
Prospects for Foamy Viral Vector Anti-HIV Gene Therapy

Abstract
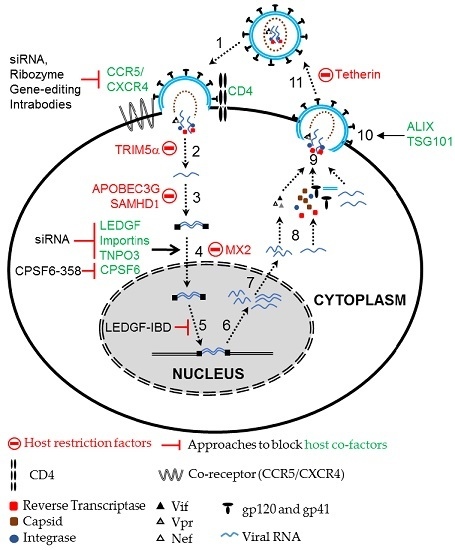
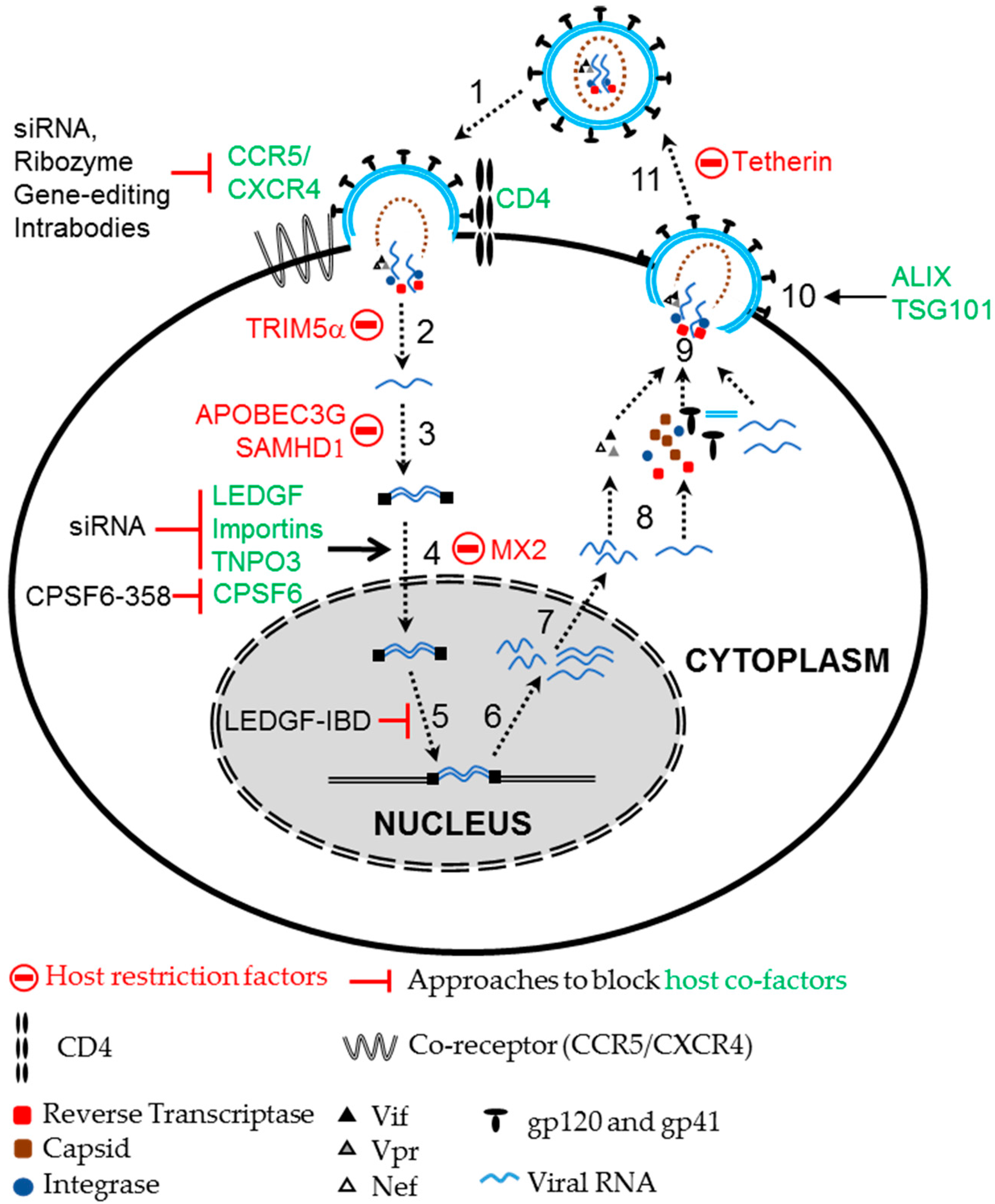
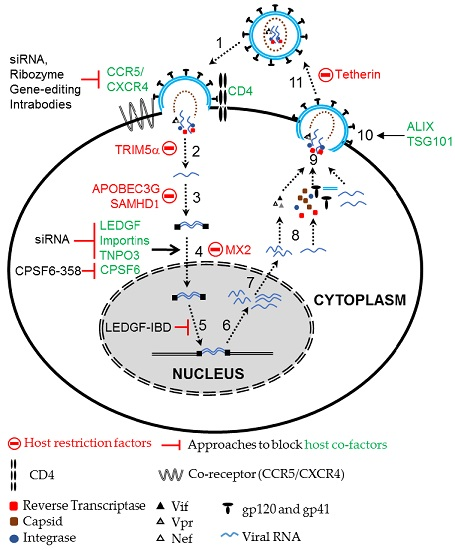
:
1. Introduction
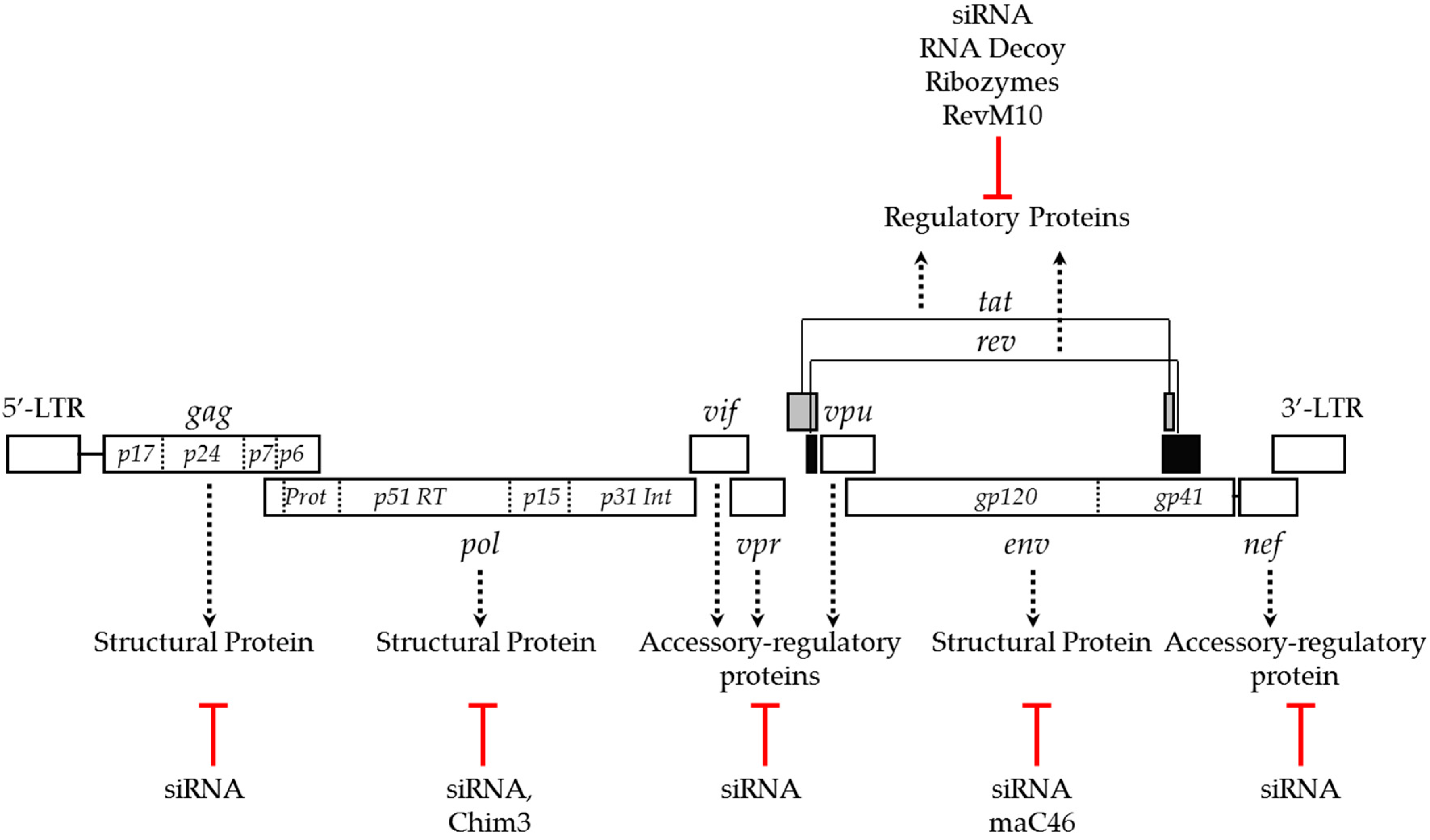
2. Anti-HIV Transgenes
2.1. Targeting Viral Genes to Inhibit HIV Infection
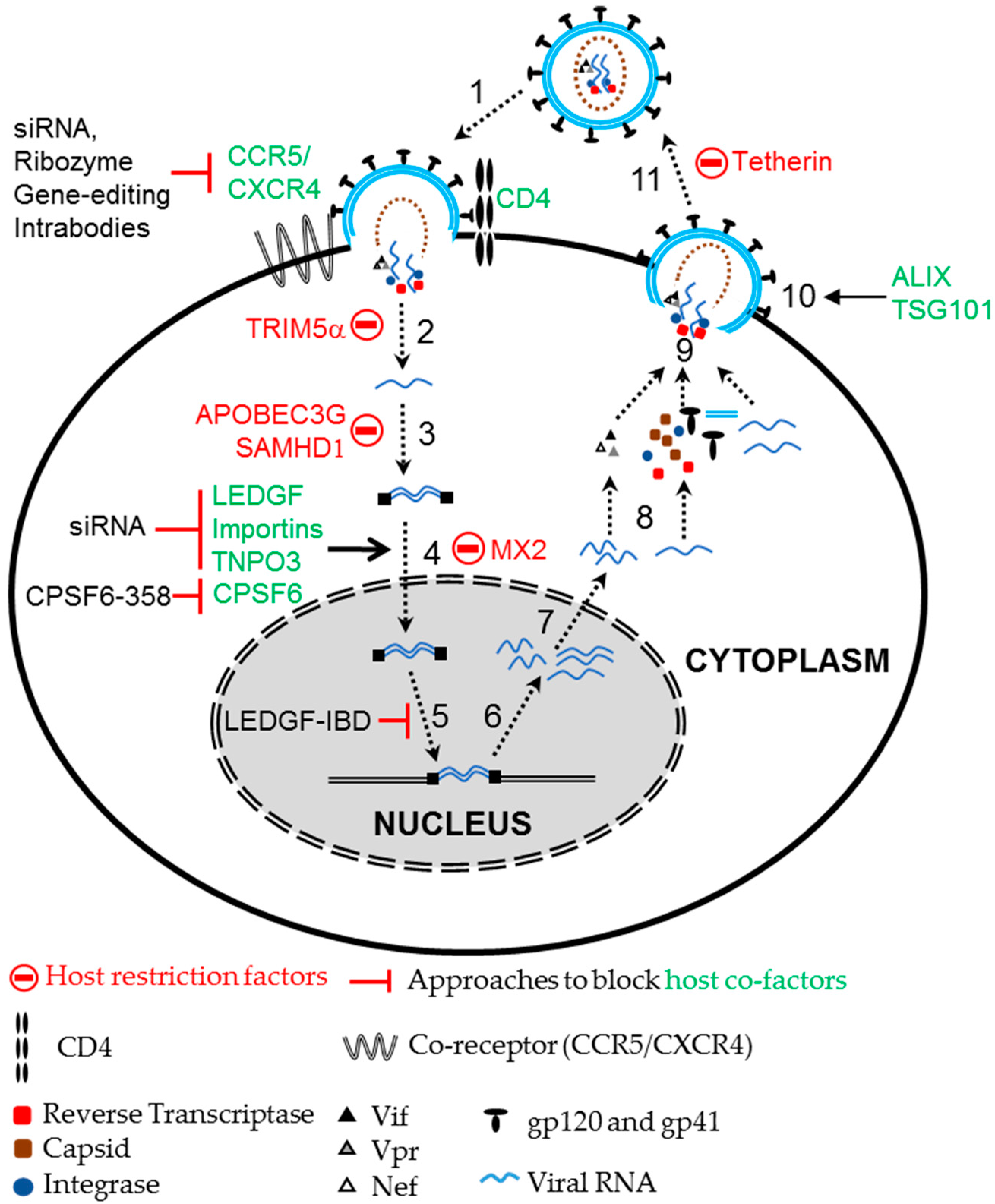
2.2. Using Host Cellular Proteins as Targets to Block HIV Infection
2.3. Efficacy of Combinatorial Approaches to Combat HIV Infection
3. Anti-HIV Gene Therapy Clinical Trails
4. Retroviral Vectors and Their Limitations in Anti-HIV Gene Therapy
4.1. Gammaretroviral Vectors
4.2. Lentiviral Vectors
4.3. Limitations of HIV Based LV Vectors for Anti-HIV Gene Therapy
5. Foamy Viral Vectors
5.1. FV Vectors Have a Promising Safety Profile
5.2. Anti-HIV Transgenes in FV Vectors
5.3. FV Vectors for Anti-HIV Gene Therapy
5.4. FV in HIV Vaccine Development
6. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Unaids Report: How AIDS Changed Everything; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Shen, L.; Siliciano, R.F. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J. Allergy Clin. Immunol. 2008, 122, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.G.; Lima, V.; Gouws, E. Modelling the impact of antiretroviral therapy on the epidemic of HIV. Curr. HIV Res. 2011, 9, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Camacho, R.; Teofilo, E. Antiretroviral therapy in treatment-naive patients with HIV infection. Curr. Opin. HIV AIDS 2011, 6 (Suppl. 1), S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Olszko, M.E.; Trobridge, G.D. Foamy virus vectors for HIV gene therapy. Viruses 2013, 5, 2585–2600. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 D32/D32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5 D32/D32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia growth and development study, multicenter aids cohort study, multicenter hemophilia cohort study, san francisco city cohort, ALIVE study. Science 1996, 273, 1856–1862. [Google Scholar] [PubMed]
- Hutter, G.; Thiel, E. Allogeneic transplantation of CCR5-deficient progenitor cells in a patient with HIV infection: An update after 3 years and the search for patient No. 2. AIDS 2011, 25, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Martinson, J.J.; Chapman, N.H.; Rees, D.C.; Liu, Y.T.; Clegg, J.B. Global distribution of the CCR5 gene 32-basepair deletion. Nat. Genet. 1997, 16, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Dhanda, R.; Bamshad, M.; Mummidi, S.; Geevarghese, R.; Catano, G.; Anderson, S.A.; Walter, E.A.; Stephan, K.T.; Hammer, M.F.; et al. Global survey of genetic variation in CCR5, RANTES, and MIP-1a: Impact on the epidemiology of the HIV-1 pandemic. Proc. Natl. Acad. Sci. USA 2001, 98, 5199–5204. [Google Scholar] [CrossRef] [PubMed]
- Cannon, P.M.; Kohn, D.B.; Kiem, H.P. HIV eradication—From Berlin to Boston. Nat. Biotechnol. 2014, 32, 315–316. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO-2 associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Hendrie, P.C.; Huo, Y.; Stolitenko, R.B.; Russell, D.W. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol. Ther. 2008, 16, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Banerjea, A.; Li, M.J.; Remling, L.; Rossi, J.; Akkina, R. Lentiviral transduction of TAR decoy and CCR5 ribozyme into CD34+ progenitor cells and derivation of HIV-1 resistant T cells and macrophages. AIDS Res. Ther. 2004, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von Laer, D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.W.; Younan, P.; Jerome, K.R.; Kiem, H.P. Combinatorial anti-HIV gene therapy: Using a multipronged approach to reach beyond HAART. Gene Ther. 2013, 20, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Mautino, M.R.; Morgan, R.A. Potent inhibition of human immunodeficiency virus type 1 replication by conditionally replicating human immunodeficiency virus-based lentiviral vectors expressing envelope antisense mRNA. Hum. Gene Ther. 2000, 11, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Bahner, I.; Sumiyoshi, T.; Kagoda, M.; Swartout, R.; Peterson, D.; Pepper, K.; Dorey, F.; Reiser, J.; Kohn, D.B. Lentiviral vector transduction of a dominant-negative Rev gene into human CD34+ hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Mol. Ther. 2007, 15, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.A.; Vojtech, L.; Bahner, I.; Kohn, D.B.; Laer, D.V.; Russell, D.W.; Richard, R.E. Foamy virus vectors expressing anti-HIV transgenes efficiently block HIV-1 replication. Mol. Ther. 2008, 16, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Von Laer, D.; Hasselmann, S.; Hasselmann, K. Impact of gene-modified T cells on HIV infection dynamics. J. Theor. Biol. 2006, 238, 60–77. [Google Scholar] [CrossRef] [PubMed]
