
Innate Lymphoid Cells in Tumor Immunity


Abstract
:
{kind=link}
{kind=link}
1. Introduction
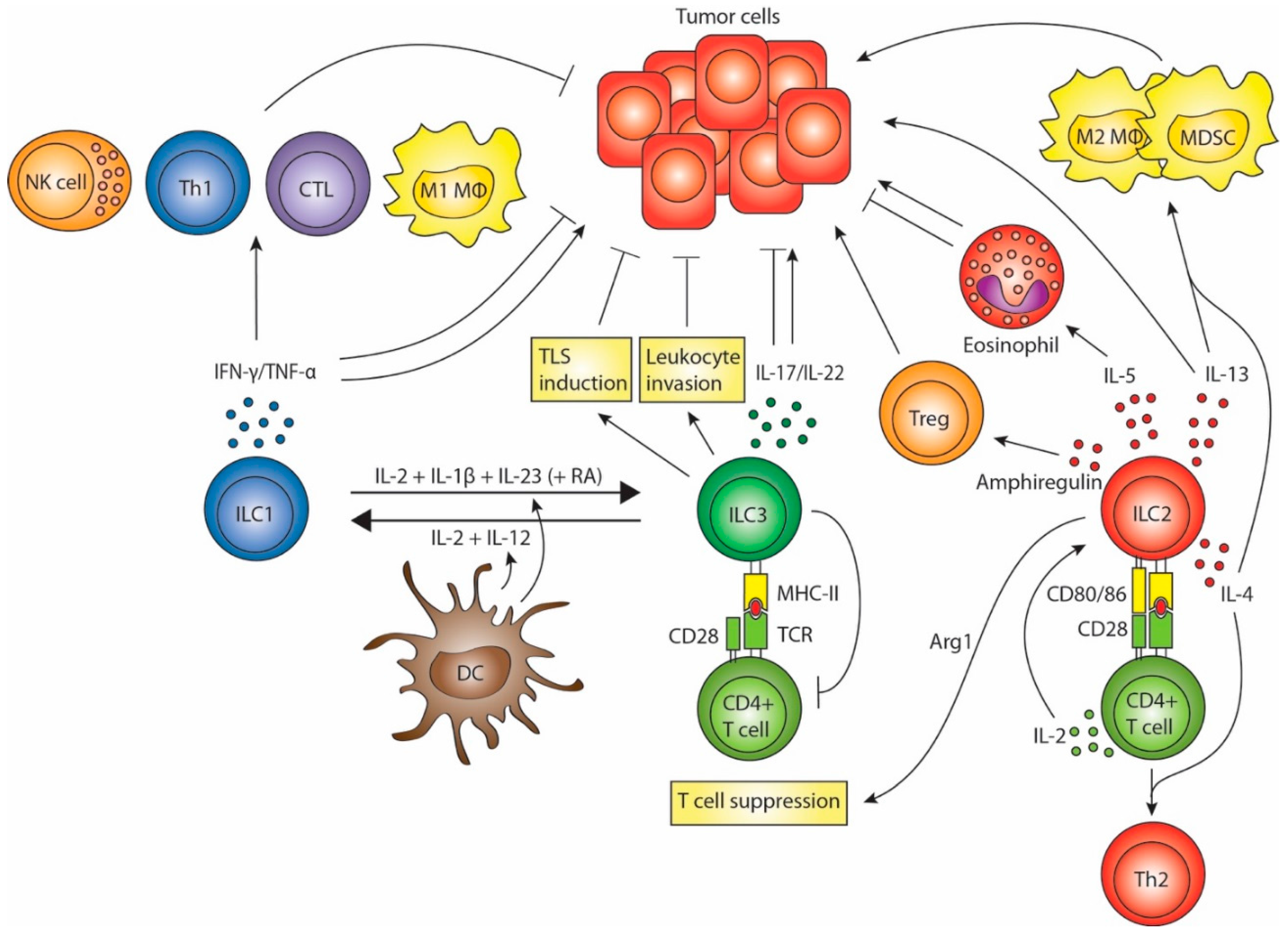
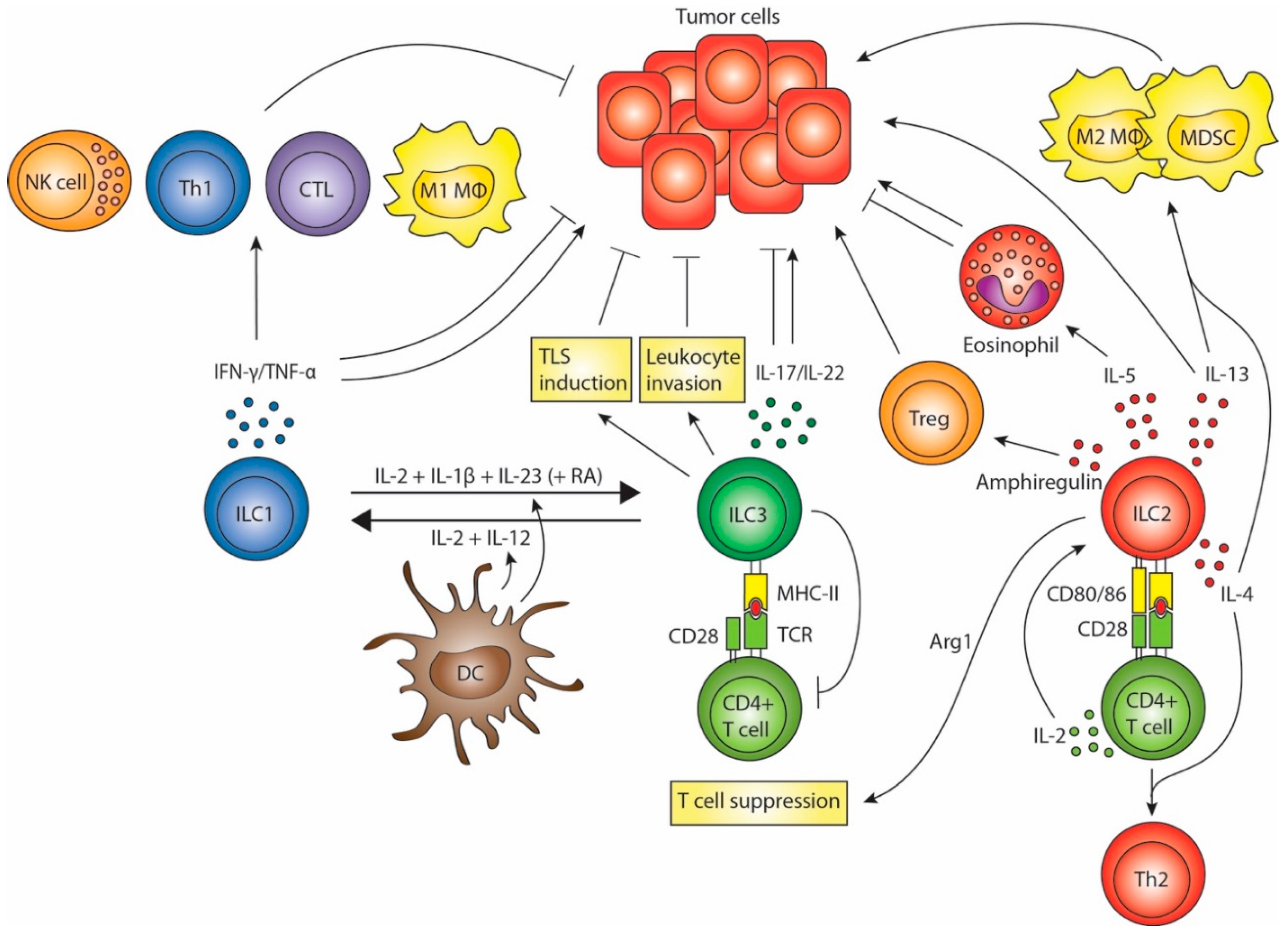
2. ILC1s in Tumor Immunity
3. ILC2s in Tumor Immunity
4. ILC3s in Tumor Immunity
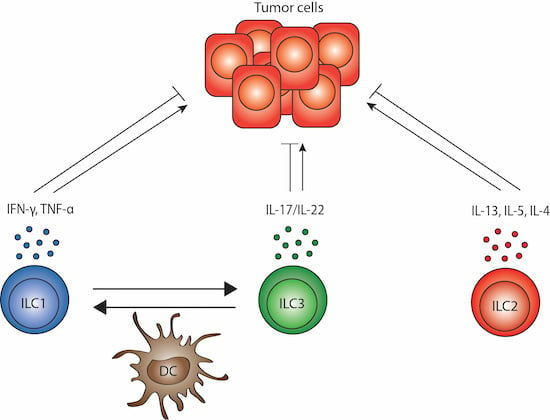
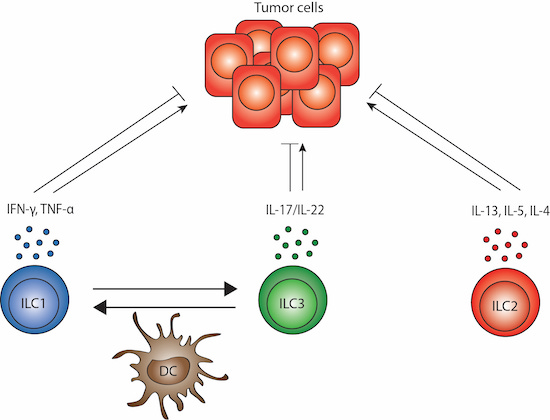
5. ILC Plasticity and Crosstalk with DCs
6. ILCs in Cancer Treatment
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; D’Hargues, Y.; Göppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Hoyler, T.; Klose, C.S.N.; Souabni, A.; Turqueti-Neves, A.; Pfeifer, D.; Rawlins, E.L.; Voehringer, D.; Busslinger, M.; Diefenbach, A. The Transcription Factor GATA-3 Controls Cell Fate and Maintenance of Type 2 Innate Lymphoid Cells. Immunity 2012, 37, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.; Bernink, J.; Golebski, K.; Karrich, J.J.; Peters, C.P.; Blom, B.; te Velde, A.A.; Fokkens, W.J.; van Drunen, C.M.; Spits, H. The Transcription Factor GATA3 Is Essential for the Function of Human Type 2 Innate Lymphoid Cells. Immunity 2012, 37, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef] [PubMed]
- Fallon, P.G.; Ballantyne, S.J.; Mangan, N.E.; Barlow, J.L.; Dasvarma, A.; Hewett, D.R.; McIlgorm, A.; Jolin, H.E.; McKenzie, A.N.J. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 2006, 203, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.-I.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.; Hirota, K.; Stieglitz, B.; Van Snick, J.; Tolaini, M.; Lahl, K.; Sparwasser, T.; Helmby, H.; Stockinger, B. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat. Immunol. 2011, 12, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.K.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Price, A.E.; Liang, H.-E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.F.; Krauß, R.H.; Sun, A.C.; Takei, F. Lung Natural Helper Cells Are a Critical Source of Th2 Cell-Type Cytokines in Protease Allergen-Induced Airway Inflammation. Immunity 2012, 36, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.F.; Steer, C.A.; Mathä, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.J.; Takei, F. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Eberl, G.; Marmon, S.; Sunshine, M.-J.; Rennert, P.D.; Choi, Y.; Littman, D.R. An essential function for the nuclear receptor RORγt in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 2004, 5, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Sanos, S.L.; Bui, V.L.; Mortha, A.; Oberle, K.; Heners, C.; Johner, C.; Diefenbach, A. RORγt and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 2009, 10, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Vonarbourg, C.; Mortha, A.; Bui, V.L.; Hernandez, P.P.; Kiss, E.A.; Hoyler, T.; Flach, M.; Bengsch, B.; Thimme, R.; Hölscher, C.; et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt(+) innate lymphocytes. Immunity 2010, 33, 736–751. