
Recent Innovations in Peptide Based Targeted Drug Delivery to Cancer Cells


Abstract
:
1. Introduction
2. Strategies for Cancer Cell Targeting Peptides Discovery
2.1. Phage Display
2.2. Synthetic Peptide Libraries—OBOC
2.3. SPOT-Synthesis
2.4. Rationally Designed Peptides
3. Targeted Delivery of Chemotherapeutics Based on Clinically Investigated Peptides: Arginine-Glycine-Aspartic Acid (RGD), Somatostatin, Luteinizing Hormone-Releasing Hormone (LHRH) and Bombesin
3.1. RGD
3.2. LHRH
3.3. Somatostatin
3.4. Bombesin
3.5. Angiopeptin-2
4. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References
- Marchetti, C.; Palaia, I.; Giorgini, M.; De Medici, C.; Iadarola, R.; Vertechy, L.; Domenici, L.; Di Donato, V.; Tomao, F.; Muzii, L.; et al. Targeted drug delivery via folate receptors in recurrent ovarian cancer: A review. OncoTargets Ther. 2014, 7, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Nagy, A. Chemotherapy targeted to cancers through tumoral hormone receptors. Trends Endocrinol. Metab. 2004, 15, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Firer, M.A.; Gellerman, G. Targeted drug delivery for cancer therapy: The other side of antibodies. J. Hematol. Oncol. 2012, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, J.W.; Kay, B.K. Filamentous phage display in the new millennium. Chem. Rev. 2005, 105, 4056–4072. [Google Scholar] [CrossRef] [PubMed]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Babickova, J.; Tothova, L.; Boor, P.; Celec, P. In vivo phage display—A discovery tool in molecular biomedicine. Biotechnol. Adv. 2013, 31, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Kathlynn, C.B. Peptidic tumor targeting agents: The road from phage display peptide selections to clinical applications. Curr. Pharm. Des. 2010, 16, 1040–1054. [Google Scholar]
- Molek, P.; Strukelj, B.; Bratkovic, T. Peptide phage display as a tool for drug discovery: Targeting membrane receptors. Molecules 2011, 16, 857–887. [Google Scholar] [CrossRef] [PubMed]
- Matochko, W.L.; Chu, K.; Jin, B.; Lee, S.W.; Whitesides, G.M.; Derda, R. Deep sequencing analysis of phage libraries using illumina platform. Methods 2012, 58, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Geysen, H.M.; Meloen, R.H.; Barteling, S.J. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc. Natl. Acad. Sci. USA 1984, 81, 3998–4002. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Beck-Sickinger, A.G. Multiple peptide synthesis methods and their applications. New synthetic methods (87). Angew. Chem. Int. Ed. Engl. 1992, 31, 367–383. [Google Scholar] [CrossRef]
- Maeji, N.J.; Bray, A.M.; Geysen, H.M. Multi-pin peptide synthesis strategy for T cell determinant analysis. J. Immunol. Methods 1990, 134, 23–33. [Google Scholar] [CrossRef]
- Frank, R. The spot-synthesis technique: Synthetic peptide arrays on membrane supports—Principles and applications. J. Immunol. Methods 2002, 267, 13–26. [Google Scholar] [CrossRef]
- Lam, K.S.; Lebl, M.; Krchňák, V. The “one-bead-one-compound” combinatorial library method. Chem. Rev. 1997, 97, 411–448. [Google Scholar] [CrossRef] [PubMed]
- Tribbick, G. Multipin peptide libraries for antibody and receptor epitope screening and characterization. J. Immunol. Methods 2002, 267, 27–35. [Google Scholar] [CrossRef]
- Fleeman, R.; LaVoi, T.M.; Santos, R.G.; Morales, A.; Nefzi, A.; Welmaker, G.S.; Medina-Franco, J.L.; Giulianotti, M.A.; Houghten, R.A.; Shaw, L.N. Combinatorial libraries as a tool for the discovery of novel, broad-spectrum antibacterial agents targeting the eskape pathogens. J. Med. Chem. 2015, 58, 3340–3355. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Hirsch, A.K. Dynamic combinatorial chemistry: A tool to facilitate the identification of inhibitors for protein targets. Chem. Soc. Rev. 2015, 44, 2455–2488. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S. Combinatorial approaches in anticancer drug discovery: Recent advances in design and synthesis. Curr. Med. Chem. 2001, 8, 1383–1404. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J. Synthetic peptide arrays and peptide combinatorial libraries for the exploration of protein-ligand interactions and the design of protein inhibitors. Comb. Chem. High Throughput Screen. 2005, 8, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Enstrom, A.M.; Lam, K.S. Combinatorial peptide library methods for immunobiology research. Exp. Hematol. 2003, 31, 11–30. [Google Scholar] [CrossRef]
- Gray, B.P.; Brown, K.C. Combinatorial peptide libraries: Mining for cell-binding peptides. Chem. Rev. 2014, 114, 1020–1081. [Google Scholar] [CrossRef] [PubMed]
- Aina, O.H.; Liu, R.; Sutcliffe, J.L.; Marik, J.; Pan, C.-X.; Lam, K.S. From combinatorial chemistry to cancer-targeting peptides. Mol. Pharm. 2007, 4, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Marani, M.M.; Martínez Ceron, M.C.; Giudicessi, S.L.; de Oliveira, E.; Côté, S.; Erra-Balsells, R.; Albericio, F.; Cascone, O.; Camperi, S.A. Screening of one-bead-one-peptide combinatorial library using red fluorescent dyes. Presence of positive and false positive beads. J. Comb. Chem. 2009, 11, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Svensen, N.; Diaz-Mochon, J.J.; Dhaliwal, K.; Planonth, S.; Dewar, M.; Armstrong, J.D.; Bradley, M. Screening of a combinatorial homing peptide library for selective cellular delivery. Angew. Chem. Int. Ed. 2011, 50, 6133–6136. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Egholm, M.; Buchardt, O.; Nielsen, P.E.; Berg, R.H. Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral peptide backbone. J. Am. Chem. Soc. 1992, 114, 1895–1897. [Google Scholar] [CrossRef]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskil, M.D.; Egholm, M.; Buchard, O.; Sönnichsen, S.H.; Nlelsen, P.E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [Google Scholar] [CrossRef]
- Egholm, M.; Behrens, C.; Christensen, L.; Berg, R.H.; Nielsen, P.E.; Buchardt, O. Peptide nucleic acids containing adenine or guanine recognize thymine and cytosine in complementary DNA sequences. J. Chem. Soc. Chem. Commun. 1993, 800–801. [Google Scholar] [CrossRef]
- Egholm, M.; Nielsen, P.E.; Buchardt, O.; Berg, R.H. Recognition of guanine and adenine in DNA by cytosine and thymine containing peptide nucleic acids (PNA). J. Am. Chem. Soc. 1992, 114, 9677–9678. [Google Scholar] [CrossRef]
- Frank, R. Spot-synthesis: An easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 1992, 48, 9217–9232. [Google Scholar] [CrossRef]
- Hilpert, K.; Winkler, D.F.H.; Hancock, R.E.W. Peptide arrays on cellulose support: Spot synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat. Protoc. 2007, 2, 1333–1349. [Google Scholar] [CrossRef] [PubMed]
- Reineke, U.; Volkmer-Engert, R.; Schneider-Mergener, J. Applications of peptide arrays prepared by the spot-technology. Curr. Opin. Biotechnol. 2001, 12, 59–64. [Google Scholar] [CrossRef]
