Abstract
Epithelial ovarian cancer (EOC) is one important cause of gynecologic cancer-related death. Currently, the mainstay of ovarian cancer treatment consists of cytoreductive surgery and platinum-based chemotherapy (introduced 30 years ago) but, as the disease is usually diagnosed at an advanced stage, its prognosis remains very poor. Clearly, there is a critical need for new treatment options, and immunotherapy is one attractive alternative. Prophylactic vaccines for prevention of infectious diseases have led to major achievements, yet therapeutic cancer vaccines have shown consistently low efficacy in the past. However, as they are associated with minimal side effects or invasive procedures, efforts directed to improve their efficacy are being deployed, with Dendritic Cell (DC) vaccination strategies standing as one of the more promising options. On the other hand, recent advances in our understanding of immunological mechanisms have led to the development of successful strategies for the treatment of different cancers, such as immune checkpoint blockade strategies. Combining these strategies with DC vaccination approaches and introducing novel combinatorial designs must also be considered and evaluated. In this review, we will analyze past vaccination methods used in ovarian cancer, and we will provide different suggestions aiming to improve their efficacy in future trials.
1. Rationale for Immunotherapy in Ovarian Cancer
Epithelial ovarian cancer (EOC) is one important cause of gynecologic cancer-related death, with an overall five-year survival rate of ~45% and an overall 10-year survival rate of 35% in the USA [1]. Globally, it is estimated that 282,741 new cases would be diagnosed with ovarian cancer in the world in 2020, with about 66% of the cases affecting women aged <65 years [2]. Currently, the mainstay of ovarian cancer treatment consists of cytoreductive surgery and platinum-based chemotherapy (introduced 30 years ago) but, as the disease is usually diagnosed at an advanced stage, its prognosis remains poor, with an overall five-year survival rate of ~45%, which drops to 28% for invasive EOC diagnosed at stage IV [1]. There is a clear unmet need for developing new treatment approaches and a potentially attractive approach for ovarian cancer treatment is immunotherapy [3].
It was recently demonstrated that EOC is an immunogenic tumor that can be recognized by the host immune system [4]. Indeed, tumor reactive T cells and antibodies can be detected in the blood, tumor and ascites of EOC patients with advanced disease [5,6]. The tumor reactive T cells collected from patients harboring advanced ovarian cancer are oligoclonal, recognize autologous tumor-associated-antigens (TAAs) and exhibit tumor-specific cytolytic activity in vitro [7]. In fact, the observed frequency of serological responses to these antigens is variable according to tumor type, stage or grade [8]. However, in most tumors the tumor-reactive lymphocyte populations show impaired antitumor function in vivo, due to several mechanisms. Ovarian tumors present multiple mechanisms of immune evasion, which consequently reduce the efficacy of immunotherapy. Thus, recruitment of Tregs in the tumor microenvironment in ovarian carcinoma confers immune privilege and is associated with poor prognosis and reduced survival [9,10]. Other mechanisms include high expression of PD-L1 and IDO production, which are independently associated with poor prognosis in EOC [11,12,13]. Furthermore, it has been demonstrated that both local and systemic dysfunction of plasmacytoid Dendritic Cells (pDCs) play a critical role in the progression of ovarian cancer via induction of immune tolerance [14].
Immunotherapies include active, passive or immunomodulatory strategies; although some overlap exists among them [15]. Active strategies (such as vaccines or adoptive-cell therapies with autologous T-cells) aim to increase the ability of the patients’ own immune system to mount an immune response against their own tumor. The first type of immunotherapy used in oncology was “Coley’s toxin” [16], an adjuvant which induces in vivo vaccination. Vaccination strategies indeed represent an attractive approach in ovarian cancer treatment, as they are associated with minimal side effects or invasive procedures.
Vaccination strategies for the prevention of infectious diseases led to major medical successes, such as global smallpox eradication [17]. Prophylactic vaccines against viruses with known oncogenic potential (such as HPV or HBV) have a demonstrated effect in preventing cancer development [18], although they do not confer any benefit on pre-existing infections or lesions [19]. For this reason, therapeutic vaccines against established lesions have been developed, yet they have a long history of low efficacy that has created a negative image on immunotherapy. First, it should be kept in mind that prophylactic and therapeutic vaccines have different goals: in prophylactic vaccines, the aim is to trigger a good humoral response, so that effective antibodies are produced, able to bind to and inactivate the targeted pathogen when it enters the blood or mucosal surfaces. Yet, a therapeutic vaccine should be able to induce cell mediated immunity, so that immune cells are activated to identify and destroy their cellular targets in the affected tissues. Then, understanding the reasons for the limited efficacy of therapeutic vaccines to date will allow the development of new approaches that may aid to overcome the barriers to adequate anti-tumor activation. Some of these new strategies are currently being developed and tested in ovarian cancer with encouraging results.