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Takayama, N.; Vosshenrich, C.A.J.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.-J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.; Becknell, B.; Freud, A.G.; McClory, S.; Briercheck, E.; Yu, J.; Mao, C.; Giovenzana, C.; Nuovo, G.; Wei, L.; et al. Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity 2010, 32, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Mebius, R.E.; Rennert, P.; Weissman, I.L. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 1997, 7, 493–504. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Monticelli, L.A.; Alenghat, T.; Fung, T.C.; Hutnick, N.A.; Kunisawa, J.; Shibata, N.; Grunberg, S.; Sinha, R.; Zahm, A.M.; et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 2012, 336, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014, 343, 1249288. [Google Scholar] [CrossRef] [PubMed]
- Hoorweg, K.; Peters, C.P.; Cornelissen, F.; Aparicio-Domingo, P.; Papazian, N.; Kazemier, G.; Mjösberg, J.M.; Spits, H.; Cupedo, T. Functional Differences between Human NKp44− and NKp44+ RORC+ Innate Lymphoid Cells. Front. Immunol. 2012, 3, 72. [Google Scholar] [CrossRef] [PubMed]
- Glatzer, T.; Killig, M.; Meisig, J.; Ommert, I.; Luetke-Eversloh, M.; Babic, M.; Paclik, D.; Blüthgen, N.; Seidl, R.; Seifarth, C.; et al. RORγt+ Innate Lymphoid Cells Acquire a Proinflammatory Program upon Engagement of the Activating Receptor NKp44. Immunity 2013, 38, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Villanova, F.; Flutter, B.; Tosi, I.; Grys, K.; Sreeneebus, H.; Perera, G.K.; Chapman, A.; Smith, C.H.; Di Meglio, P.; Nestle, F.O. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J. Investig. Dermatol. 2014, 134, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 Control Plasticity of CD127+ Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Testi, M.G.; Pasetto, M.; Picchio, M.C.; Innamorati, G.; Mazzocco, M.; Ugel, S.; Cingarlini, S.; Bronte, V.; Zanovello, P.; et al. IFN-gamma-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine 2010, 28, 3548–3557. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Horvath, C.M.; Wen, Z.; Schreiber, R.D.; Darnell, J.E. Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc. Natl. Acad. Sci. USA 1996, 93, 7673–7678. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.; Paterson, Y. IFN-gamma-dependent inhibition of tumor angiogenesis by tumor-infiltrating CD4+ T cells requires tumor responsiveness to IFN-gamma. J. Immunol. 2001, 166, 2276–2282. [Google Scholar] [CrossRef] [PubMed]