- Frank, R.; Overwin, H. Spot Synthesis. In Epitope Mapping Protocols; Morris, G.E., Ed.; Humana Press: Totowa, NJ, USA, 1996; pp. 149–169. [Google Scholar]
- Ast, T.; Heine, N.; Germeroth, L.; Schneider-Mergener, J.; Wenschuh, H. Efficient assembly of peptomers on continuous surfaces. Tetrahedron Lett. 1999, 40, 4317–4318. [Google Scholar] [CrossRef]
- Weiler, J.; Gausepohl, H.; Hauser, N.; Jensen, O.N.; Hoheisel, J.D. Hybridisation based DNA screening on peptide nucleic acid (PNA) oligomer arrays. Nucleic Acids Res. 1997, 25, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Scharn, D.; Wenschuh, H.; Reineke, U.; Schneider-Mergener, J.; Germeroth, L. Spatially addressed synthesis of amino- and amino-oxy-substituted 1,3,5-triazine arrays on polymeric membranes. J. Comb. Chem. 2000, 2, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Zander, N. New planar substrates for the in situ synthesis of peptide arrays. Mol. Divers. 2004, 8, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J.; Bienert, M.; Stierandova, A.; Lebl, M. Evaluation of cotton as a carrier for solid-phase peptide synthesis. Pept. Res. 1991, 4, 296–307. [Google Scholar] [PubMed]
- Wang, Z.; Laursen, R.A. Multiple peptide synthesis on polypropylene membranes for rapid screening of bioactive peptides. Pept. Res. 1992, 5, 275–280. [Google Scholar] [PubMed]
- Volkmer-Engert, R.; Hoffmann, B.; Schneider-Mergener, J. Stable attachment of the hmb-linker to continuous cellulose membranes for parallel solid phase spot synthesis. Tetrahedron Lett. 1997, 38, 1029–1032. [Google Scholar] [CrossRef]
- Licha, K.; Bhargava, S.; Rheinländer, C.; Becker, A.; Schneider-Mergener, J.; Volkmer-Engert, R. Highly parallel nano-synthesis of cleavable peptide-dye conjugates on cellulose membranes. Tetrahedron Lett. 2000, 41, 1711–1715. [Google Scholar] [CrossRef]
- Frank, R. Simultaneous and combinatorial chemical synthesis techniques for the generation and screening of molecular diversity. J. Biotechnol. 1995, 41, 259–272. [Google Scholar] [CrossRef]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Powell, M.F.; Stewart, T.; Otvos, L., Jr.; Urge, L.; Gaeta, F.C.A.; Sette, A.; Arrhenius, T.; Thomson, D.; Soda, K.; Colon, S.M. Peptide stability in drug development. II. Effect of single amino acid substitution and glycosylation on peptide reactivity in human serum. Pharm. Res. 1993, 10, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.G. Somatostatin and somatostatin analogs: Pharmacokinetics and pharmacodynamic effects. Gut 1994, 35, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Vanhee, P.; van der Sloot, A.M.; Verschueren, E.; Serrano, L.; Rousseau, F.; Schymkowitz, J. Computational design of peptide ligands. Trends Biotechnol. 2011, 29, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ballester, G.; Beltrao, P.; Gonzalez, J.M.; Song, Y.-H.; Wilmanns, M.; Valencia, A.; Serrano, L. Structure-based prediction of the saccharomyces cerevisiae SH3–ligand interactions. J. Mol. Biol. 2009, 388, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Kiel, C.; Wohlgemuth, S.; Rousseau, F.; Schymkowitz, J.; Ferkinghoff-Borg, J.; Wittinghofer, F.; Serrano, L. Recognizing and defining true ras binding domains II: In silico prediction based on homology modelling and energy calculations. J. Mol. Biol. 2005, 348, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulos, D.; Poulos, C.; Gatos, D.; Cordopatis, P.; Escher, E.; Mizrahi, J.; Regoli, D.; Dalietos, D.; Furst, A.; Lee, T.D. Conformationally restricted C-terminal peptides of substance P. Synthesis, mass spectral analysis and pharmacological properties. J. Med. Chem. 1985, 28, 1536–1539. [Google Scholar] [CrossRef] [PubMed]
- Darman, P.S.; Landis, G.C.; Smits, J.R.; Hirning, L.D.; Gulya, K.; Yamamura, H.I.; Burks, T.F.; Hruby, V.J. Conformationally restricted cyclic analogues of substance P: Insight into the receptor binding process. Biochem. Biophys. Res. Commun. 1985, 127, 656–662. [Google Scholar] [CrossRef]
- Gazal, S.; Gelerman, G.; Ziv, O.; Karpov, O.; Litman, P.; Bracha, M.; Afargan, M.; Gilon, C. Human somatostatin receptor specificity of backbone-cyclic analogues containing novel sulfur building units. J. Med. Chem. 2002, 45, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Gazal, S.; Gellerman, G.; Glukhov, E.; Gilon, C. Synthesis of novel protected Nα(ω-thioalkyl) amino acid building units and their incorporation in backbone cyclic disulfide and thioetheric bridged peptides. J. Pept. Res. Off. J. Am. Pept. Soc. 2001, 58, 527–539. [Google Scholar] [CrossRef]
- Jiang, S.; Gazal, S.; Gelerman, G.; Ziv, O.; Karpov, O.; Litman, P.; Bracha, M.; Afargan, M.; Gilon, C.; Goodman, M. A bioactive somatostatin analog without a type ii' beta-turn: Synthesis and conformational analysis in solution. J. Pept. Sci. 2001, 7, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Gazal, S.; Gellerman, G.; Gilon, C. Novel gly building units for backbone cyclization: Synthesis and incorporation into model peptides. Peptides 2003, 24, 1847–1852. [Google Scholar] [CrossRef] [PubMed]
- Gellerman, G.; Elgavi, A.; Salitra, Y.; Kramer, M. Facile synthesis of orthogonally protected amino acid building blocks for combinatorial n-backbone cyclic peptide chemistry. J. Pept. Res. 2001, 57, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Dooley, C.T.; Houghten, R.A. The use of positional scanning synthetic peptide combinatorial libraries for the rapid determination of opioid receptor ligands. Life Sci. 1993, 52, 1509–1517. [Google Scholar] [CrossRef]
- Bionda, N.; Fleeman, R.M.; de la Fuente-Núñez, C.; Rodriguez, M.C.; Reffuveille, F.; Shaw, L.N.; Pastar, I.; Davis, S.C.; Hancock, R.E.W.; Cudic, P. Identification of novel cyclic lipopeptides from a positional scanning combinatorial library with enhanced antibacterial and antibiofilm activities. Eur. J. Med. Chem. 2016, 108, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Variants of the cell recognition site of fibronectin that retain attachment-promoting activity. Proc. Natl. Acad. Sci. USA 1984, 81, 5985–5988. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Kennedy, D.W. Dualistic nature of adhesive protein function: Fibronectin and its biologically active peptide fragments can autoinhibit fibronectin function. J. Cell Biol. 1984, 99, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Gartner, T.K.; Bennett, J.S. The tetrapeptide analogue of the cell attachment site of fibronectin inhibits platelet aggregation and fibrinogen binding to activated platelets. J. Biol. Chem. 1985, 260, 11891–11894. [Google Scholar] [PubMed]
- Plow, E.F.; Pierschbacher, M.D.; Ruoslahti, E.; Marguerie, G.A.; Ginsberg, M.H. The effect of arg-gly-asp-containing peptides on fibrinogen and von willebrand factor binding to platelets. Proc. Natl. Acad. Sci. USA 1985, 82, 8057–8061. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Oldberg, A.; Hayman, E.G.; Pierschbacher, M.D.; Ruoslahti, E. Complete amino acid sequence of human vitronectin deduced from cdna. Similarity of cell attachment sites in vitronectin and fibronectin. EMBO J. 1985, 4, 2519–2524. [Google Scholar] [PubMed]