2. Therapeutic Vaccines in Ovarian Cancer
Cancer vaccines can be classified in different categories, according to the method of choice to deliver the selected TAAs; thus, cell-based vaccines, peptide/protein, epigenetic, and genetic vaccines have been developed so far [20] (see Table 1). Vaccines can be given alone or in combination with different adjuvants, such as cytokines or other stimulatory factors [21]. Furthermore, different routes for immunization can be used, which contribute differently to immune cells activation: vaccine injections can be subcutaneous, intradermal, intranodal, intraperitoneal, or intravenous [22]. Finally, another consideration is the vaccination schedule, in terms of dose, number of injections and time, to initiate and maintain the appropriate immune response. More studies are warranted to determine which of those elements (alone and in combination) would offer optimal clinical efficacy [23]. Here, we will summarize the results obtained to date with all types of vaccination strategies in ovarian cancer with the intent to provide a general overview allowing undertaking more rational decisions (Table 1).

Table 1.
Published results from therapeutic vaccines tested in ovarian cancer from 2000 to date.
2.1. Cell-Based Vaccines
Cell-based vaccines can use DCs, which play a critical role in the interface between innate and adaptive immunity [59]. Their function consists in the uptake, process and presentation of antigenic peptides (either from pathogens or host-derived) to naïve T cells in peripheral tissues, and they constitute the most important antigen-presenting cell (APC) population for activating antitumor T-cell responses. For this reason, they are the most frequently used cellular therapeutics in clinical trials, also because DC vaccination has demonstrated a good safety profile, rarely presenting immune-related toxicities [60], and it is associated with preserved quality of life of cancer patients [61]. DCs can be given alone after cytotoxic therapy (either chemo or radiotherapy, which increase antigen availability in vivo [62]), or alternatively they can be loaded ex vivo with different antigens, such as whole tumor lysate, peptides, proteins, or genetic material delivering the desired antigen (transfected/electroporated DNA, RNA or transduced virus), prior to reinfusion into the patient.
The advantage of whole cells as a source for antigens is that they will present to the immune system the complete repertoire of TAAs from that particular tumor, including the specific neo-antigens, therefore predicting a better immune response [22]. In this direction, in a meta-analysis including about 1800 patients, those who were immunized with whole tumor vaccines had a significantly higher objective response (8.1%) than patients who were immunized with defined tumor antigens (3.6%) [63]. Ovarian cancer lends itself to surgical intervention during the course of disease, including for the recurrent setting. This enables whole tumor vaccination approaches, since tumor cells can be easily recovered through cytoreductive surgery. Indeed, we as well as others have shown clinical benefit in recurrent advanced ovarian cancer patients vaccinated with DCs loaded with whole tumor lysate [24,25]. In our study, we treated five recurrent ovarian cancer patients with DCs loaded with hypochlorus acid-oxidized whole tumor lysate (inducing primary necrosis and enhancing the immunogenicity of lysed tumor cells), administered intranodally. We observed potent T-cell responses against known ovarian tumor antigens, and two patients presented durable PFS of 24 months or more, with only few grade 1 toxicities. We believe this is an approach that warrants further exploration [25]; thereby, our group has continued this pilot clinical study in heavily pretreated recurrent ovarian cancer patients to test vaccine injected intranodally, alone or in combination with immunomodulatory therapy (NCT01132014).
An example of a whole tumor cell vaccine in ovarian cancer is gemogenovatucel-T (FANG vaccine); it consists of autologous tumor cells eletroporated with FANG vector, a plasmid encoding GM-CSF (granulocyte macrophage-colony stimulating factor; a potent stimulator of dendritic cell maturation) and a bi-shRNA targeting furin convertase, thereby downregulating endogenous immunosuppressive growth factors TGF-β1 and β2 [33]. This vaccine has been tested in a Phase I study including 27 patients with advanced or metastatic non-curable solid tumors of different types (5 ovarian cancer), and in 26 of 27 patients evaluable for tumor response, 23 achieved SD at Month 2 or later as best response. This vaccine is currently being tested in a Phase II/III study in women with stages III/IV high grade serous/endometrioid ovarian, fallopian tube or primary peritoneal cancer (NCT02346747).