- Detjen, K.M.; Farwig, K.; Welzel, M.; Wiedenmann, B.; Rosewicz, S. Interferon γ inhibits growth of human pancreatic carcinoma cells via caspase-1 dependent induction of apoptosis. Gut 2001, 49, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Whitmire, J.K.; Tan, J.T.; Whitton, J.L. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J. Exp. Med. 2005, 201, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Reiter, Z. Interferon—A major regulator of natural killer cell-mediated cytotoxicity. J. Interferon Res. 1993, 13, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chakravarty, S.D.; Ivashkiv, L.B. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008, 226, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Haabeth, O.A.W.; Lorvik, K.B.; Hammarström, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun. 2011, 2, 240. [Google Scholar] [CrossRef] [PubMed]
- Hoepner, S.; Loh, J.M.S.; Riccadonna, C.; Derouazi, M.; Maroun, C.Y.; Dietrich, P.-Y.; Walker, P.R. Synergy between CD8 T cells and Th1 or Th2 polarised CD4 T cells for adoptive immunotherapy of brain tumours. PLoS ONE 2013, 8, e63933. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-γ in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, J.; Tremblay, P.; Vallières, L. Tumor necrosis factor reduces brain tumor growth by enhancing macrophage recruitment and microcyst formation. Cancer Res. 2005, 65, 3928–3936. [Google Scholar] [CrossRef] [PubMed]
- Larmonier, N.; Cathelin, D.; Larmonier, C.; Nicolas, A.; Merino, D.; Janikashvili, N.; Audia, S.; Bateman, A.; Thompson, J.; Kottke, T.; et al. The inhibition of TNF-α anti-tumoral properties by blocking antibodies promotes tumor growth in a rat model. Exp. Cell Res. 2007, 313, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Calzascia, T.; Pellegrini, M.; Hall, H.; Sabbagh, L.; Ono, N.; Elford, A.R.; Mak, T.W.; Ohashi, P.S. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Investig. 2007, 117, 3833–3845. [Google Scholar] [PubMed]
- Dace, D.S.; Chen, P.W.; Niederkorn, J.Y. CD8+ T cells circumvent immune privilege in the eye and mediate intraocular tumor rejection by a TNF-alpha-dependent mechanism. J. Immunol. 2007, 178, 6115–6122. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Boyer, C.M.; Whitaker, R.S.; Berchuck, A.; Wiener, J.R.; Weinberg, J.B.; Bast, R.C. Tumor necrosis factor alpha as an autocrine and paracrine growth factor for ovarian cancer: Monokine induction of tumor cell proliferation and tumor necrosis factor alpha expression. Cancer Res. 1993, 53, 1939–1944. [Google Scholar] [PubMed]
