expressing siRNAs. Virology 2005, 343, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Mühle, M.; Hoffmann, K.; Löchelt, M.; Denner, J. Immunisation with foamy virus bet fusion proteins as novel strategy for HIV-1 epitope delivery. Immunol. Res. 2013, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Osen, W.; Gardyan, A.; Hotz-Wagenblatt, A.; Wei, G.; Gissmann, L.; Eichmüller, S.; Löchelt, M. Replication-competent foamy virus vaccine vectors as novel epitope scaffolds for immunotherapy. PLoS ONE 2015, 10, e0138458. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).