- Kimpel, J.; Braun, S.E.; Qiu, G.; Wong, F.E.; Conolle, M.; Schmitz, J.E.; Brendel, C.; Humeau, L.M.; Dropulic, B.; Rossi, J.J.; et al. Survival of the fittest: Positive selection of CD4+ T cells expressing a membrane-bound fusion inhibitor following HIV-1 infection. PLoS ONE 2010, 5, e12357. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Allers, K.; Schneider, T. The additional use of viral entry inhibitors during autologous hematopoietic stem cell transplantation in patients with non-hodgkin lymphoma and HIV-1 infection. Biol. Blood Marrow Transpl. 2011, 17, 586–587. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Butticaz, C.; Ciuffi, A.; Munoz, M.; Thomas, J.; Bridge, A.; Pebernard, S.; Iggo, R.; Meylan, P.; Telenti, A. Protection from HIV-1 infection of primary CD4 T cells by CCR5 silencing is effective for the full spectrum of CCR5 expression. Antivir. Ther. 2003, 8, 373–377. [Google Scholar] [PubMed]
- Martinez, M.A.; Gutierrez, A.; Armand-Ugon, M.; Blanco, J.; Parera, M.; Gomez, J.; Clotet, B.; Este, J.A. Suppression of chemokine receptor expression by RNA interference allows for inhibition of HIV-1 replication. AIDS 2002, 16, 2385–2390. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Banerjea, A.; Akkina, R. Bispecific short hairpin siRNA constructs targeted to CD4, CXCR4, and CCR5 confer HIV-1 resistance. Oligonucleotides 2003, 13, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Swan, C.H.; Buhler, B.; Steinberger, P.; Tschan, M.P.; Barbas, C.F., 3rd; Torbett, B.E. T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther. 2006, 13, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Leavitt, M.; Tritz, R.; Duarte, E.; Kang, D.; Mamounas, M.; Gilles, P.; Wong-Staal, F.; Kennedy, S.; Merson, J.; et al. Inhibition of CCR5-dependent HIV-1 infection by hairpin ribozyme gene therapy against CC-chemokine receptor 5. Virology 2000, 276, 271–278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cagnon, L.; Rossi, J.J. Downregulation of the CCR5 beta-chemokine receptor and inhibition of HIV-1 infection by stable VA1-ribozyme chimeric transcripts. Antisense Nucleic Acid Drug Dev. 2000, 10, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Hong, P.; Arumugam, B.; Pokomo, L.; Boyer, J.; Koizumi, N.; Kittipongdaja, P.; Chen, A.; Bristol, G.; Galic, Z.; et al. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood 2010, 115, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Kamata, M.; Chen, K.N.; Pariente, N.; An, D.S.; Chen, I.S. Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J. Gene Med. 2010, 12, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Myburgh, R.; Ivic, S.; Pepper, M.S.; Gers-Huber, G.; Li, D.; Audige, A.; Rochat, M.A.; Jaquet, V.; Regenass, S.; Manz, M.G.; et al. Lentivector knockdown of CCR5 in hematopoietic stem and progenitor cells confers functional and persistent HIV-1 resistance in humanized mice. J. Virol. 2015, 89, 6761–6772. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5D32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Ferreira, L.M.; Collins, R.; Meissner, T.B.; Boutwell, C.L.; Friesen, M.; Vrbanac, V.; Garrison, B.S.; Stortchevoi, A.; Bryder, D.; et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 2014, 15, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ye, C.; Liu, J.; Zhang, D.; Kimata, J.T.; Zhou, P. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS ONE 2014, 9, e115987. [Google Scholar] [CrossRef] [PubMed]
- Steen, A.; Schwartz, T.W.; Rosenkilde, M.M. Targeting CXCR4 in HIV cell-entry inhibition. Mini Rev. Med. Chem. 2009, 9, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Banerjea, A.; Planelles, V.; Akkina, R. Potent suppression of HIV type 1 infection by a short hairpin anti-CXCR4 siRNA. AIDS Res. Hum. Retroviruses 2003, 19, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Chen, S.; Wang, S.; Yu, X.; Chen, Y.; Jiang, M.; Zhuang, K.; Ho, W.; Hou, W.; Huang, J.; et al. Genome editing of CXCR4 by CRISPR/Cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015, 5, 15577. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.; Urak, K.; Veillette, M.; Nepveu-Traversy, M.E.; Pham, Q.T.; Hamel, S.; Rossi, J.J.; Berthoux, L. Preclinical assessment of mutant human TRIM5a a as an anti-HIV-1 transgene. Hum. Gene Ther. 2015, 26, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.R.; Ziegler, P.; Pertel, T.; Strambio-De-Castillia, C.; Grutter, C.; Martinetti, G.; Mazzucchelli, L.; Grutter, M.; Manz, M.G.; Luban, J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J. Clin. Investig. 2009, 119, 3035–3047. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Vets, S.; Kimpel, J.; Volk, A.; De Rijck, J.; Schrijvers, R.; Verbinnen, B.; Maes, W.; Von Laer, D.; Debyser, Z.; Gijsbers, R. Lens epithelium-derived growth factor/p75 qualifies as a target for HIV gene therapy in the NSG mouse model. Mol. Ther. 2012, 20, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Geerts, D.; Jeeninga, R.E.; Berkhout, B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antivir. Res. 2011, 89, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Sagnier, S.; Geerts, D.; Jeeninga, R.E.; Biard-Piechaczyk, M.; Berkhout, B. Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol. J. 2012, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Mulky, A.; Yuen, W.; Martin, T.D.; Meyerson, N.R.; Choi, L.; Yu, H.; Sawyer, S.L.; Kewalramani, V.N. HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. J. Virol. 2012, 86, 3851–3860. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Egelhofer, M.; Brandenburg, G.; Martinius, H.; Schult-Dietrich, P.; Melikyan, G.; Kunert, R.; Baum, C.; Choi, I.; Alexandrov, A.; von Laer, D. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J. Virol. 2004, 78, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Riley, J.L.; Carroll, R.G.; von Laer, D.; June, C.H. Suppression of HIV-1 infection in primary CD4 T cells transduced with a self-inactivating lentiviral vector encoding a membrane expressed gp41-derived fusion inhibitor. Clin. Immunol. 2005, 115, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bonyhadi, M.L.; Moss, K.; Voytovich, A.; Auten, J.; Kalfoglou, C.; Plavec, I.; Forestell, S.; Su, L.; Bohnlein, E.; Kaneshima, H. RevM10-expressing T cells derived in vivo from transduced human hematopoietic stem-progenitor cells inhibit human immunodeficiency virus replication. J. Virol. 1997, 71, 4707–4716. [Google Scholar] [PubMed]
- Su, L.; Lee, R.; Bonyhadi, M.; Matsuzaki, H.; Forestell, S.; Escaich, S.; Bohnlein, E.; Kaneshima, H. Hematopoietic stem cell-based gene therapy for acquired immunodeficiency syndrome: Efficient transduction and expression of RevM10 in myeloid cells in vivo and in vitro. Blood 1997, 89, 2283–2290. [Google Scholar] [PubMed]
- Bauer, G.; Valdez, P.; Kearns, K.; Bahner, I.; Wen, S.F.; Zaia, J.A.; Kohn, D.B. Inhibition of human immunodeficiency virus-1 (HIV-1) replication after transduction of granulocyte colony-stimulating factor-mobilized CD34+ cells from HIV-1-infected donors using retroviral vectors containing anti-HIV-1 genes. Blood 1997, 89, 2259–2267. [Google Scholar] [PubMed]
- Lee, S.W.; Gallardo, H.F.; Gilboa, E.; Smith, C. Inhibition of human immunodeficiency virus type 1 in human T cells by a potent rev response element decoy consisting of the 13-nucleotide minimal rev-binding domain. J. Virol. 1994, 68, 8254–8264. [Google Scholar] [PubMed]
- Smith, C.; Lee, S.W.; Wong, E.; Gallardo, H.; Page, K.; Gaspar, O.; Lebkowski, J.; Gilboa, E. Transient protection of human T-cells from human immunodeficiency virus type 1 infection by transduction with adeno-associated viral vectors which express RNA decoys. Antivir. Res. 1996, 32, 99–115. [Google Scholar] [CrossRef]
- Michienzi, A.; Li, S.; Zaia, J.A.; Rossi, J.J. A nucleolar TAR decoy inhibitor of HIV-1 replication. Proc. Natl. Acad. Sci. USA 2002, 99, 14047–14052. [Google Scholar] [CrossRef] [PubMed]
- Michienzi, A.; De Angelis, F.G.; Bozzoni, I.; Rossi, J.J. A nucleolar localizing rev binding element inhibits HIV replication. AIDS Res. Ther. 2006, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Bauer, G.; Michienzi, A.; Yee, J.K.; Lee, N.S.; Kim, J.; Li, S.; Castanotto, D.; Zaia, J.; Rossi, J.J. Inhibition of HIV-1 infection by lentiviral vectors expressing pol III-promoted anti-HIV RNAs. Mol. Ther. 2003, 8, 196–206. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Coburn, G.A.; Cullen, B.R. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J. Virol. 2002, 76, 9225–9231. [Google Scholar] [CrossRef] [PubMed]