- Gardner, J.M.; Hynes, R.O. Interaction of fibronectin with its receptor on platelets. Cell 1985, 42, 439–448. [Google Scholar] [CrossRef]
- Damsky, C.H.; Wylie, D.E.; Buck, C.A. Studies on the function of cell surface glycoproteins. II. Possible role of surface glycoproteins in the control of cytoskeletal organization and surface morphology. J. Cell Biol. 1979, 80, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.A.; Horwitz, A.F.; Buck, C.A. A monoclonal antibody identifies a glycoprotein complex involved in cell-substratum adhesion. Exp. Cell Res. 1985, 157, 218–226. [Google Scholar] [CrossRef]
- Neff, N.T.; Lowrey, C.; Decker, C.; Tovar, A.; Damsky, C.; Buck, C.; Horwitz, A.F. A monoclonal antibody detaches embryonic skeletal muscle from extracellular matrices. J. Cell Biol. 1982, 95, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Greve, J.M.; Gottlieb, D.I. Monoclonal antibodies which alter the morphology of cultured chick myogenic cells. J. Cell. Biochem. 1982, 18, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Tarone, G.; Galetto, G.; Prat, M.; Comoglio, P.M. Cell surface molecules and fibronectin-mediated cell adhesion: Effect of proteolytic digestion of membrane proteins. J. Cell Biol. 1982, 94, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Juliano, R.L. Selective inhibition of fibronectin-mediated cell adhesion by monoclonal antibodies to a cell-surface glycoprotein. Science 1985, 228, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.E. Characterization of a 140Kd cell surface glycoprotein involved in myoblast adhesion. J. Cell. Biochem. 1984, 25, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Comoglio, P.M.; Tarone, G. Fibronectin-plasma membrane interaction in the adhesion of hemopoietic cells. J. Cell Biol. 1986, 103, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Pytela, R.; Pierschbacher, M.D.; Ruoslahti, E. Identification and isolation of a 140 kd cell surface glycoprotein with properties expected of a fibronectin receptor. Cell 1985, 40, 191–198. [Google Scholar] [CrossRef]
- Tamkun, J.W.; DeSimone, D.W.; Fonda, D.; Patel, R.S.; Buck, C.; Horwitz, A.F.; Hynes, R.O. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 1986, 46, 271–282. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Schwartz, M.A.; Schaller, M.D.; Ginsberg, M.H. Integrins: Emerging paradigms of signal transduction. Annu. Rev. Cell Dev. Biol. 1995, 11, 549–599. [Google Scholar] [CrossRef] [PubMed]
- Taverna, D.; Crowley, D.; Connolly, M.; Bronson, R.T.; Hynes, R.O. A direct test of potential roles for β3 and β5 integrins in growth and metastasis of murine mammary carcinomas. Cancer Res. 2005, 65, 10324–10329. [Google Scholar] [CrossRef] [PubMed]
- Danen, E.H.J.; van Kraats, A.A.; Cornelissen, I.M.H.A.; Ruiter, D.J.; van Muijen, G.N.P. Integrin β3 cDNA transfection into a highly metastatic αvβ3-negative human melanoma cell line inhibits invasion and experimental metastasis. Biochem. Biophys. Res. Commun. 1996, 226, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Ishii, S.; Ikeda, T.; Masamura, S.; Doi, M.; Kitajima, M. The relationship between bone metastasis from human breast cancer and integrin αvβ3 expression. Anticancer Res. 2005, 25, 79–83. [Google Scholar] [PubMed]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Vellon, L.; Menendez, J.A.; Lupu, R. αvβ3 integrin regulates heregulin (HRG)-induced cell proliferation and survival in breast cancer. Oncogene 2005, 24, 3759–3773. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. Src Phosphorylates Tyr284 in TGF-β Type II Receptor and Regulates TGF-β Stimulation of p38 MAPK during Breast Cancer Cell Proliferation and Invasion. Canser Res. 2007, 67, 3752–3758. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Manning, C.; Millar, H.; McCabe, F.; Ferrante, C.; Sharp, C.; Shahied-Arruda, L.; Doshi, P.; Nakada, M.; Anderson, G.M. Cnto 95, a fully human anti αv integrin antibody, inhibits cell signaling, migration, invasion, and spontaneous metastasis of human breast cancer cells. Clin. Exp. Metastasis 2008, 25, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Felding-Habermann, B.; Habermann, R.; Saldívar, E.; Ruggeri, Z.M. Role of 3 integrins in melanoma cell adhesion to activated platelets under flow. J. Biol. Chem. 1996, 271, 5892–5900. [Google Scholar] [CrossRef] [PubMed]
- White, D.P.; Caswell, P.T.; Norman, J.C. Avβ3 and α5β1 integrin recycling pathways dictate downstream rho kinase signaling to regulate persistent cell migration. J. Cell Biol. 2007, 177, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Albelda, S.M.; Mette, S.A.; Elder, D.E.; Stewart, R.; Damjanovich, L.; Herlyn, M.; Buck, C.A. Integrin distribution in malignant melanoma: Association of the β3 subunit with tumor progression. Cancer Res. 1990, 50, 6757–6764. [Google Scholar] [PubMed]
- Seftor, R.E.; Seftor, E.A.; Gehlsen, K.R.; Stetler-Stevenson, W.G.; Brown, P.D.; Ruoslahti, E.; Hendrix, M.J. Role of the alpha v beta 3 integrin in human melanoma cell invasion. Proc. Natl. Acad. Sci. USA 1992, 89, 1557–1561. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Brennan, M.; Moran, N. Integrins as therapeutic targets: Lessons and opportunities. Nat. Rev. Drug Discov. 2010, 9, 804–820. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, X. Integrin targeted delivery of chemotherapeutics. Theranostics 2011, 1, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, D.J.; Kalet, B.T.; Coleman, M.P.; Post, G.C.; Koch, T.H. Doxorubicin-formaldehyde conjugates targeting αvβ3 integrin. Mol. Cancer Ther. 2004, 3, 1593–1604. [Google Scholar] [PubMed]
- Chen, X.; Plasencia, C.; Hou, Y.; Neamati, N. Synthesis and biological evaluation of dimeric RGD peptide–paclitaxel conjugate as a model for integrin-targeted drug delivery. J. Med. Chem. 2005, 48, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Vroman, B.; Lecouturier, N.; Crokart, N.; Pourcelle, V.; Freichels, H.; Jérôme, C.; Marchand-Brynaert, J.; Feron, O.; Préat, V. Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles loaded with paclitaxel. J. Control. Release 2009, 140, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hou, Y.; Tohme, M.; Park, R.; Khankaldyyan, V.; Gonzales-Gomez, I.; Bading, J.R.; Laug, W.E.; Conti, P.S. Pegylated Arg-Gly-Asp peptide: 64Cu labeling and pet imaging of brain tumor αvβ3-integrin expression. J. Nucl. Med. 2004, 45, 1776–1783. [Google Scholar] [PubMed]
- Wu, Y.; Zhang, X.; Xiong, Z.; Cheng, Z.; Fisher, D.R.; Liu, S.; Gambhir, S.S.; Chen, X. Micropet imaging of glioma integrin αvβ3 expression using 64cu-labeled tetrameric rgd peptide. J. Nucl. Med. 2005, 46, 1707–1718. [Google Scholar] [PubMed]