Other approaches use DCs loaded with specific antigens, either peptides or proteins. The first validation of active immunotherapy as a viable approach to cancer treatment was the FDA approval of Provenge® (sipuleucel-T) for advanced prostate cancer. Provenge® is an autologous DC-based vaccine (developed by Dendreon), in which autologous peripheral blood mononuclear cells (PBMC) are matured with a cytokine and a tumor-derived differentiation agent, and then pulsed with a fusion protein composed of prostatic acid phosphatase (PAP; a tumor-associated differentiation antigen) linked to GM-CSF prior to reinfusion into patients [64]. In the pivotal Phase III trial of this vaccine in men with metastatic castration-resistant prostate cancer, clinical results showed little evidence of tumor shrinkage or delay in disease progression [65]. Nevertheless, a 4.1 month improvement in median survival was achieved (25.8 vs. 21.7 months), which was considered significant by the FDA in a patient population that has almost no other effective therapeutic option. A similar vaccine was prepared and tested by Dendreon (lapuleucel-T or Neuvenge), consisting of DCs loaded with a fusion protein of HER-2/neu linked to GM-CSF. This vaccine was targeted to patients with advanced adenocarcinomas of the breast, ovary, endometrium, or gastrointestinal tract with HER-2/neu positive tumors, yet in a Phase I study including 18 patients, only two (11%) experienced stable disease lasting more than 48 weeks [29].
2.2. Peptide/Protein-Based Vaccines
The broad use of autologous cancer vaccines, including DCs or whole tumor cells, is limited by both the availability of patient’s samples or specimens and the complex procedure of preparing individualized vaccines and, from this point of view, recombinant vaccines have a clear advantage. Peptide/protein-based vaccines are usually based on defined TAAs and administered together with an adjuvant or immune modulator to improve their uptake by endogenous DCs. Unfortunately, many initial attempts were compromised by a poor understanding of the mechanism of immunization: frequently patients were treated with vaccines consisting of short peptides, binding exactly to HLA class I molecules (usually HLA-A*02), which do not induce CD4+ T cells, resulting in short-lived CD8+ T cell responses, often even without an effective adjuvant [66]. A better understanding of the importance and function of DCs in stimulating T cell responses has led to a more rational design of current vaccines. For instance, the use of peptides (~20 mer) somewhat longer than the optimal MHC class I-binding molecules (10–12 mer), in the presence of a suitable DC-activating adjuvant, are thought to be more efficient at generating effector T cells, due to additional processing required for long peptides to allow loading in DC HLA molecules, leading to potential dual stimulation of CD4+ T as well as CD8+ T cells [67]. However, results observed with this type of long peptides are still far from clinically relevant [45,48,49,50]. On the other hand, the use of full length proteins (in principle available for complete processing by DCs) has given mixed results to date [42,46]
In ovarian cancer, many different peptides targeting HER-2/neu have been tested. HER-2/neu is a member of the epidermal growth factor receptor (HER/EGFR/ERBB) family, whose amplification in breast cancer is associated with increased aggressiveness, therefore becoming an important target of therapy for about 20%–30% of patients [68]. HER-2/neu overexpression/amplification has been reported in ovarian cancer [69], and consequently it was considered a potential target for cancer vaccination. However, in most studies using single or mixed HER-2/neu peptides, no immunogenicity was observed [38,39,40,41], and no clinical data was obtained. Other vaccine targets in ovarian cancers using peptides have been tested [43,44,45,47,48,49,50], generally with low efficacy in the clinic. The best results in ovarian cancer using peptide-based vaccines have been obtained using a personalized peptide vaccine (PPV), in which a mixture of 4 peptides (from a panel of 31) previously tested for immunity in each patient was admixed in Montanide ISA51VG and subcutaneously administrated. In this study [51], median survival time (MST) was 39.2 months in platinum-sensitive patients, compared to 16.2 months in platinum-resistant patients, whereas the corresponding values in standard of care patients are 18–30 months (platinum-sensitive) vs. 8–12 (platinum-resistant). Interestingly, it was observed that PPV induced not only peptide-specific immunological boosting in response to the vaccinated peptides but also promoted the spreading of immune responses to the other TAA-derived peptides, which together resulted in the prolongation of OS. Results here indicate that vaccination strategies in which the vaccine antigens are selected and administered based on the pre-existing host immunity before vaccination can prolong OS in advanced ovarian cancer patients.