- Jacque, J.M.; Triques, K.; Stevenson, M. Modulation of HIV-1 replication by RNA interference. Nature 2002, 418, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.J.; Liu, X.; He, J. Lentiviral siRNAs targeting multiple highly conserved RNA sequences of human immunodeficiency virus type 1. Gene Ther. 2005, 12, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.S.; Li, Y.; Kameoka, M.; Ng, T.B.; Wan, D.C. Suppression of HIV replication using RNA interference against HIV-1 integrase. FEBS Lett. 2007, 581, 3253–3259. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Berkhout, B. Lentiviral vectors that carry anti-HIV shRNAs: Problems and solutions. J. Gene Med. 2007, 9, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; ’t Hooft, K.; Liu, Y.P.; Centlivre, M.; von Eije, K.J.; Berkhout, B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol. Ther. 2008, 16, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, S.; Alberici, L.; Gubinelli, F.; Lupo, R.; Olgiati, C.; Rizzardi, G.P.; Bovolenta, C. The F12-Vif derivative Chim3 inhibits HIV-1 replication in CD4+ T lymphocytes and CD34+-derived macrophages by blocking HIV-1 DNA integration. Blood 2009, 113, 3443–3452. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, S.; Gubinelli, F.; Alberici, L.; Piovani, B.M.; Rizzardi, G.P.; Bovolenta, C. Chim3 confers survival advantage to CD4+ T cells upon HIV-1 infection by preventing HIV-1 DNA integration and HIV-1-induced G2 cell-cycle delay. Blood 2010, 115, 4021–4029. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Vink, M.A.; Westerink, J.T.; Ramirez de Arellano, E.; Konstantinova, P.; Ter Brake, O.; Berkhout, B. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 2010, 16, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Podsakoff, G.M.; Engel, B.C.; Carbonaro, D.A.; Choi, C.; Smogorzewska, E.M.; Bauer, G.; Selander, D.; Csik, S.; Wilson, K.; Betts, M.R.; et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34+ cells. Mol. Ther. 2005, 12, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, J.L.; Boyd, M.P.; Arndt, A.J.; Todd, A.V.; Fanning, G.C.; Ely, J.A.; Elliott, F.; Knop, A.; Raponi, M.; Murray, J.; et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J. Gene Med. 2005, 7, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Harris, R.S.; Neuberger, M.S. The vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 2003, 13, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.M.; Coolen, C.; Davis, A.J.; Burrell, C.J.; Li, P. Human immunodeficiency virus 1 (HIV-1) virion infectivity factor (Vif) is part of reverse transcription complexes and acts as an accessory factor for reverse transcription. Virology 2008, 372, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Vallanti, G.; Lupo, R.; Federico, M.; Mavilio, F.; Bovolenta, C. T lymphocytes transduced with a lentiviral vector expressing F12-vif are protected from HIV-1 infection in an APOBEC3G-independent manner. Mol. Ther. 2005, 12, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.E.; Sanders, R.W.; Deng, Y.; Jurriaans, S.; Lange, J.M.; Lu, M.; Berkhout, B. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J. Virol. 2004, 78, 12428–12437. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.; Berkhout, B. Mechanistic studies of a T20-dependent human immunodeficiency virus type 1 variant. J. Virol. 2008, 82, 7735–7740. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Cai, L.; Lu, H.; Qi, Z.; Jiang, S. Combinations of the first and next generations of human immunodeficiency virus (HIV) fusion inhibitors exhibit a highly potent synergistic effect against enfuvirtide-sensitive and -resistant HIV type 1 strains. J. Virol. 2009, 83, 7862–7872. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.; Kawaji, K.; Miyamoto, F.; Shimane, K.; Shimura, K.; Sakagami, Y.; Hattori, T.; Watanabe, K.; Oishi, S.; Fujii, N.; et al. Mechanism of resistance to S138A substituted enfuvirtide and its application to peptide design. Int. J. Biochem. Cell Biol. 2013, 45, 908–915. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cheng, J.; Lu, H.; Li, J.; Hu, J.; Qi, Z.; Liu, Z.; Jiang, S.; Dai, Q. Potent HIV fusion inhibitors against enfuvirtide-resistant HIV-1 strains. Proc. Natl. Acad. Sci. USA 2008, 105, 16332–16337. [Google Scholar] [CrossRef] [PubMed]