- Marinelli, L.; Lavecchia, A.; Gottschalk, K.-E.; Novellino, E.; Kessler, H. Docking studies on αvβ3 integrin ligands: Pharmacophore refinement and implications for drug design. J. Med. Chem. 2003, 46, 4393–4404. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.-P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Goldshaid, L.; Rubinstein, E.; Brandis, A.; Segal, D.; Leshem, N.; Brenner, O.; Kalchenko, V.; Eren, D.; Yecheskel, T.; Salitra, Y.; et al. Novel design principles enable specific targeting of imaging and therapeutic agents to necrotic domains in breast tumors. Breast Cancer Res. 2010, 12, R29. [Google Scholar] [CrossRef] [PubMed]
- Hölig, P.; Bach, M.; Völkel, T.; Nahde, T.; Hoffmann, S.; Müller, R.; Kontermann, R.E. Novel RGD lipopeptides for the targeting of liposomes to integrin-expressing endothelial and melanoma cells. Protein Eng. Des. Sel. 2004, 17, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Jonczyk, A.; Goodman, S.; Diefenbach, B.; Sutter, A.; Holzemann, G.; Kessler, H.; Dechantsreiter, M. Cyclic Adhesion Inhibitors. U.S. Patent US5866540 A, 2 February 1999. [Google Scholar]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. n-methylated cyclic RGD peptides as highly active and selective αvβ3 integrin antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Tabatabai, G.; Weller, M.; Nabors, B.; Picard, M.; Reardon, D.; Mikkelsen, T.; Ruegg, C.; Stupp, R. Targeting integrins in malignant glioma. Target. Oncol. 2010, 5, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Gratias, R.; Diefenbach, B.; Goodman, S.L.; Jonczyk, A.; Kessler, H. Structural and functional aspects of RGD-containing cyclic pentapeptides as highly potent and selective integrin αvβ3 antagonists. J. Am. Chem. Soc. 1996, 118, 7461–7472. [Google Scholar] [CrossRef]
- Gilad, Y.; Noy, E.; Senderowitz, H.; Albeck, A.; Firer, M.A.; Gellerman, G. Synthesis, biological studies and molecular dynamics of new anticancer rgd-based peptide conjugates for targeted drug delivery. Bioorg. Med. Chem. 2016, 24, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Haft, D.H.; Getzoff, E.D.; Tainer, J.A.; Lerner, R.A.; Brenner, S. Identical short peptide sequences in unrelated proteins can have different conformations: A testing ground for theories of immune recognition. Proc. Natl. Acad. Sci. USA 1985, 82, 5255–5259. [Google Scholar] [CrossRef] [PubMed]
- Kessler, H. Conformation and biological activity of cyclic peptides. Angew. Chem. Int. Ed. 1982, 21, 512–523. [Google Scholar] [CrossRef]
- Gilad, Y.; Noy, E.; Senderowitz, H.; Albeck, A.; Firer, M.A.; Gellerman, G. Dual-drug rgd conjugates provide enhanced cytotoxicity to melanoma and non-small lung cancer cells. Biopolymers 2015. [Google Scholar] [CrossRef] [PubMed]
- Gellerman, G.; Baskin, S.; Galia, L.; Gilad, Y.; Firer, M.A. Drug resistance to chlorambucil in murine B-cell leukemic cells is overcome by its conjugation to a targeting peptide. Anti-Cancer Drugs 2013, 24, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Comaru-Schally, A.M.; Plonowski, A.; Nagy, A.; Halmos, G.; Rekasi, Z. Peptide analogs in the therapy of prostate cancer. Prostate 2000, 45, 158–166. [Google Scholar] [CrossRef]
- Miller, W.R.; Scott, W.N.; Morris, R.; Fraser, H.M.; Sharpe, R.M. Growth of human breast cancer cells inhibited by a luteinizing hormone-releasing hormone agonist. Nature 1985, 313, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Szepeshazi, K.; Schally, A.V.; Nagy, A. Effective treatment of advanced estrogen-independent MXT mouse mammary cancers with targeted cytotoxic LH-RH analogs. Breast Cancer Res. Treat. 1999, 56, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Bajo, A.M.; Schally, A.V.; Halmos, G.; Nagy, A. Targeted doxorubicin-containing luteinizing hormone-releasing hormone analogue AN-152 inhibits the growth of doxorubicin-resistant MX-1 human breast cancers. Clin. Cancer Res. 2003, 9, 3742–3748. [Google Scholar] [PubMed]
- Kahan, Z.; Nagy, A.; Schally, A.V.; Halmos, G.; Arencibia, J.M.; Groot, K. Complete regression of MX-1 human breast carcinoma xenografts after targeted chemotherapy with a cytotoxic analog of luteinizing hormone-releasing hormone, AN-207. Cancer 1999, 85, 2608–2615. [Google Scholar] [CrossRef]
- Letsch, M.; Schally, A.V.; Szepeshazi, K.; Halmos, G.; Nagy, A. Preclinical evaluation of targeted cytotoxic luteinizing hormone-releasing hormone analogue AN-152 in androgen-sensitive and insensitive prostate cancers. Clin. Cancer Res. 2003, 9, 4505–4513. [Google Scholar] [PubMed]
- Plonowski, A.; Schally, A.V.; Nagy, A.; Groot, K.; Krupa, M.; Navone, N.M.; Logothetis, C. Inhibition of in vivo proliferation of MDA-PCa-2b human prostate cancer by a targeted cytotoxic analog of luteinizing hormone-releasing hormone AN-207. Cancer Lett. 2002, 176, 57–63. [Google Scholar] [CrossRef]
- Grundker, C.; Volker, P.; Griesinger, F.; Ramaswamy, A.; Nagy, A.; Schally, A.V.; Emons, G. Antitumor effects of the cytotoxic luteinizing hormone-releasing hormone analog AN-152 on human endometrial and ovarian cancers xenografted into nude mice. Am. J. Obstet. Gynecol. 2002, 187, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Nagy, A.; Schally, A.V.; Lamharzi, N.; Halmos, G.; Szepeshazi, K.; Groot, K.; Armatis, P. Growth inhibition of human ovarian cancers by cytotoxic analogues of luteinizing hormone-releasing hormone. J. Natl. Cancer Inst. 1997, 89, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Schally, A.V.; Nagy, A.; Lamharzi, N.; Halmos, G.; Szepeshazi, K.; Armatis, P. Targeted cytotoxic analog of luteinizing hormone-releasing hormone AN-207 inhibits growth of OV-1063 human epithelial ovarian cancers in nude mice. Am. J. Obstet. Gynecol. 1999, 180, 1095–1103. [Google Scholar] [CrossRef]
- Arencibia, J.M.; Bajo, A.M.; Schally, A.V.; Krupa, M.; Chatzistamou, I.; Nagy, A. Effective treatment of experimental ES-2 human ovarian cancers with a cytotoxic analog of luteinizing hormone-releasing hormone AN-207. Anticancer Drugs 2002, 13, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Koppan, M.; Nagy, A.; Schally, A.V.; Plonowski, A.; Halmos, G.; Arencibia, J.M.; Groot, K. Targeted cytotoxic analog of luteinizing hormone-releasing hormone AN-207 inhibits the growth of PC-82 human prostate cancer in nude mice. Prostate 1999, 38, 151–158. [Google Scholar] [CrossRef]
- Nagy, A.; Schally, A.V.; Armatis, P.; Szepeshazi, K.; Halmos, G.; Kovacs, M.; Zarandi, M.; Groot, K.; Miyazaki, M.; Jungwirth, A.; et al. Cytotoxic analogs of luteinizing hormone-releasing hormone containing doxorubicin or 2-pyrrolinodoxorubicin, a derivative 500–1000 times more potent. Proc. Natl. Acad. Sci. USA 1996, 93, 7269–7273. [Google Scholar] [CrossRef] [PubMed]