2.3. Genetic Vaccines
Genetic vaccines (based on DNA, RNA or virus) can be used to induce expression in vivo of the selected TAAs in somatic (keratinocytes, myocytes) or dendritic cells infiltrating muscle or skin at the vaccination site, resulting in cross-priming or direct antigen-presentation to infiltrating T-cells. Genetic vaccines present the advantage of easy delivery of multiple antigens in one immunization and activation of various arms of immunity, in combination with cheaper and more standardized manufacturing [70].
In ovarian cancer, two viral vaccines have been tested so far. One group has focused on the “cancer-testis” antigen NY-ESO-1, engineered into vaccinia (rV) as prime and fowlpox (rF) as booster vaccination. In a Phase II study including 22 patients with advanced NY-ESO-1-expressing ovarian cancer at high risk of recurrence, showed encouraging results, as the median time to disease progression or recurrence was 21 months (95% CI, 16–29 months) and median OS was 48 months (CI non-estimable) [55]. A second genetic vaccine tested in ovarian cancer (PANVAC-C + PANVAC-V) is a Poxviral vaccine, in which the CEA-MUC1-TRICOM (B7.1, ICAM-1, LFA-3) was engineered into vaccinia (PANVAC-V) as prime and fowlpox (PANVAC-C) as booster vaccination. However, clinical results from a Phase I clinical trial including 25 patients with CEA- or MUC1-expressing metastatic cancers who had progressive disease following standard chemotherapy (three of them with ovarian cancer) showed limited evidence of clinical activity [53]. Currently, several clinical trials are testing different genetic vaccines in ovarian cancer treatments (see Table 2).
Table 2.
Vaccines for ovarian cancer in clinical development (January 2016).
2.4. Epigenetic Vaccines
Glycosylation is the most diverse post-translational protein modification, playing a key role in a wide range of biological processes. It has been shown that epigenetic regulation of glycosyltransferases in cancer cells results in the creation of novel glycan structures [71,72,73], and this correlates with the fact that most tumor cells present altered glycosylation relative to the normal tissue from which they derive [74]. Altered glycosylation has been proposed to be one of the mechanisms used by cancer cells to evade the host immune response, since cellular presentation of glycopeptide and glycolipid antigens can be potent modulators of T cells [75]. Antiglycan vaccination strategies against tumors were proposed early [76], and they have subsequently been developed, mostly against the epithelial MUC1. However, due to poor immunogenicity, glycan-based vaccines need to be conjugated to helper T epitopes, such as keyhole limpet hemocyanin (KLH) [77].
In ovarian cancer, two antiglycan vaccines have been tested: one against Lewis(y) (Ley), and the Theratope vaccine, targeting Syalyl-Tn (STn). Ley is a carbohydrate antigen overexpressed in ovarian cancer [78]. In a phase I study, 25 patients with persistent or recurrent ovarian, fallopian tube, or peritoneal cancer of any stage or grade at diagnosis were vaccinated with a synthetic Ley pentasaccharide coupled to KLH carrier protein, together with the QS-21 immunological adjuvant. At a median of 18 months follow-up, 19 of 24 patients had either biochemical or measurable disease recurrence. The median TTP was six months (range 2–17 months), with five patients in CR at 18 months of follow-up. Theratope® is a Sialyl-Tn—keyhole limpet hemocyanin (STn-KLH) vaccine that incorporates a synthetic STn antigen mimicking the unique tumor-associated STn carbohydrate, designed to stimulate tumor antigen-specific immune responses in patients with mucin-expressing tumors. STn expression is associated with a poor prognosis in metastatic breast [79] and ovarian cancer patients [80], among others. Theratope® vaccine was tested in a Phase II/III trial including 70 patients with either advanced breast or ovarian cancer expressing mucin. Vaccination was performed after autologous stem cell transplantation, considering that patients with low tumor burden would be more likely to respond immunologically to a cancer vaccine. Interestingly, it was observed that vaccinated patients had a decrease in the risk for relapse and death (p = 0.07 and p = 0.10, respectively), as compared to patients who underwent transplantation during the same period, but were not vaccinated [56]. However, later on this vaccine failed to show any improvement in TTP nor patient survival in a big Phase III trial reported in 2011 for more than 1000 breast cancer patients, despite a vigorous and specific humoral response to the STn antigen [81]. A different epigenetic vaccine using the Globo H hexasaccharide 1 (Globo H) epitope linked to KLH is currently being tested in a Phase II study including patients with non-progressive epithelial ovarian, fallopian tube, or primary peritoneal cancer after cytoreductive surgery and platinum-based chemotherapy (NCT02132988). Globo H is an antigen that was identified on a variety of epithelial cell tumors of ovarian, gastric, pancreatic, endometrial, prostate, and lung (small cell and non-small cell) origin, and selected as potential target for cancer immunotherapy [82].