- Leibovich, S.J.; Polverini, P.J.; Shepard, H.M.; Wiseman, D.M.; Shively, V.; Nuseir, N. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-alpha. Nature 1987, 329, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Arnott, C.H.; Robinson, S.C.; Moore, R.J.; Thompson, R.G.; Marshall, J.F.; Balkwill, F.R. TNF-alpha regulates epithelial expression of MMP-9 and integrin alphavbeta6 during tumour promotion. A role for TNF-alpha in keratinocyte migration? Oncogene 2004, 23, 6954–6966. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.-S.; Zhou, B.-H.; Li, C.-L.; Zhang, F.; Wang, X.-F.; Zhang, G.; Bu, X.-Z.; Cai, S.-H.; Du, J. Epithelial-mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated stabilization of snail in colorectal cancer. PLoS ONE 2013, 8, e56664. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Moore, R.J.; Arnott, C.H.; East, N.; Thompson, R.G.; Scallon, B.J.; Shealy, D.J.; Balkwill, F.R. An anti-tumor necrosis factor-alpha antibody inhibits the development of experimental skin tumors. Mol. Cancer Ther. 2003, 2, 445–451. [Google Scholar] [PubMed]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [PubMed]
- Trabanelli, S.; Curti, A.; Lecciso, M.; Salomé, B.; Riether, C.; Ochsenbein, A.; Romero, P.; Jandus, C. CD127+ innate lymphoid cells are dysregulated in treatment naïve acute myeloid leukemia patients at diagnosis. Haematologica 2015, 100, e257–e260. [Google Scholar] [CrossRef] [PubMed]
- Dadi, S.; Chhangawala, S.; Whitlock, B.M.; Franklin, R.A.; Luo, C.T.; Oh, S.A.; Toure, A.; Pritykin, Y.; Huse, M.; Leslie, C.S.; et al. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell 2016, 164, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Pantic, J.M.; Milovanovic, M.Z.; Arsenijevic, N.N.; Lukic, M.L. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int. J. Cancer 2014, 134, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Matsui, S.; Park, J.-M.; Mamura, M.; Noben-Trauth, N.; Donaldson, D.D.; Chen, W.; Wahl, S.M.; Ledbetter, S.; Pratt, B.; et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: Abrogation prevents tumor recurrence. J. Exp. Med. 2003, 198, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, M.; Hardaway, J.C.; Guloglu, F.B.; Miller, M.M.; Hoeman, C.M.; Zaghouani, A.A.; Wan, X.; Rowland, L.M.; Cascio, J.A.; Sherman, M.P.; et al. IL-13Rα1 is a surface marker for M2 macrophages influencing their differentiation and function. Eur. J. Immunol. 2014, 44, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Marvie, P.; Lisbonne, M.; L’helgoualc'h, A.; Rauch, M.; Turlin, B.; Preisser, L.; Bourd-Boittin, K.; Théret, N.; Gascan, H.; Piquet-Pellorce, C.; et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J. Cell. Mol. Med. 2010, 14, 1726–1739. [Google Scholar] [CrossRef] [PubMed]
- Mchedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.J.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-Dependent Innate Lymphoid Cells Mediate Hepatic Fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular Carcinoma in Cirrhosis: Incidence and Risk Factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Razumilava, N.; Gores, G.J.; Walters, S.; Mizuochi, T.; Mourya, R.; Bessho, K.; Wang, Y.-H.; Glaser, S.S.; Shivakumar, P.; et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J. Clin. Investig. 2014, 124, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- Ikutani, M.; Yanagibashi, T.; Ogasawara, M.; Tsuneyama, K.; Yamamoto, S.; Hattori, Y.; Kouro, T.; Itakura, A.; Nagai, Y.; Takaki, S.; et al. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J. Immunol. 2012, 188, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Zaynagetdinov, R.; Sherrill, T.P.; Gleaves, L.A.; McLoed, A.G.; Saxon, J.A.; Habermann, A.C.; Connelly, L.; Dulek, D.; Peebles, R.S.; Fingleton, B.; et al. Interleukin-5 facilitates lung metastasis by modulating the immune microenvironment. Cancer Res. 2015, 75, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Liu, L.-B.; Shang, W.-Q.; Chang, K.-K.; Meng, Y.-H.; Mei, J.; Yu, J.-J.; Li, D.-J.; Li, M.-Q. The infiltration and functional regulation of eosinophils induced by TSLP promote the proliferation of cervical cancer cell. Cancer Lett. 2015, 364, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Bie, Q.; Zhang, P.; Su, Z.; Zheng, D.; Ying, X.; Wu, Y.; Yang, H.; Chen, D.; Wang, S.; Xu, H. Polarization of ILC2s in peripheral blood might contribute to immunosuppressive microenvironment in patients with gastric cancer. J. Immunol. Res. 2014, 2014, 923135. [Google Scholar] [CrossRef] [PubMed]
- Zea, A.H.; Rodriguez, P.C.; Culotta, K.S.; Hernandez, C.P.; DeSalvo, J.; Ochoa, J.B.; Park, H.-J.; Zabaleta, J.; Ochoa, A.C. l-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes. Cell. Immunol. 2004, 232, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Munder, M.; Eichmann, K.; Modolell, M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: Competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J. Immunol. 1998, 160, 5347–5354. [Google Scholar] [PubMed]
- Bronte, V.; Serafini, P.; De Santo, C.; Marigo, I.; Tosello, V.; Mazzoni, A.; Segal, D.M.; Staib, C.; Lowel, M.; Sutter, G.; et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J. Immunol. 2003, 170, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Bando, J.K.; Nussbaum, J.C.; Liang, H.; Locksley, R.M. Type 2 innate lymphoid cells constitutively express arginase-I in the naive and inflamed lung. J. Leukoc. Biol. 2013, 94, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.W.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.W.; van Loosdregt, J.; Gorlani, A.; Bekker, C.P.J.; Gröne, A.; Sibilia, M.; van Bergen en Henegouwen, P.M.P.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J.A.M. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 2013, 38, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Tanaka, T.; Shi, W.; Matsumoto, M.; Minami, M.; Kashiwamura, S.; Nakanishi, K.; Yoshida, N.; Kishimoto, T.; Akira, S. Essential role of Stat6 in IL-4 signalling. Nature 1996, 380, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.Y.; Iijima, K.; Kita, H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy 2014, 69, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, C.J.; Hwang, Y.Y.; Walker, J.A.; Salimi, M.; Wong, S.H.; Brewer, J.M.; Englezakis, A.; Barlow, J.L.; Hams, E.; Scanlon, S.T.; et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Mirchandani, A.S.; Besnard, A.-G.; Yip, E.; Scott, C.; Bain, C.C.; Cerovic, V.; Salmond, R.J.; Liew, F.Y. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J. Immunol. 2014, 192, 2442–2448. [Google Scholar] [CrossRef] [PubMed]
- Cătană, C.-S.; Berindan Neagoe, I.; Cozma, V.; Magdaş, C.; Tăbăran, F.; Dumitraşcu, D.L. Contribution of the IL-17/IL-23 axis to the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 5823–5830. [Google Scholar] [PubMed]
- Ermann, J.; Staton, T.; Glickman, J.N.; de Waal Malefyt, R.; Glimcher, L.H. Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet–/–.Rag2–/– (TRUC) mice. Proc. Natl. Acad. Sci. USA 2014, 111, E2559–E2566. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Arancibia-Cárcamo, C.V.; Fleming, M.P.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.L.; Powrie, F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Kvedaraite, E.; Lourda, M.; Ideström, M.; Chen, P.; Olsson-Åkefeldt, S.; Forkel, M.; Gavhed, D.; Lindforss, U.; Mjösberg, J.; Henter, J.-I.; et al. Tissue-infiltrating neutrophils represent the main source of IL-23 in the colon of patients with IBD. Gut 2015. [Google Scholar] [CrossRef] [PubMed]
- Triantafillidis, J.K.; Nasioulas, G.; Kosmidis, P.A. Colorectal cancer and inflammatory bowel disease: Epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009, 29, 2727–2737. [Google Scholar] [PubMed]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.H.; Jain, R.; Tessmer, M.S.; Gorman, D.; Mangadu, R.; Sathe, M.; Vives, F.; Moon, C.; Penaflor, E.; Turner, S.; et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol. 2014, 7, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Duan, Y.; Cheng, X.; Chen, X.; Xie, W.; Long, H.; Lin, Z.; Zhu, B. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem. Biophys. Res. Commun. 2011, 407, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Mulcahy, L.A.; Mohammed, R.A.; Lee, A.H.; Franks, H.A.; Kilpatrick, L.; Yilmazer, A.; Paish, E.C.; Ellis, I.O.; Patel, P.M.; et al. IL-17 expression by breast-cancer-associated macrophages: IL-17 promotes invasiveness of breast cancer cell lines. Breast Cancer Res. 2008, 10, R95. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Shen, J.; Cao, J.; Zhou, Y.; Shang, L.; Jin, S.; Cao, S.; Che, D.; Liu, F.; Yu, Y. Interleukin-17 promotes angiogenesis by stimulating VEGF production of cancer cells via the STAT3/GIV signaling pathway in non-small-cell lung cancer. Sci. Rep. 2015, 5, 16053. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-P.; Yan, J.; Xu, J.; Pang, X.-H.; Chen, M.-S.; Li, L.; Wu, C.; Li, S.-P.; Zheng, L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J. Hepatol. 2009, 50, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, T.; Liu, X.; Guo, J.N.; Xie, T.; Ding, Y.; Lin, M.; Yang, H. IL-17 promotes tumor angiogenesis through Stat3 pathway mediated upregulation of VEGF in gastric cancer. Tumor Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.D.; Xu, H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.J.; Bériou, G.; Heslan, M.; Chauvin, C.; Utriainen, L.; Aumeunier, A.; Scott, C.L.; Mowat, A.; Cerovic, V.; Houston, S.A.; et al. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol. 2014, 7, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Kobold, S.; Völk, S.; Clauditz, T.; Küpper, N.J.; Minner, S.; Tufman, A.; Düwell, P.; Lindner, M.; Koch, I.; Heidegger, S.; et al. Interleukin-22 is frequently expressed in small- and large-cell lung cancer and promotes growth in chemotherapy-resistant cancer cells. J. Thorac. Oncol. 2013, 8, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Kirchberger, S.; Royston, D.J.; Boulard, O.; Thornton, E.; Franchini, F.; Szabady, R.L.; Harrison, O.; Powrie, F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013, 210, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Von Burg, N.; Chappaz, S.; Baerenwaldt, A.; Horvath, E.; Bose Dasgupta, S.; Ashok, D.; Pieters, J.; Tacchini-Cottier, F.; Rolink, A.; Acha-Orbea, H.; et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc. Natl. Acad. Sci. USA 2014, 111, 12835–12840. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 2015, 348, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Benchetrit, F.; Ciree, A.; Vives, V.; Warnier, G.; Gey, A.; Sautès-Fridman, C.; Fossiez, F.; Haicheur, N.; Fridman, W.H.; Tartour, E.; et al. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood 2002, 99, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Szeliga, W.; Vatan, L.; Zou, W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood 2009, 114, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.F.; Gaertner, F.C.; Erl, W.; Janssen, K.-P.; Blechert, B.; Holzmann, B.; Weighardt, H.; Essler, M. IL-22-mediated tumor growth reduction correlates with inhibition of ERK1/2 and AKT phosphorylation and induction of cell cycle arrest in the G2-M phase. J. Immunol. 2006, 177, 8266–8272. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Loiacono, F.; Di Carlo, E.; Scaramuccia, A.; Mora, M.; Conte, R.; Benelli, R.; Spaggiari, G.M.; Cantoni, C.; Campana, S.; et al. NCR+ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun. 2015, 6, 8280. [Google Scholar] [CrossRef] [PubMed]