- Chatzistamou, L.; Schally, A.V.; Nagy, A.; Armatis, P.; Szepeshazi, K.; Halmos, G. Effective treatment of metastatic MDA-MB-435 human estrogen-independent breast carcinomas with a targeted cytotoxic analogue of luteinizing hormone-releasing hormone AN-207. Clin. Cancer Res. 2000, 6, 4158–4165. [Google Scholar] [PubMed]
- Kahan, Z.; Nagy, A.; Schally, A.V.; Halmos, G.; Arencibia, J.M.; Groot, K. Administration of a targeted cytotoxic analog of luteinizing hormone-releasing hormone inhibits growth of estrogen-independent MDA-MB-231 human breast cancers in nude mice. Breast Cancer Res. Treat. 2000, 59, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Emons, G.; Pinski, J.; Schally, A.V. AEZS-108: A targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin. Investig. Drugs 2012, 21, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Hohla, F.; Winder, T.; Greil, R.; Rick, F.G.; Block, N.L.; Schally, A.V. Targeted therapy in advanced metastatic colorectal cancer: Current concepts and perspectives. World J. Gastroenterol. 2014, 20, 6102–6112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, S.V.; Tsao-Wei, D.D.; Xiong, S.; Groshen, S.; Dorff, T.B.; Quinn, D.I.; Tai, Y.-C.; Engel, J.; Hawes, D.; Schally, A.V.; et al. Phase I, dose-escalation study of the targeted cytotoxic LHRH analog AEZS-108 in patients with castration- and taxane-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 6277–6283. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, C.; Enright, F.M.; Gawronska–Kozak, B.; Hansel, W. Human prostate cancer cells and xenografts are targeted and destroyed through luteinizing hormone releasing hormone receptors. Prostate 2003, 56, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Schlick, J.-L.; Dulieu, P.; Desvoyes, B.; Adami, P.; Radom, J.; Jouvenot, M. Cytotoxic activity of a recombinant GnRH-PAP fusion toxin on human tumor cell lines. FEBS Lett. 2000, 472, 241–246. [Google Scholar] [CrossRef]
- Dharap, S.S.; Qiu, B.; Williams, G.C.; Sinko, P.; Stein, S.; Minko, T. Molecular targeting of drug delivery systems to ovarian cancer by BH3 and LHRH peptides. J. Control. Release 2003, 91, 61–73. [Google Scholar] [CrossRef]
- Dharap, S.S.; Wang, Y.; Chandna, P.; Khandare, J.J.; Qiu, B.; Gunaseelan, S.; Sinko, P.J.; Stein, S.; Farmanfarmaian, A.; Minko, T. Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc Natl Acad Sci USA 2005, 102, 12962–12967. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.C. Somatostatin and its receptor family. Front. Neuroendocrinol. 1999, 20, 157–198. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yuan, X.M.; Lei, P.; Wu, S.; Xing, W.; Lan, X.L.; Zhu, H.F.; Huang, T.; Wang, G.B.; An, R.; et al. The antiproliferative effects of somatostatin receptor subtype 2 in breast cancer cells. Acta Pharmacol. Sin. 2009, 30, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Herlin, G.; Kolbeck, K.G.; Menzel, P.L.; Svensson, L.; Aspelin, P.; Capitanio, A.; Axelsson, R. Quantitative assessment of 99mTc-depreotide uptake in patients with non-small-cell lung cancer: Immunohistochemical correlations. Acta Radiol. 2009, 50, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yi, C.; Hao, Z.; Cao, S.; Li, H.; Shao, X.; Zhang, J.; Qiao, T.; Fan, D. The effect of somatostatin and sstr3 on proliferation and apoptosis of gastric cancer cells. Cancer Biol. Ther. 2004, 3, 726–730. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mazzucchelli, R.; Morichetti, D.; Santinelli, A.; Scarpelli, M.; Bono, A.V.; Lopez-Beltran, A.; Cheng, L.; Montironi, R. Immunohistochemical expression and localization of somatostatin receptor subtypes in androgen ablated prostate cancer. Anal. Cell. Pathol. 2010, 33, 27–36. [Google Scholar] [CrossRef]
- Sclafani, F.; Carnaghi, C.; Di Tommaso, L.; Rodari, M.; Destro, A.; Rimassa, L.; Giordano, L.; Chiti, A.; Roncalli, M.; Santoro, A. Detection of somatostatin receptor subtypes 2 and 5 by somatostatin receptor scintigraphy and immunohistochemistry: Clinical implications in the diagnostic and therapeutic management of gastroenteropancreatic neuroendocrine tumors. Tumori 2011, 97, 620–628. [Google Scholar] [PubMed]
- Ji, X.Q.; Ruan, X.J.; Chen, H.; Chen, G.; Li, S.Y.; Yu, B. Somatostatin analogues in advanced hepatocellular carcinoma: An updated systematic review and meta-analysis of randomized controlled trials. Med. Sci. Monit. 2011, 17, RA169–RA176. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krenning, E.P.; Kwekkeboom, D.J.; Bakker, W.H.; Breeman, W.A.; Kooij, P.P.; Oei, H.Y.; van Hagen, M.; Postema, P.T.; de Jong, M.; Reubi, J.C.; et al. Somatostatin receptor scintigraphy with [111In-DTPA-d-Phe1]- and [123I-Tyr3]-octreotide: The rotterdam experience with more than 1000 patients. Eur. J. Nuclear Med. 1993, 20, 716–731. [Google Scholar] [CrossRef]
- Krenning, E.P.; Kwekkeboom, D.J.; Reubi, J.C.; Van Hagen, P.M.; van Eijck, C.H.; Oei, H.Y.; Lamberts, S.W. 111In-octreotide scintigraphy in oncology. Metab. Clin. Exp. 1992, 41, 83–86. [Google Scholar] [CrossRef]
- De Jong, M.; Breeman, W.A.; Kwekkeboom, D.J.; Valkema, R.; Krenning, E.P. Tumor imaging and therapy using radiolabeled somatostatin analogues. Acc. Chem. Res. 2009, 42, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Menda, Y.; Kahn, D. Somatostatin receptor imaging of non-small cell lung cancer with 99mTc depreotide. Semin. Nucl. Med. 2002, 32, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, M.; Decristoforo, C.; Donnemiller, E.; Ulmer, H.; Watfah Rychlinski, C.; Mather, S.J.; Moncayo, R. An intrapatient comparison of 99mTc-EDDA/HYNIC-TOC with 111In-DTPA-octreotide for diagnosis of somatostatin receptor-expressing tumors. J. Nucl. Med. 2003, 44, 708–716. [Google Scholar] [PubMed]
- Sun, L.C.; Coy, D.H. Somatostatin receptor-targeted anti-cancer therapy. Curr. Drug Deliv. 2011, 8, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fuselier, J.A.; Coy, D.H. Effects of camptothecin conjugated to a somatostatin analog vector on growth of tumor cell lines in culture and related tumors in rodents. Drug Deliv. 2004, 11, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.C.; Luo, J.; Mackey, L.V.; Fuselier, J.A.; Coy, D.H. A conjugate of camptothecin and a somatostatin analog against prostate cancer cell invasion via a possible signaling pathway involving PI3K/AKT, αvβ3/αvβ5 and MMP-2/-9. Cancer Lett. 2007, 246, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.C.; Mackey, L.V.; Luo, J.; Fuselier, J.A.; Coy, D.H. Targeted chemotherapy using a cytotoxic somatostatin conjugate to inhibit tumor growth and metastasis in nude mice. Clin. Med. Oncol. 2008, 2, 491–499. [Google Scholar] [PubMed]
- Huang, C.-M.; Wu, Y.-T.; Chen, S.-T. Targeting delivery of paclitaxel into tumor cells via somatostatin receptor endocytosis. Chem. Biol. 2000, 7, 453–461. [Google Scholar] [CrossRef]