3. Improving Vaccination Strategies in Ovarian Cancer
Cancer vaccines have shown, in general, low therapeutic efficacy, probably associated with their inability to elicit a rapid and strong T-cell response, and this has generated a great deal of criticism [83]. Similar criticisms have been addressed specifically to DC-vaccination [84]. However, a systematic review analyzing all published clinical trials performed to document the proportion of patients who had an objective response rate after DC vaccination in melanoma, prostate cancer, malignant glioma, and renal cell carcinoma [85] demonstrated that, in melanoma, DC therapy had similar objective response (8.5%) than dacarbazine (standard of care), or ipilimumab (5%–15%). In prostate cancer patients, objective response rate was 7.1% after DC vaccination, similar to 10% of the population treated with conventional chemotherapeutic drugs. Comparably, objective response after DC therapy was 15.6% in patients with malignant glioma, and 11.5% in advanced RCC. Interestingly, in most studies using DC therapies an increase of at least 20% in overall survival has been documented [85], although many of these studies were early phase, in which survival was not the main endpoint. Therefore, given the positive toxicity profile associated with vaccines, it is important to identify novel strategies to increase their efficacy, ideally without inducing significant changes in toxicity. Basically, an ideal vaccination strategy should include: (i) an adequate mixture of immunogenic antigens (ex vivo or in vivo); (ii) either a selected maturation signal for the target DC population in vivo, or the targeted matured DC population loaded with those antigens ex vivo; (iii) a defined route of administration to enhance presentation to T cells; and (iv) at least one immunomodulatory agent aiming to reduce the immunosuppressor environment imposed by the tumor [66].
3.1. Choosing the Right Antigen
In ovarian cancer, the scarcity of well characterized tumor antigens and the elevated molecular heterogeneity of the disease [86] have represented an important limitation to finding an adequate target antigen for vaccination. Additionally, even when a defined target is known, and vaccination induces an immune response, evolution of the tumor selecting antigen-loss variants following vaccination by the process of immunoediting may hinder long-term benefit [43]. This proves that single-target immunization can result in tumor variants facilitating immune escape following initial response, and therefore vaccination with multiple defined antigens seems crucial for achieving significant clinical benefit.
Another possible reason for reduced efficacy is that, to date, most vaccines were targeted against defined non-mutated self-antigens [87]. Tumors express non-mutated self-antigens (the so-called “public” antigens) as a result of tissue or lineage-specific gene expression or gene deregulation induced by transformation. These tumor self-antigens are male germline antigens (such as NY-ESO-1), overexpressed antigens (as HER-2/neu) or tissue/lineage-specific antigens, which are shared both by the tumor and the tissue they originated from (as gp100 in melanoma). For most self-antigens, T cell reactivity against them is low by definition, due to the development of tolerance toward them, designed to avoid undesired autoimmune events.