- Lohrengel, S.; Hermann, F.; Hagmann, I.; Oberwinkler, H.; Scrivano, L.; Hoffmann, C.; von Laer, D.; Dittmar, M.T. Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J. Virol. 2005, 79, 10237–10246. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Baler-Bitterlich, G.; Ragheb, J.A.; Wong-Staal, F.; Gallo, R.C.; Anderson, W.F. Further evaluation of soluble CD4 as an anti-HIV type 1 gene therapy: Demonstration of protection of primary human peripheral blood lymphocytes from infection by HIV type 1. AIDS Res. Hum. Retroviruses 1994, 10, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Fessel, W.J.; Anderson, B.; Follansbee, S.E.; Winters, M.A.; Lewis, S.T.; Weinheimer, S.P.; Petropoulos, C.J.; Shafer, R.W. The efficacy of an anti-CD4 monoclonal antibody for HIV-1 treatment. Antivir. Res. 2011, 92, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Patel, B.; Ghanem, M.H.; Bundoc, V.; Zheng, Z.; Morgan, R.A.; Rosenberg, S.A.; Dey, B.; Berger, E.A. Novel CD4-based bispecific chimeric antigen receptor designed for enhanced anti-HIV potency and absence of HIV entry receptor activity. J. Virol. 2015, 89, 6685–6694. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34+ cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36ra43. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Waite, J.; Brewer, F.; Sunshine, M.J.; Littman, D.R.; Zou, Y.R. The role of CXCR4 in maintaining peripheral B-cell compartments and humoral immunity. J. Exp. Med. 2004, 200, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An essential role for LEDGF/p75 in HIV integration. Science 2006, 314, 461–464. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J.; Vandekerckhove, L.; Gijsbers, R.; Hombrouck, A.; Hendrix, J.; Vercammen, J.; Engelborghs, Y.; Christ, F.; Debyser, Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J. Virol. 2006, 80, 11498–11509. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Fassati, A.; Görlich, D.; Harrison, I.; Zaytseva, L.; Mingot, J.M. Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. EMBO J. 2003, 22, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
- Zielske, S.P.; Stevenson, M. Importin 7 may be dispensable for human immunodeficiency virus type 1 and simian immunodeficiency virus infection of primary macrophages. J. Virol. 2005, 79, 11541–11546. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Thys, W.; De Rijck, J.; Gijsbers, R.; Albanese, A.; Arosio, D.; Emiliani, S.; Rain, J.C.; Benarous, R.; Cereseto, A.; et al. Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 2008, 18, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- De Iaco, A.; Luban, J. Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 2011, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.; Valle-Casuso, J.C.; White, T.E.; Brandariz-Nuñez, A.; Bosche, W.J.; Reszka, N.; Gorelick, R.; Diaz-Griffero, F. The ability of TNPO3-depleted cells to inhibit HIV-1 infection requires CPSF6. Retrovirology 2013, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Bloch, N.; Landau, N.R. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 2015, 16, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5a restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Lynch, C.; Stoye, J.P.; Yap, M.W. A TRIM5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. USA 2004, 101, 13324–13328. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Perron, M.; Welikala, S.; Sodroski, J. Species-specific variation in the B30.2(SPRY) domain of TRIM5a determines the potency of human immunodeficiency virus restriction. J. Virol. 2005, 79, 3139–3145. [Google Scholar] [CrossRef] [PubMed]
- Pham, Q.T.; Bouchard, A.; Grutter, M.G.; Berthoux, L. Generation of human TRIM5a mutants with high HIV-1 restriction activity. Gene Ther. 2010, 17, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Stremlau, M.; Lee, M.; Sodroski, J. Removal of Arginine 332 allows human TRIM5a to bind human immunodeficiency virus capsids and to restrict infection. J. Virol. 2006, 80, 6738–6744. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.W.; Nisole, S.; Stoye, J.P. A single amino acid change in the SPRY domain of human TRIM5a leads to HIV-1 restriction. Curr. Biol. 2005, 15, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Schaller, T.; Eddaoudi, A.; Zhan, H.; Tan, C.P.; Jacobsen, M.; Thrasher, A.J.; Towers, G.J.; Qasim, W. Lentiviral gene therapy against human immunodeficiency virus type 1, using a novel human TRIM21-cyclophilin a restriction factor. Hum. Gene Ther. 2012, 23, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Bitzegeio, J.; Sampias, M.; Bieniasz, P.D.; Hatziioannou, T. Adaptation to the interferon-induced antiviral state by human and simian immunodeficiency viruses. J. Virol. 2013, 87, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Malim, M.H. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 2010, 84, 9254–9266. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Delobel, P.; Sandres-Saune, K.; Cazabat, M.; Pasquier, C.; Marchou, B.; Massip, P.; Izopet, J. R5 to X4 switch of the predominant HIV-1 population in cellular reservoirs during effective highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2005, 38, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Hermann, F.G.; Egerer, L.; Brauer, F.; Gerum, C.; Schwalbe, H.; Dietrich, U.; von Laer, D. Mutations in gp120 contribute to the resistance of human immunodeficiency virus type 1 to membrane-anchored C-peptide maC46. J. Virol. 2009, 83, 4844–4853. [Google Scholar] [CrossRef] [PubMed]
- Desmezieres, E.; Gupta, N.; Vassell, R.; He, Y.; Peden, K.; Sirota, L.; Yang, Z.; Wingfield, P.; Weiss, C.D. Human immunodeficiency virus (HIV) gp41 escape mutants: Cross-resistance to peptide inhibitors of HIV fusion and altered receptor activation of gp120. J. Virol. 2005, 79, 4774–4781. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Li, M.J.; Palmer, B.; Remling, L.; Li, S.; Yam, P.; Yee, J.K.; Rossi, J.; Zaia, J.; Akkina, R. Safety and efficacy of a lentiviral vector containing three anti-HIV genes—CCR5 ribozyme, tat-rev siRNA, and TAR decoy in SCID—Hu mouse-derived T cells. Mol. Ther. 2007, 15, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Javien, J.; Nolta, J.A.; Bauer, G. Preintegration HIV-1 inhibition by a combination lentiviral vector containing a chimeric TRIMa protein, a CCR5 shRNA, and a TAR decoy. Mol. Ther. 2009, 17, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; von Eije, K.J.; Schopman, N.C.; Westerink, J.T.; ter Brake, O.; Haasnoot, J.; Berkhout, B. Combinatorial RNAi against HIV-1 using extended short hairpin RNAs. Mol. Ther. 2009, 17, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Wolstein, O.; Boyd, M.; Millington, M.; Impey, H.; Boyer, J.; Howe, A.; Delebecque, F.; Cornetta, K.; Rothe, M.; Baum, C.; et al. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol. Ther. Methods Clin. Dev. 2014, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.P.; Levin, B.R.; Zhang, J.; Sahakyan, A.; Boyer, J.; Carroll, M.V.; Colon, J.C.; Keech, N.; Rezek, V.; Bristol, G.; et al. Engineering cellular resistance to HIV-1 infection in vivo using a dual therapeutic lentiviral vector. Mol. Ther. Nucleic Acids 2015, 4, e236. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Bauer, G.; Rice, C.R.; Rothschild, J.C.; Carbonaro, D.A.; Valdez, P.; Hao, Q.; Zhou, C.; Bahner, I.; Kearns, K.; et al. A clinical trial of retroviral-mediated transfer of a REV-responsive element decoy gene into CD34+ cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood 1999, 94, 368–371. [Google Scholar] [PubMed]
- Kang, E.M.; De Witte, M.; Malech, H.; Morgan, R.A.; Carter, C.; Leitman, S.F.; Childs, R.; Barrett, A.J.; Little, R.; Tisdale, J.F. Gene therapy-based treatment for HIV-positive patients with malignancies. J. Hematother. Stem Cell Res. 2002, 11, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Mitsuyasu, R.T.; Rosenblatt, J.D.; Ngok, F.K.; Bakker, A.; Cole, S.; Chorn, N.; Lin, L.S.; Bristol, G.; Boyd, M.P.; et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: Myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum. Gene Ther. 2004, 15, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Van Lunzen, J.; Glaunsinger, T.; Stahmer, I.; von Baehr, V.; Baum, C.; Schilz, A.; Kuehlcke, K.; Naundorf, S.; Martinius, H.; Hermann, F.; et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol. Ther. 2007, 15, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.L.; Humeau, L.M.; Boyer, J.; MacGregor, R.R.; Rebello, T.; Lu, X.; Binder, G.K.; Slepushkin, V.; Lemiale, F.; Mascola, J.R.; et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17372–17377. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Stan, R.; Krishnan, A.; Li, H.; Rossi, J.J.; Zaia, J.A. Development of hematopoietic stem cell based gene therapy for HIV-1 infection: Considerations for proof of concept studies and translation to standard medical practice. Viruses 2013, 5, 2898–2919. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Thrasher, A.J. Gene therapy for PIDS: Progress, pitfalls and prospects. Gene 2013, 525, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. Integration of murine leukemia virus dna depends on mitosis. EMBO J. 1993, 12, 2099–2108. [Google Scholar] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, S.I.; Schambach, A.; Howe, S.J.; Ulaganathan, M.; Grassman, E.; Williams, D.; Schiedlmeier, B.; Sebire, N.J.; Gaspar, H.B.; Kinnon, C.; et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. 