- Engel, J.B.; Schally, A.V.; Dietl, J.; Rieger, L.; Hönig, A. Targeted therapy of breast and gynecological cancers with cytotoxic analogues of peptide hormones. Mol. Pharm. 2007, 4, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Nagy, A. Cancer chemotherapy based on targeting of cytotoxic peptide conjugates to their receptors on tumors. Eur. J. Endocrinol. 1999, 141, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.B.; Schally, A.V.; Halmos, G.; Baker, B.; Nagy, A.; Keller, G. Targeted cytotoxic bombesin analog AN-215 effectively inhibits experimental human breast cancers with a low induction of multi-drug resistance proteins. Endocr. Relat. Cancer 2005, 12, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M. Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr. Opin. Pharmacol. 2005, 5, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Govindan, S.V.; Goldenberg, D.M. Designing immunoconjugates for cancer therapy. Expert Opin. Biol. Ther. 2012, 12, 873–890. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody–drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012, 161, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Antibody-drug conjugate targets. Curr. Cancer Drug Targets 2009, 9, 982–1004. [Google Scholar] [CrossRef] [PubMed]
- Redko, B.; Ragozin, E.; Andreii, B.; Helena, T.; Amnon, A.; Talia, S.Z.; Mor, O.-H.; Genady, K.; Gary, G. Synthesis, drug release, and biological evaluation of new anticancer drug–bioconjugates containing somatostatin backbone cyclic analog as a targeting moiety. Pept. Sci. 2015, 104, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Kostenich, G.; Livnah, N.; Bonasera, T.A.; Yechezkel, T.; Salitra, Y.; Litman, P.; Kimel, S.; Orenstein, A. Targeting small-cell lung cancer with novel fluorescent analogs of somatostatin. Lung Cancer 2005, 50, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Wenger, S.; Schmuckli-Maurer, J.; Schaer, J.-C.; Gugger, M. Bombesin receptor subtypes in human cancers: Detection with the universal radioligand 125I-[d-Tyr6, β-Ala11, Phe13, Nle14] bombesin(6–14). Clin. Cancer Res. 2002, 8, 1139–1146. [Google Scholar] [PubMed]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R.V. International union of pharmacology. LXVIII. Mammalian bombesin receptors: Nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Cuttitta, F.; Carney, D.N.; Mulshine, J.; Moody, T.W.; Fedorko, J.; Fischler, A.; Minna, J.D. Bombesin-like peptides can function as autocrine growth factors in human small-cell lung cancer. Nature 1985, 316, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Pert, C.B.; Gazdar, A.F.; Carney, D.N.; Minna, J.D. High levels of intracellular bombesin characterize human small-cell lung carcinoma. Science 1981, 214, 1246–1248. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Carney, D.N.; Cuttitta, F.; Quattrocchi, K.; Minna, J.D.I. High affinity receptors for bombesin/GRP-like peptides on human small cell lung cancer. Life Sci. 1985, 37, 105–113. [Google Scholar] [CrossRef]
- Moody, T.W.; Sun, L.C.; Mantey, S.A.; Pradhan, T.; Mackey, L.V.; Gonzales, N.; Fuselier, J.A.; Coy, D.H.; Jensen, R.T. In vitro and in vivo antitumor effects of cytotoxic camptothecin-bombesin conjugates are mediated by specific interaction with cellular bombesin receptors. J. Pharmacol. Exp. Ther. 2006, 318, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Safavy, A.; Raisch, K.P.; Matusiak, D.; Bhatnagar, S.; Helson, L. Single-drug multiligand conjugates: Synthesis and preliminary cytotoxicity evaluation of a paclitaxel−dipeptide “scorpion” molecule. Bioconj. Chem. 2006, 17, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Armatis, P.; Cai, R.-Z.; Szepeshazi, K.; Halmos, G.; Schally, A.V. Design, synthesis, and in vitro evaluation of cytotoxic analogs of bombesin-like peptides containing doxorubicin or its intensely potent derivative, 2-pyrrolinodoxorubicin. Proc. Natl. Acad. Sci. USA 1997, 94, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Pradhan, T.; Mantey, S.A.; Jensen, R.T.; Dyba, M.; Moody, D.; Tarasova, N.I.; Michejda, C.J. Bombesin marine toxin conjugates inhibit the growth of lung cancer cells. Life Sci. 2008, 82, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Yang, H.; Xiang, B.; Li, S.; Liu, S.; Wan, L.; Zhang, J.; Li, Y.; Cheng, J.; Lu, X. Selective apoptotic killing of solid and hematologic tumor cells by bombesin-targeted delivery of mitochondria-disrupting peptides. Mol. Pharm. 2010, 7, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Fleischmann, A.; Macke, H.R.; Reubi, J.C. Targeting grpr in urological cancers—from basic research to clinical application. Nat. Rev. Urol. 2013, 10, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Alvarez, I.; Moreno, P.; Mantey, S.A.; Nakamura, T.; Nuche-Berenguer, B.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Insights into bombesin receptors and ligands: Highlighting recent advances. Peptides 2015, 72, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Ciudad, S.; Guiu, M.; Arranz-Gibert, P.; Garcia, C.; Gomis, R.R.; Cecchelli, R.; García, J.; Giralt, E.; et al. Miniap-4: A venom-inspired peptidomimetic for brain delivery. Angew. Chem. Int. Ed. 2016, 55, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Gabrail, N.; Chandhasin, C.; Moulder, S.; Smith, C.; Brenner, A.; Sankhala, K.; Mita, A.; Elian, K.; Bouchard, D.; et al. Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol. Cancer Ther. 2012, 11, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Drappatz, J.; Brenner, A.; Wong, E.T.; Eichler, A.; Schiff, D.; Groves, M.D.; Mikkelsen, T.; Rosenfeld, S.; Sarantopoulos, J.; Meyers, C.A.; et al. Phase I study of GRN1005 in recurrent malignant glioma. Clin. Cancer Res. 2013, 19, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Elamrawy, F.; Othman, A.A.; Adkins, C.; Helmy, A.; Nounou, M.I. Tailored nanocarriers and bioconjugates for combating glioblastoma and other brain tumors. J. Cancer Metastasis Treat. 2016, 2, 112–122. [Google Scholar] [CrossRef]
- Che, C.; Yang, G.; Thiot, C.; Lacoste, M.C.; Currie, J.C.; Demeule, M.; Regina, A.; Beliveau, R.; Castaigne, J.P. New angiopep-modified doxorubicin (ANG1007) and etoposide (ANG1009) chemotherapeutics with increased brain penetration. J. Med. Chem. 2010, 53, 2814–2824. [Google Scholar] [CrossRef] [PubMed]
- Regina, A.; Demeule, M.; Tripathy, S.; Lord-Dufour, S.; Currie, J.-C.; Iddir, M.; Annabi, B.; Castaigne, J.-P.; Lachowicz, J.E. ANG4043, a novel brain-penetrant peptide–mAb conjugate, is efficacious against HER2-positive intracranial tumors in mice. Mol. Cancer Ther. 2015, 14, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.C.; Taskar, K.; Rudraraju, V.; Goda, S.; Thorsheim, H.R.; Gaasch, J.A.; Mittapalli, R.K.; Palmieri, D.; Steeg, P.S.; Lockman, P.R.; et al. Uptake of ANG1005, a novel paclitaxel derivative, through the blood-brain barrier into brain and experimental brain metastases of breast cancer. Pharm. Res. 2009, 26, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Jiang, X.; Gu, J.; Sha, X.; Chen, L.; Law, K.; Chen, Y.; Wang, X.; Jiang, Y.; Fang, X. Angiopep-conjugated poly(ethylene glycol)-co-poly(epsilon-caprolactone) nanoparticles as dual-targeting drug delivery system for brain glioma. Biomaterials 2011, 32, 4293–4305. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhan, C.; Xie, C.; Meng, Q.; Gu, B.; Li, C.; Zhang, Y.; Lu, W. Poly(ethylene glycol)-block-poly(d,l-lactide acid) micelles anchored with angiopep-2 for brain-targeting delivery. J. Drug Target. 2011, 19, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Pang, Z.; Ye, H.; Qiu, B.; Guo, L.; Li, J.; Ren, J.; Qian, Y.; Zhang, Q.; Chen, J.; et al. Co-delivery of pEGFP-hTRAIL and paclitaxel to brain glioma mediated by an angiopep-conjugated liposome. Biomaterials 2012, 33, 916–924. [Google Scholar] [PubMed]