On the other hand, tumors express a second class of TAAs that can be recognized by T cells: mutated neo-antigens. These neo-antigens result from the large number of mutations that happen in tumor cells as a consequence of their inherent genetic instability; therefore, they are fully tumor specific. Deep sequencing analysis of tumor cells has revealed that they harbor usually between 10 and few thousand private somatic mutations; most of these mutations are different even among tumors of the same histotype [88,89]. In contrast to self-antigens, T-cell reactivity towards neo-antigens shows a functional avidity similar to the avidity observed in anti-viral T-cells [90]. Furthermore, T-cell response against neo-antigens is not expected to induce any autoimmune toxicity against healthy tissues, making vaccination toward neo-antigens a very attractive option. However, neo-antigens are mostly patient-specific, since the individual mutations found in any part of the tumors are essentially distinct [91]. Currently, it is possible to identify the repertoire of mutation-derived epitopes (the mutanome) present in one tumor using state-of-the-art technology, such as next-generation sequencing (NGS) to produce a list of individual cancer mutations or prediction of epitopes binding to the patient’s haplotype using bioinformatics’ tools (e.g., NetMHC). This implies that, based on current knowledge, no vaccine can be designed to target shared neo-antigens in a large group of patients. Furthermore, identification of these “private-mutations” is currently laborious, and major automation is required for adaptation in routine practice [86]. In spite of this, a recent pilot clinical trial has demonstrated that vaccination with DCs pulsed with neo-antigen peptides is safe and effective in boosting neo-antigen-specific T-cells in 3 melanoma patients [92]. For these reasons, targeting neo-antigens is a potential source for vaccine improvement, and therefore several Phase I clinical trials have been started to test the concept of neo-antigen vaccines in cancer patients, in melanoma (NCT01970358, NCT02035956), glioblastoma (NCT02287428, NCT02510950, NCT02149225), and breast cancer (NCT02316457), using peptides (admixed with adjuvants) in some trials, or RNA to deliver the neo-antigens. This principle is also applicable to ovarian cancer.
Alternatively to the use of selected antigens, tumor antigens can be obtained directly from tumor cells or lysates, which will include both the public and the private antigens (the neo-antigens), without the hurdles of neo-antigen identification and preparation. In most cases, autologous tumor lysates used in vaccine preparation are subjected to multiple freeze–thaw cycles to induce primary necrosis of cancer cells. However, freeze–thaw induced necrosis has not demonstrated high immunogenicity, and it has been shown to even inhibit TLR-induced maturation and function of DCs [93]. It was also shown that induction of tumor cell stress before lysis could partially reverse lysate-induced DC suppression, and only DCs loaded with stressed lysates afforded protection against tumor challenge in vivo. Other approaches to improve the immunogenicity of whole tumor lysate vaccination have demonstrated some successes in the clinic. DC vaccines using whole lysate from irradiated tumor cells have been successfully implemented in clinical trials including melanoma, high-grade glioma, and prostate cancer patients [94,95,96]. Interestingly, in a clinical trial including 18 patients with relapsed B-cell lymphoma patients treated with a DC vaccine loaded with whole autologous tumor lysate treated by heat shock, γ-radiation, and UV ray, 6 patients (33%) showed clinical and immunological responses, which were positively correlated with the extent of calreticulin and heat shock protein 90 (HSP90) surface expression in the DC antigenic cargo [97]. Consistently, we are currently running two trials in ovarian cancer where we use autologous hypochlorous acid (HOCl)-oxidized whole tumor cells lysate vaccines after demonstrating in mouse models that HOCl-based oxidation induces primary necrosis of tumor cells, showing superior immunogenicity as compared to UVB irradiation and freeze–thaw cycles [25].
A potential alternative to increase antigen immunogenicity would be the combined use of DC vaccination with oncolytic viruses, given their potential to induce Immunogenic Cell Death (ICD). The beneficial effect of intratumoral delivery of oncolytic viruses prior to DC vaccination has been demonstrated in different murine tumor models [98,99]. These oncolytic viruses, active in ovarian cancer models, could be either directly injected into tumors prior to DC vaccination, or alternatively, oncolysates could be prepared from whole tumor cells, that could be loaded into DCs for more efficient vaccination. Finally, when using whole tumor cell extracts (either loading DCs or by direct vaccination), immunogenicity could be enhanced by adding specific molecules, such as a fusion protein of single-chain antibody variable fragment (scFv) mesothelin (MSLN), to Mycobacterium tuberculosis (MTB) heat shock protein 70 (HSP70), which is a potent immune activator able to stimulate monocytes and DCs, enhancing DC maturation and aggregation, and improving cross-priming of T cells. Intraperitoneal injection of this bifunctional fusion protein in murine models of ovarian cancer and mesothelioma increased tumor-specific CD8+ T-cell dependent tumor responses, significantly enhancing survival and slowing tumor growth [100].