- Eisenring, M.; Berg, J.; Kristiansen, G.; Saller, E.; Becher, B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol. 2010, 11, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Moskalenko, M.; Pan, M.; Fu, Y.; de Moll, E.H.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Jayaraman, P.; Bernardo, S.; Sikora, A.G.; et al. Requirement for innate immunity and CD90+ NK1.1− lymphocytes to treat established melanoma with chemo-immunotherapy. Cancer Immunol. Res. 2015, 3, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Monticelli, L.A.; Elloso, M.M.; Fouser, L.A.; Artis, D. CD4+ lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 2011, 34, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, J.J.P.; Wimmers, F.; Hato, S.V.; de Vries, I.J.M.; Sköld, A.E. Dendritic cell cross talk with innate and innate-like effector cells in antitumor immunity: Implications for DC vaccination. Crit. Rev. Immunol. 2014, 34, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.; Walker, A.W.; Stolarczyk, E.; Canavan, J.B.; Gökmen, M.R.; Marks, E.; Jackson, I.; Hashim, A.; Curtis, M.A.; Jenner, R.G.; et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 2012, 37, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Kinnebrew, M.A.; Buffie, C.G.; Diehl, G.E.; Zenewicz, L.A.; Leiner, I.; Hohl, T.M.; Flavell, R.A.; Littman, D.R.; Pamer, E.G. Interleukin 23 Production by Intestinal CD103+CD11b+ Dendritic Cells in Response to Bacterial Flagellin Enhances Mucosal Innate Immune Defense. Immunity 2012, 36, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Spits, H. Human innate lymphoid cells. Blood 2014, 124, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [PubMed]
- Galvin, K.C.; Dyck, L.; Marshall, N.A.; Stefanska, A.M.; Walsh, K.P.; Moran, B.; Higgins, S.C.; Dungan, L.S.; Mills, K.H.G. Blocking retinoic acid receptor-α enhances the efficacy of a dendritic cell vaccine against tumours by suppressing the induction of regulatory T cells. Cancer Immunol. Immunother. 2013, 62, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Beek, J.J.P.; Martens, A.W.J.; Bakdash, G.; De Vries, I.J.M. Innate Lymphoid Cells in Tumor Immunity. Biomedicines 2016, 4, 7. https://doi.org/10.3390/biomedicines4010007
Van Beek JJP, Martens AWJ, Bakdash G, De Vries IJM. Innate Lymphoid Cells in Tumor Immunity. Biomedicines. 2016; 4(1):7. https://doi.org/10.3390/biomedicines4010007
Chicago/Turabian StyleVan Beek, Jasper J. P., Anne W. J. Martens, Ghaith Bakdash, and I. Jolanda M. De Vries. 2016. "Innate Lymphoid Cells in Tumor Immunity" Biomedicines 4, no. 1: 7. https://doi.org/10.3390/biomedicines4010007
APA StyleVan Beek, J. J. P., Martens, A. W. J., Bakdash, G., & De Vries, I. J. M. (2016). Innate Lymphoid Cells in Tumor Immunity. Biomedicines, 4(1), 7. https://doi.org/10.3390/biomedicines4010007