- Nagy, A.; Schally, A.V. Targeting cytotoxic conjugates of somatostatin, luteinizing hormone-releasing hormone and bombesin to cancers expressing their receptors: A "smarter" chemotherapy. Curr. Pharm. Des. 2005, 11, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ndinguri, M.W.; Solipuram, R.; Wakamatsu, N.; Hammer, R.P.; Ingram, D.; Hansel, W. [DLys6]-luteinizing hormone releasing hormone-curcumin conjugate inhibits pancreatic cancer cell growth in vitro and in vivo. Int. J. Cancer 2011, 129, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Temming, K.; Schiffelers, R.M.; Molema, G.; Kok, R.J. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Res. Updates 2005, 8, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Breton, A.L.; Préat, V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, K.; Cai, W.; Li, Z.; He, L.; Kashefi, A.; Chen, X. Integrin-targeted imaging and therapy with RGD4C-TNF fusion protein. Mol. Cancer Ther. 2008, 7, 1044–1053. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Groot, F.M.; Broxterman, H.J.; Adams, H.P.; van Vliet, A.; Tesser, G.I.; Elderkamp, Y.W.; Schraa, A.J.; Kok, R.J.; Molema, G.; Pinedo, H.M.; et al. Design, synthesis, and biological evaluation of a dual tumor-specific motive containing integrin-targeted plasmin-cleavable doxorubicin prodrug. Mol. Cancer Ther. 2002, 1, 901–911. [Google Scholar] [PubMed]
- He, X.; Alves, C.S.; Oliveira, N.; Rodrigues, J.; Zhu, J.; Bányai, I.; Tomás, H.; Shi, X. RGD peptide-modified multifunctional dendrimer platform for drug encapsulation and targeted inhibition of cancer cells. Colloids Surf. B Biointerfaces 2015, 125, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Takenaka, T.; Toh, K.; Wu, S.; Nishihara, H.; Kano, M.R.; Ino, Y.; Nomoto, T.; Matsumoto, Y.; Koyama, H.; et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood–brain tumor barrier. ACS Nano 2013, 7, 8583–8592. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.A.; Majeti, B.K.; Barnes, L.A.; Makale, M.; Weis, S.M.; Lutu-Fuga, K.; Wrasidlo, W.; Cheresh, D.A. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 9343–9348. [Google Scholar] [CrossRef] [PubMed]
- Dal Pozzo, A.; Ni, M.-H.; Esposito, E.; Dallavalle, S.; Musso, L.; Bargiotti, A.; Pisano, C.; Vesci, L.; Bucci, F.; Castorina, M.; et al. Novel tumor-targeted rgd peptide–camptothecin conjugates: Synthesis and biological evaluation. Bioorg. Med. Chem. 2010, 18, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Gu, B.; Xie, C.; Li, J.; Liu, Y.; Lu, W. Cyclic rgd conjugated poly(ethylene glycol)-co-poly(lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J. Control. Release 2010, 143, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Boock, A.; Miller, K.; Sanchis, J.; Lupu, R.; Vicent, M.J.; Satchi-Fainaro, R. Integrin-assisted drug delivery of nano-scaled polymer therapeutics bearing paclitaxel. Biomaterials 2011, 32, 3862–3874. [Google Scholar] [CrossRef] [PubMed]
- Ryppa, C.; Mann-Steinberg, H.; Fichtner, I.; Weber, H.; Satchi-Fainaro, R.; Biniossek, M.L.; Kratz, F. In vitro and in vivo evaluation of doxorubicin conjugates with the divalent peptide E-[c(RGDfK)2] that targets integrin αvβ3. Bioconj. Chem. 2008, 19, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Ryppa, C.; Mann-Steinberg, H.; Biniossek, M.L.; Satchi-Fainaro, R.; Kratz, F. In vitro and in vivo evaluation of a paclitaxel conjugate with the divalent peptide E-[c(RGDfK)2] that targets integrin αvβ3. Int. J. Pharm. 2009, 368, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Schally, A.V.; Halmos, G.; Armatis, P.; Cai, R.-Z.; Csernus, V.; Kovács, M.; Koppán, M.; Szepesházi, K.; Kahán, Z. Synthesis and biological evaluation of cytotoxic analogs of somatostatin containing doxorubicin or its intensely potent derivative, 2-pyrrolinodoxorubicin. Proc. Natl. Acad. Sci. USA 1998, 95, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, S.; Nagy, A.; Szoke, B.; Schally, A.V. Cytotoxic analog of somatostatin containing methotrexate inhibits growth of mia paca-2 human pancreatic cancer xenografts in nude mice. Cancer Lett. 1992, 62, 263–271. [Google Scholar] [CrossRef]
- Kiaris, H.; Schally, A.V.; Nagy, A.; Sun, B.; Armatis, P.; Szepeshazi, K. Targeted cytotoxic analogue of bombesin/gastrin-releasing peptide inhibits the growth of H-69 human small-cell lung carcinoma in nude mice. Br. J. Cancer 1999, 81, 966–971. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Safavy, A.; Raisch, K.P.; Khazaeli, M.B.; Buchsbaum, D.J.; Bonner, J.A. Paclitaxel derivatives for targeted therapy of cancer: Toward the development of smart taxanes. J. Med. Chem. 1999, 42, 4919–4924. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.S. Antibody-drug conjugates for cancer therapy: The technological and regulatory challenges of developing drug-biologic hybrids. Biologicals 2015, 43, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Liang, R.; An, X.; Wang, K.; Shen, G.; Tu, Y.; Zhu, J.; Tao, J. Recent advances in targeted nanoparticles drug delivery to melanoma. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 769–794. [Google Scholar] [CrossRef] [PubMed]
- Raucher, D.; Ryu, J.S. Cell-penetrating peptides: Strategies for anticancer treatment. Trends Mol. Med. 2015, 21, 560–570. [Google Scholar] [CrossRef] [PubMed]
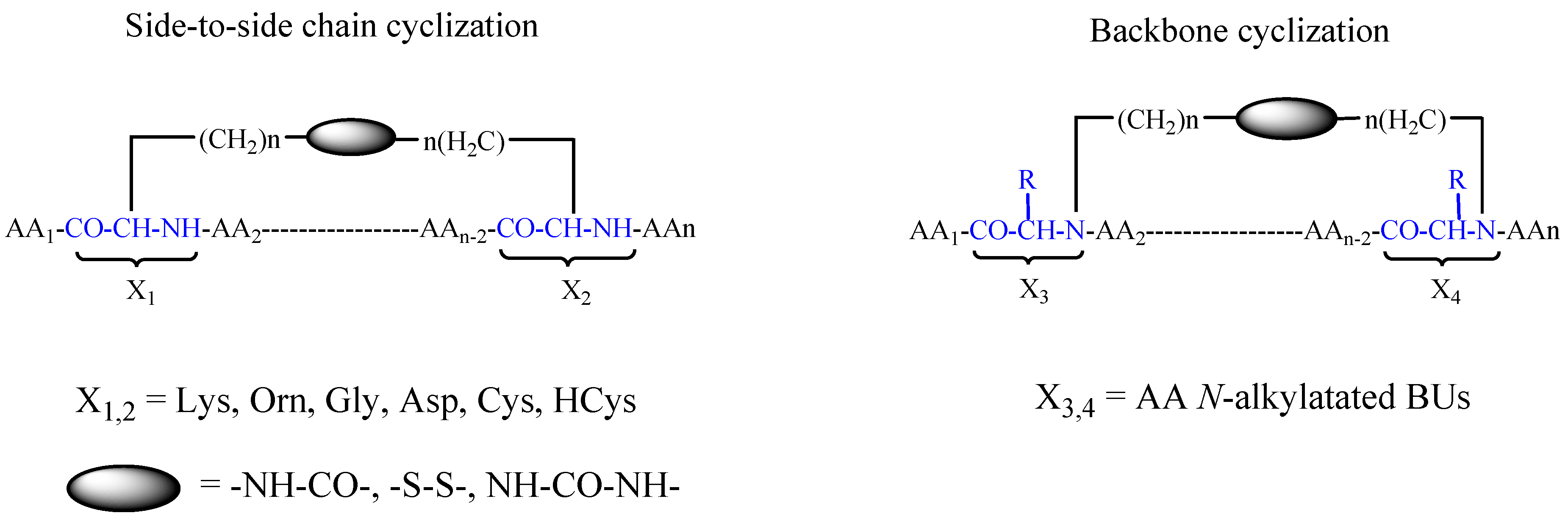

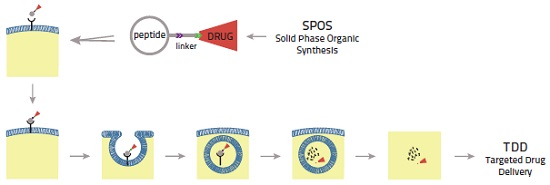{kind=link}
{kind=link}
{kind=link}
| Sequence of the Targeting Peptide | Name of the Targeting Peptide | Formulation | Therapeutic Agent | Target | In-Vitro Model | In-Vivo Model | Ref. |
|---|---|---|---|---|---|---|---|
| GlpHWSYKLRPG-NH2 (Glp = Pyroglutamic acid) | (D-Lys6)LH-RH | PDC | DOX | LHRH | * Human breast cancer cells MCF7 a, Mouse mammary carcinoma cells MXT a, Human breast cancer cells MX-1 b | a [126] b [118] | |
| GlpHWSYKLRPG-NH2 (Glp = Pyroglutamic acid) | (D-Lys6)LH-RH | PDC | 2-pyrrolino-DOX | LHRH | * Human breast cancer cells MX-1 a/ MDA-MB-231 b/ MDA-MB-435 c | * MX-1 a/ MDA-MB-231 b/ MDA-MB-435 c tumor in mice | a [126] b [127] c [128] |