3.2. Providing DC Maturation Signals to Enhance T Cell Activation
One important consideration regarding T cell activation is that, in the absence of adequate DC maturation signals, presentation of antigens to T cells may induce tolerance by production of regulatory T cells (Treg) [101,102,103]. Consequently, in vaccination strategies using peptides or proteins (single or mixtures; synthetic or whole cell lysates), either directly or pulsed onto DCs, an appropriate adjuvant must be incorporated, in order to provide the required activation/maturation signal to DCs that will allow them to differentiate, as well as to process and present tumor-antigen derived peptides to T cells [104,105]. For this reason, a number of different adjuvants are currently available, such as Toll-like receptor ligands, which play a key role in DC maturation [21]. However, the best adjuvant choice is still not defined. To this end, we are currently running a trial in ovarian cancer (NCT02452775) comparing OC-L vaccine alone with the addition of either Montanide (a water-in-oil emulsion possessing an immune stimulatory effect) or poly-ICLC alone (a TLR-3 agonist able to upregulate genes involved in innate immune pathways including IFN-α, IFN-β, IFN-γ upon administration in healthy volunteers [106]). In a previous pilot clinical study with ovarian cancer patients, we have also demonstrated that LPS-activated DCs produced high levels of Th-1 polarizing cytokines including IL-12p70 and CXCL10, as well as stimulated potent polyclonal tumor T cells in patients [25]. Other TLR agonists with potential interest in ovarian cancer vaccines are imiquimod, motolimod, or CpG-oligodeoxynucleotides (ODNs). Imiquimod is a TLR7 agonist approved by the FDA for topical use in basal cell skin cancer, which has been shown to induce IFN-α and other cytokines [107], as well as to efficiently activate DC maturation [108]; it has been successfully used as an adjuvant for NY-ESO-1 protein for treating metastatic melanoma [109]. Motolimod (VTX-2337) is a TLR8 agonist which is able to stimulate production of TNF-α and IL-12 from monocytes and myeloid DCs, and stimulates IFN-γ production from NK cells [110]; motolimod has been tested for dose finding in a clinical trial including patients with advanced solid tumors and lymphoma [111], and is currently being tested in patients with recurrent ovarian cancer, in combination with doxorubicin (NCT01666444).
Unmethylated cytosine-phosphate-guanine (CpG) dinucleotides, which are relatively common in bacterial and viral DNA but are suppressed and methylated in vertebrate DNA, are recognized by and able to activate TLR9. Several synthetic CpG-ODNs have been developed for cancer treatment [112]. In an ovarian preclinical model, CpG-ODNs have been tested as adjuvants in combination with DCs electroporated with whole tumor cell RNA, and found to enhance their efficacy [113]. Similarly, in a murine glioblastoma model, 55% of mice vaccinated with CpG/lysate combination demonstrated over two times greater median survival compared to mice treated with CpG only, tumor lysate only or no treatment (p < 0.05) [114]. Furthermore, CpG-ODNs can synergize with other TLR agonists to activate more than one DC subset, such as flagelin (a TLR5 agonist, leading to activation of pDCs and dermal DCs) [115], or poly-ICLC, a combination leading to Langerhans cells activation with strong production of IL-6 and IL-12 and enhanced antitumor immunity [116].
3.3. Targeting the Right DC
In DC vaccination strategies, another important consideration to increase immunogenicity is the choice of the DC subset. In most immunotherapy trials, monocyte-derived DCs are used, which are differentiated ex vivo with recombinant GM-CSF and IL-4 [117]. These mature DCs are efficient phagocytes of antigens, able to produce high IL-12 upon activation. Their major advantage is that they can be easily manipulated before infusing them into patients. However, the ex vivo production of these DCs is labor intensive and costly. One attractive alternative is to specifically target DCs in vivo with appropriate tumor antigens, activating them to elicit potent anti-tumor T cell responses. In the immune system, hematopoietic stem cells (HSCs) differentiate into common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs). CMPs subsequently differentiate into monocytes and pre-DCs in the bone marrow. Both monocytes and pre-DCs enter the blood and migrate to lymphoid organs and peripheral tissues, where they can differentiate into lymphoid DCs and tissue-resident DCs [118]. Differentiated DCs can be classified in different subsets, according to their phenotype, receptor expression, chemokine and cytokine production, tissue location, and the type of immune response they induce. Ideally, we could use specific adjuvants, as previously discussed, to selectively activate in vivo one or more DC subsets by their surface receptors. For instance, TLR7 and TLR9 are uniquely expressed in plasmacytoid DCs (pDCs) [119], and they can be activated to become potent secretors of IFN-α and -β with imiquimod and CpG-ODNs, respectively [120,121]. Langerhans cells present TLR3, and therefore can be activated using poly-ICLC; and dermal DCs present TLR4, thereby they can be activated with monophosphoril A (MPLA), which is a derivative of LPS with lower toxicity. Lastly, blood and lymph node DCs expressing TLR8 can be activated using motolimod. However, targeting DCs in vivo one would have less control over the quality and magnitude of the anti-tumor response.