2008, 16, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Avedillo Diez, I.; Zychlinski, D.; Coci, E.G.; Galla, M.; Modlich, U.; Dewey, R.A.; Schwarzer, A.; Maetzig, T.; Mpofu, N.; Jaeckel, E.; et al. Development of novel efficient sin vectors with improved safety features for Wiskott-Aldrich syndrome stem cell based gene therapy. Mol. Pharm. 2011, 8, 1525–1537. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human b-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.T.; Garcia, J.V. Lentivirus vector mobilization and spread by human immunodeficiency virus. Hum. Gene Ther. 2000, 11, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Klimatcheva, E.; Planelles, V.; Day, S.L.; Fulreader, F.; Renda, M.J.; Rosenblatt, J. Defective lentiviral vectors are efficiently trafficked by HIV-1 and inhibit its replication. Mol. Ther. 2001, 3, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.D.; Hu, W.S. RNAs from genetically distinct retroviruses can copackage and exchange genetic information in vivo. J. Virol. 1997, 71, 6237–6242. [Google Scholar] [PubMed]
- Rizvi, T.A.; Panganiban, A.T. Simian immunodeficiency virus RNA is efficiently encapsidated by human immunodeficiency virus type 1 particles. J. Virol. 1993, 67, 2681–2688. [Google Scholar] [PubMed]
- White, S.M.; Renda, M.; Nam, N.Y.; Klimatcheva, E.; Zhu, Y.; Fisk, J.; Halterman, M.; Rimel, B.J.; Federoff, H.; Pandya, S.; et al. Lentivirus vectors using human and simian immunodeficiency virus elements. J. Virol. 1999, 73, 2832–2840. [Google Scholar] [PubMed]
- Browning, M.T.; Schmidt, R.D.; Lew, K.A.; Rizvi, T.A. Primate and feline lentivirus vector RNA packaging and propagation by heterologous lentivirus virions. J. Virol. 2001, 75, 5129–5140. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D. Foamy virus vectors for gene transfer. Expert Opin. Biol. Ther. 2009, 9, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar] [PubMed]
- Trobridge, G.; Josephson, N.; Vassilopoulos, G.; Mac, J.; Russell, D.W. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 2002, 6, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, G.; Trobridge, G.; Josephson, N.C.; Russell, D.W. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood 2001, 98, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.P.; Allen, J.; Trobridge, G.; Olson, E.; Keyser, K.; Peterson, L.; Russell, D.W. Foamy-virus-mediated gene transfer to canine repopulating cells. Blood 2007, 109, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Zucali, J.R.; Ciccarone, T.; Kelley, V.; Park, J.; Johnson, C.M.; Mergia, A. Transduction of umbilical cord blood CD34+ NOD/SCID-repopulating cells by simian foamy virus type 1 (SFV-1) vector. Virology 2002, 302, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Josephson, N.C.; Trobridge, G.; Russell, D.W. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Hum. Gene Ther. 2004, 15, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Mergia, A.; Chari, S.; Kolson, D.L.; Goodenow, M.M.; Ciccarone, T. The efficiency of simian foamy virus vector type-1 (SFV-1) in nondividing cells and in human PBLS. Virology 2001, 280, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.; Russell, D.W. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J. Virol. 2004, 78, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Allen, J.; Peterson, L.; Ironside, C.; Russell, D.W.; Kiem, H.P. Foamy and lentiviral vectors transduce canine long-term repopulating cells at similar efficiency. Hum. Gene Ther. 2009, 20, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Nasimuzzaman, M.; Kim, Y.S.; Wang, Y.D.; Persons, D.A. High-titer foamy virus vector transduction and integration sites of human CD34+ cell-derived SCID-repopulating cells. Mol. Ther. Methods Clin. Dev. 2014, 1, 14020. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Beard, B.C.; Wu, R.A.; Ironside, C.; Malik, P.; Kiem, H.P. Stem cell selection in vivo using foamy vectors cures canine pyruvate kinase deficiency. PLoS ONE 2012, 7, e45173. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.R., Jr.