| Ac-D-Nal(2)-f(4Cl)-D-Pal(3)-SYkLRPa-NH2 [where Nal(2) = 3-(2-naphthyl)alanine, Pal(3) = 3-(3-pyridyl)alanine, and f(4CI) = 4-chloro-d-phenylalanine) | Antagonistic analog | PDC | 2-pyrrolino-DOX | LHRH | * Human breast cancer cells MCF7 a, Mouse mammary carcinoma cells MXT a | a [126] | |
| QHWSYkLRP-NH–Et | (D-Lys6)LH-RH Des-Gly10, Pro-NHEt9 | PEGylated carrier system | CPT | LHRH | Human ovarian cancer cells A2780 | [134] | |
| GlpHWSYKLRPG-NH2 (Glp = Pyroglutamic acid) | (D-Lys6)LH-RH | PDC | Curcumin | LHRH | Human pancreatic cancer cells MIAPaCa-2, BxPC-3 and Panc-1 | Pancreas cancer-MIA PaCa-2-tumor in mice | [187] |
| Name of the Targeting Peptide | Sequence of the Targeting Peptide | Formulation | Therapeutic Agent | Target | In-Vitro Model | In-Vivo Model | Ref. |
|---|---|---|---|---|---|---|---|
| RGD4C | Cyclic CDCRGDCFC | * Fusion protein | TNFα | αVβ3 | Human glioblastoma cells U87MG, Human breast cancer cells MDA-MB-435, Rat glioma cells C6, Mouse fibroblast cells L929 | U87MG tumor in mice, MDAMB-435 tumor in mice | [190] |
| RGD4C | Cyclic CDCRGDCFC | PDC | DOX | αVβ3 | MDAMB-435 tumor in mice | [94] | |
| RGD4C | Bicyclic CDCRGDCFC | Drug conjugate with plasmin self immolative linker vFK | DOX | αVβ3 | Human fibroblast cells HT1080, Human endothelial cells HUVEC | [191] | |
| Acyclic RGD4C | Acyclic CDCRGDCFC | PDC | Doxsaliform | αVβ3 | Human breast cancer cells MDA-MB-435 | [95] | |
| c(RGDfK) | c(RGDfK) | PDC | CLB, CPT | αVβ3 | Human non-small lung carcinoma cells H1299, Murine melanoma cells B16-F10, Human embryonic kidney cells HEK-293 | [109] | |
| c(RGDfK) | c(RGDfK) | Drug conjugate with dual drug payload | CLB, CPT | αVβ3 | Human non-small lung carcinoma cells H1299, Murine melanoma cells B16-F10, Human embryonic kidney cells HEK-293 | [112] | |
| c(RGDfK) | c(RGDfK) | PAMAM Drug loaded PEGylated dendrimers | DOX | αVβ3 | Human glioblastoma cells U87MG | [192] | |
| c(RGDfK) | c(RGDfK) | PEG polymeric micelles | (DACHPt) | αVβ3/5 | Human glioblastoma cells U87MG | U87MG tumor in mice | [193] |
| c(RGDfK) | c(RGDfK) | ** Nanoparticles | DOX | αVβ3/5 | Human endothelial cells HUVEC | Pancreas tumor in mice-murine R40P cells | [194] |
| c(RGDfK) | c(RGDfK) | PDC | CPT | αVβ3 | Human prostate cancer cells PC3, Human renal carcinoma cells A498, Human ovarian cancer cells A2780 | A2780 tumor in mice | [195] |
| Celingitide | cyclic-(N-Me-VRGDf-NH) | PDC | Doxsaliform | αVβ3 | Human breast cancer cells MDA-MB-435 | [95] | |
| c(RGDyK) | c(RGDyK) | Drug loaded PEG-PLA micelles | PTX | αVβ3 | Human glioblastoma cells U87MG | U87MG tumor in mice | [196] |
| c(RGDfS) | c(RGDfS) | PDC | CLB | αVβ3 | Human non-small lung carcinoma cells H1299, Murine melanoma cells B16-F10, Human embryonic kidney cells HEK-293 | [109] | |
| E[c(RGDyK)]2 | E[c(RGDyK)]2 | PDC | PTX | αVβ3 | Human breast cancer cells MDA-MB-435 | MDA-MB-435 tumor in mice | [96] |
| E-[c(RGDfK)2] | divalent cyclic peptide E-[c(RGDfK)2] | PGA nano-scaled conjugate | PTX | Human glioblastoma cells U87MG, Murine breast cancer cells 4T1, Human endothelial cells HUVEC | [197] | ||
| E-[c(RGDfK)2] | E-[c(RGDfK)2] | Peptide drug conjugate with the MMP2/9 sensitive linker GPLGILG | DOX | αVβ3 | Human endothelial cells HUVEC, Human ovarian cancer cells OVCAR-3 | OVCAR-3 tumor in mice | [198] |
| E-[c(RGDfK)2] | divalent cyclic peptide E-[c(RGDfK)2] | PDC | PTX | αVβ3 | Human endothelial cells HUVEC | ovarian cancer-OVCAR-3-tumor in mice | [199] |
| Name of the Targeting Peptide | Sequence of the Targeting Peptide | Formulation | Therapeutic Agent | Target | In-Vitro Model | In-Vivo Model | Ref. |
|---|---|---|---|---|---|---|---|
| RC-160 | Cyclic fCYwKVCW-NH2 | PDC | 2-Pyrrolino-DOX, DOX | SSTRs | * MDA-MB-435 tumor in mice, mouse mammary carcinoma-MXT in mice, Dunning AT-1 prostate cancers in rat | [201] | |
| RC-121 | Cyclic fCYwKVCT-NH2 | PDC | MTX | SSTRs | Pancreas cancer-MIA PaCa-2-tumor in mice | [202] | |
| RC-121 | Cyclic fCYwKVCT-NH2 | PDC | 2-Pyrrolino-DOX, DOX | SSTRs | * Human gastric cancer cells MKN-45, Human breast cancer cells MDA-MB-231, Human prostate cancer cells-PC-3, Human pancreatic cancer cells-MIA PaCa2, Human SCLC cells H-345 | * MDA-MB-435 tumor in mice, mouse mammary carcinoma-MXT in mice, Dunning AT-1 prostate cancer in rat | [201] |
| 3207-86 | PDC | Amonafide, ABT-751, CPT, COMB, CLB | SSTR2 | Human non-small lung carcinoma cells H1299, Human embryonic kidney cells HEK-293, Human colon cancer cells HTC 116, Human prostate cancer cells TRAMP C2 | [161] |
| Name of the Targeting Peptide | Sequence of the Targeting Peptide | Formulation | Therapeutic Agent | Target | In-Vitro Model | In-Vivo Model | Ref. |
|---|---|---|---|---|---|---|---|
| RC-3094 | ** QWAVGHL–Ψ(CH2-NH)–L-NH2 | PDC | 2-pyrrolino-DOX, DOX | Bombesin | * Human pancreatic cancer cells CFPAC-1, Human lung cancer cells DMS-53, Human prostate cancer cells PC-3, Human gastric cancer cells MKN-45 | [170] | |
| RC-3094 | QWAVGHL–Ψ(CH2-NH)–L-NH2 | PDC | 2-pyrrolino-DOX, DOX | Bombesin | * Human SCLC cells NCI-H-69 | * NCI-H-69 tumor in mice | [203] |
| BBN(7-13) | WAVGHL-NH2 | PDC with PEGylated linker | PTX | Bombesin | Human SCLC cells NCI-H-69 | [204] |
| Name of the Targeting Peptide | Sequence of the Targeting Peptide | Formulation | Therapeutic Agent | Target | In-Vitro Model | In-Vivo Model | Ref. |
|---|---|---|---|---|---|---|---|
| Angiopep-2 | TFFYGGSRGKRNNFKTEEY | PDC | 3 × PXT | Low-density lipoprotein receptor (LDLr) | U87 glioma | [180] | |
| Angiopep-2 | TFFYGGSRGKRNNFKTEEY | PDC | 3 × DOX | Low-density lipoprotein receptor (LDLr) | Glioblastoma (U87 MG) Hepatocarcinoma (SK-Hep-1) Lung carcinoma (NCI-H460) | U87 glioma | [180] |
| Angiopep-2 | TFFYGGSRGKRNNFKTEEY | PDC | dimethylglycine etoposide (ETO) | Low-density lipoprotein receptor (LDLr) | Glioblastoma (U87 MG) Hepatocarcinoma (SK-Hep-1) Lung carcinoma (NCI-H460) | U87 glioma | [180] |
| Angiopep-2 | TFFYGGSRGKRNNFKTEEY | Peptide–Ab Conjugate | Trastuzumab | Low-density lipoprotein receptor (LDLr) | HER2-positive BT-474 breast ductal carcinoma cells | HER2-positive intracranial tumors in mice | [181] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilad, Y.; Firer, M.; Gellerman, G. Recent Innovations in Peptide Based Targeted Drug Delivery to Cancer Cells. Biomedicines 2016, 4, 11. https://doi.org/10.3390/biomedicines4020011
Gilad Y, Firer M, Gellerman G. Recent Innovations in Peptide Based Targeted Drug Delivery to Cancer Cells. Biomedicines. 2016; 4(2):11. https://doi.org/10.3390/biomedicines4020011
Chicago/Turabian StyleGilad, Yosi, Michael Firer, and Gary Gellerman. 2016. "Recent Innovations in Peptide Based Targeted Drug Delivery to Cancer Cells" Biomedicines 4, no. 2: 11. https://doi.org/10.3390/biomedicines4020011
APA StyleGilad, Y., Firer, M., & Gellerman, G. (2016). Recent Innovations in Peptide Based Targeted Drug Delivery to Cancer Cells. Biomedicines, 4(2), 11. https://doi.org/10.3390/biomedicines4020011