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.W.; et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.R., Jr.; Tuschong, L.M.; Calvo, K.R.; Shive, H.R.; Burkholder, T.H.; Karlsson, E.K.; West, R.R.; Russell, D.W.; Hickstein, D.D. Long-term follow-up of foamy viral vector-mediated gene therapy for canine leukocyte adhesion deficiency. Mol. Ther. 2013, 21, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Nowrouzi, A.; Dittrich, M.; Klanke, C.; Heinkelein, M.; Rammling, M.; Dandekar, T.; von Kalle, C.; Rethwilm, A. Genome-wide mapping of foamy virus vector integrations into a human cell line. J. Gen. Virol. 2006, 87, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Olszko, M.E.; Adair, J.E.; Linde, I.; Rae, D.T.; Trobridge, P.; Hocum, J.D.; Rawlings, D.J.; Kiem, H.P.; Trobridge, G.D. Foamy viral vector integration sites in SCID-repopulating cells after MGMTP140K-mediated in vivo selection. Gene Ther. 2015, 22, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Nadeau, P.E.; Mergia, A. Activity of TAR in inducible inhibition of HIV replication by foamy virus vector expressing siRNAs under the control of HIV LTR. Virus Res. 2009, 140, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Nadeau, P.; Zucali, J.R.; Johnson, C.M.; Mergia, A. Inhibition of simian immunodeficiency virus by foamy virus vectors expressing siRNAs. Virology 2005, 343, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Mühle, M.; Hoffmann, K.; Löchelt, M.; Denner, J. Immunisation with foamy virus bet fusion proteins as novel strategy for HIV-1 epitope delivery. Immunol. Res. 2013, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Osen, W.; Gardyan, A.; Hotz-Wagenblatt, A.; Wei, G.; Gissmann, L.; Eichmüller, S.; Löchelt, M. Replication-competent foamy virus vaccine vectors as novel epitope scaffolds for immunotherapy. PLoS ONE 2015, 10, e0138458. [Google Scholar] [CrossRef] [PubMed]
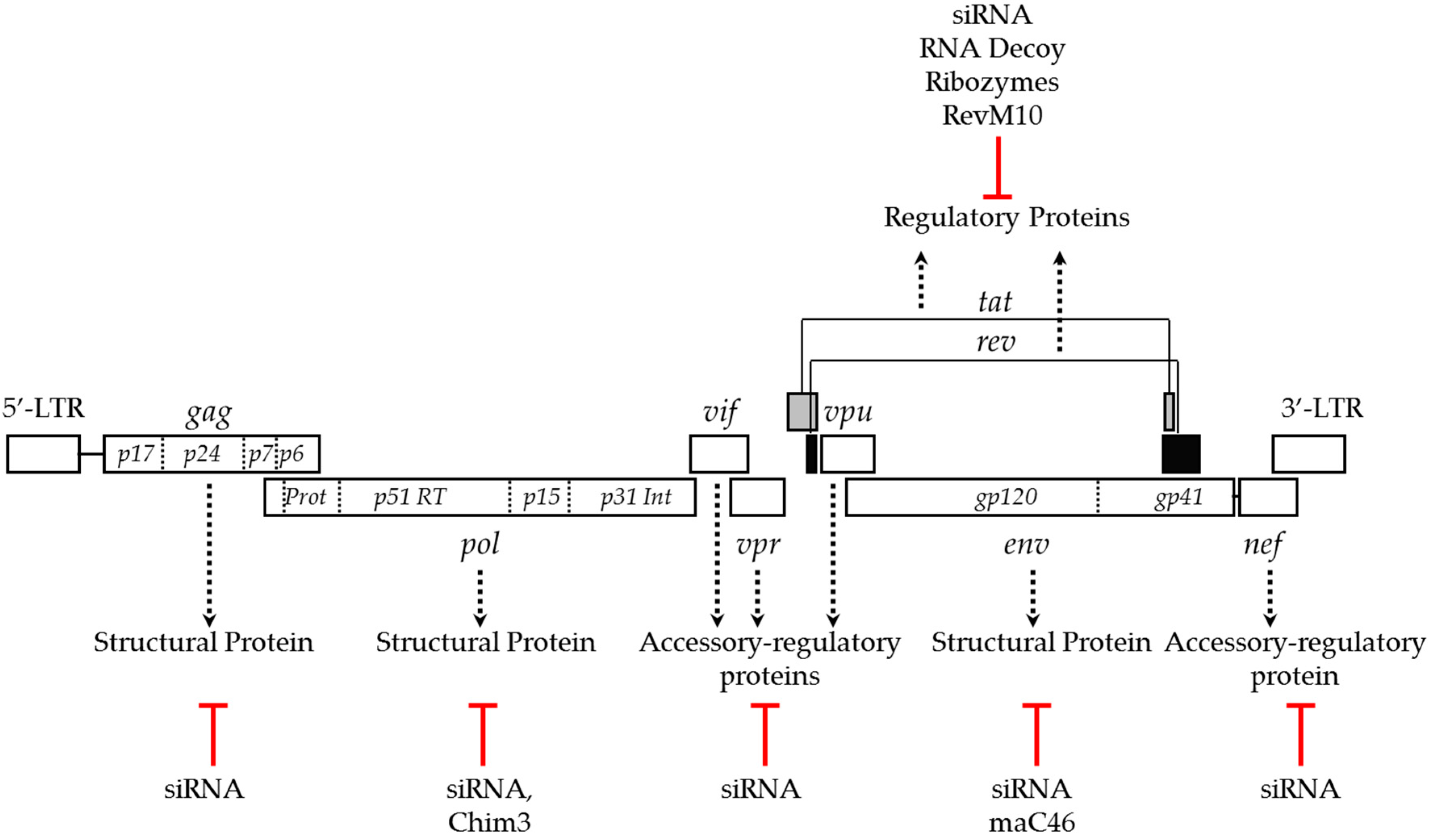

{kind=link}
{kind=link}
{kind=link}
| Target Type | Gene | Function Related to HIV Life Cycle | Interacting Stage | Inhibition Strategies | Inhibitory Class | References |
|---|---|---|---|---|---|---|
| Host | CCR5 | Entry coreceptor | Entry | siRNA, Ribozymes, Gene-editing, Intrabodies | I | [31,32,33,34,35,36,37,38,39,40,41,42,43,44] |
| CXCR4 | Entry coreceptor | Entry | siRNA, Gene-editing | I | [45,46,47] | |
| TRIM5 | Host restriction factor | Uncoating | Protein expression | I | [48,49] | |
| LEDGF | Interacts with viral integrase and transport pre-integration complex into nucleus | Nuclear import, Pre-integration | siRNA, Expression of LEDGF-IBD | I | [50,51,52] | |
| Atg-5 & Atg-16 | Autophage related | Entry or early RT | siRNA | I | [53,54] | |
| CPSF6 | Host cofactor bind to viral capsid protein | Nuclear import | expression of CPSF6-358 | I | [55,56] | |
| MX2 | Interferon stimulated protein | Uncoating, nuclear import | Protein expression | I | [57,58] | |
| Viral | maC46 | Membrane anchored protein-fusion interferes with gp41 and block viral fusion to cell membrane | Entry | Protein expression | I | [23,27,29,59,60] |
| RevM10 | Dominant negative mutant competes with viral rev | Post-integration | Protein expression | II | [61,62] | |
| rev | Translocation of viral transcripts from nucleus to cytoplasm | Post-integration | siRNA, Ribozymes, RNA decoy, CRISPR/Cas | II | [61,62,63,64,65,66,67,68,69] | |
| tat | Viral transcription regulatory protein | Post-integration | siRNA, Ribozymes, RNA decoy, CRISPR/Cas | II | [61,62,63,64,65,66,67,68,69] | |
| gag-pol | Viral enzymes: reverse transcriptase, integrase, RNaseH and protease | Reverse transcription and pre-integration and assembly | siRNA, CRISPR/Cas | I | [69,70,71,72,73,74,75,76] | |
| nef | Accessory protein, modulate CD4 expression and stimulate HIV infectivity | Assembly, Budding | siRNA CRISPR/Cas | III | [69,76,77] | |
| Chim3 | Derivative of a mutant viral infectivity factor, (F12-Vif) | Pre-integration | Protein expression | I | [78,79] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nalla, A.K.; Trobridge, G.D. Prospects for Foamy Viral Vector Anti-HIV Gene Therapy. Biomedicines 2016, 4, 8. https://doi.org/10.3390/biomedicines4020008
Nalla AK, Trobridge GD. Prospects for Foamy Viral Vector Anti-HIV Gene Therapy. Biomedicines. 2016; 4(2):8. https://doi.org/10.3390/biomedicines4020008
Chicago/Turabian StyleNalla, Arun K., and Grant D. Trobridge. 2016. "Prospects for Foamy Viral Vector Anti-HIV Gene Therapy" Biomedicines 4, no. 2: 8. https://doi.org/10.3390/biomedicines4020008
APA StyleNalla, A. K., & Trobridge, G. D. (2016). Prospects for Foamy Viral Vector Anti-HIV Gene Therapy. Biomedicines, 4(2), 8. https://doi.org/10.3390/biomedicines4020008