Additionally, selection of the administration route for vaccination can also have an effect in the effectiveness of DC maturation process. Traditionally, the routes used for vaccination against many infectious diseases have been subcutaneous (s.c.) and intramuscular (i.m.). However, different studies demonstrate that the intradermal (i.d.) route is more effective in inducing protective immunity, even inducing seroconversion in subjects unresponsive to i.m. HBV vaccination [122]. This is probably due to the fact that the dermis contains a much larger population of DCs compared to subcutaneous fat and muscles, as well as the extensive network of lymphatic vessels present in the dermis, which will favor an effective loading of antigen on DCs and their subsequent transport to draining lymph nodes for presentation to T cells. Alternatively, direct administration of antigens into the regional lymph node (intranodally, i.n.) is perhaps the most effective way to ensure maximum display of antigens to DCs, which will not have to migrate after antigen loading. Different routes of antigen-coding mRNA delivery has been analyzed in a mouse model, showing that following intranodal injection, resident DCs in the nodes selectively took up the mRNA, inducing potent CD4+ and CD8+ T cell responses after repeated i.n. injections in tumor-bearing mice, which was not observed with subcutaneous, intradermal, or near nodal administrations [123]. However, a different possibility is direct antigen administration to tumor-infiltrating DCs (TIDCs), which have been documented in different types of cancers, including ovarian cancer [124], although their function is not yet well characterized. Interestingly, in a review of 54 trials using DC vaccines in melanoma performed to evaluate the relationship between clinical effects and vaccine parameters, it was concluded that the objective (11.7%) and clinical (27.7%) responses for the i.n. route were the highest, but the type of response did not differ significantly among the injection routes (p = 0.40 and p = 0.64, respectively, by Kruskal–Wallis test) [125].
Another option in DC vaccination strategies to increase immunogenicity while keeping control over the anti-tumor response would be using a different subset of DCs to be loaded ex vivo. For instance, in some cases the use of naturally occurring DCs has been tested, such as antigen-loaded purified plasmacytoid DCs [126]. However, whether this strategy is more efficacious than the use of monocyte-derived DC vaccine remains to be determined [127]. Other groups are exploring the use of Langerhans-like cells (named “IL-15 DCs”) as sources for the DC vaccines, given their strong potential to stimulate cytotoxic T cell responses [128,129]. IL-15 DCs can be obtained by culturing monocytes from blood in IL-15 instead of IL-4 in standard protocols, and the resulting DCs have an increased capacity to stimulate NK cells cytotoxicity, which could be a crucial contribution in anti-tumor efficacy of DC vaccines [130]. In this respect, we (in collaboration with other leading groups in the field) are planning a clinical trial in ovarian cancer patients to receive an autologous vaccine comprised of selected professional crosspriming autologous dendritic cells (XP-DC), loaded in vitro with lysate from autologous oxidized tumor cells, administered intranodally. XP-DC are a rare subset of human myeloid DCs, representing 0.2%–0.3% of total mononuclear monocytes and expressing BDCA-3 (a.k.a. CD141 or thromobomodulin), which have been confirmed as functional equivalents to mouse CD8a+ family [131]. BDCA-3+ DC (XP-DC) are superior in cross-presentation of cell-associated antigens to CD8+ T cells to induce CTL [132,133,134], and this cross-priming capacity of XP-DC has proven essential for the initiation of effective CTL cell responses against tumors.
3.4. Improving the Immunocompetent Status of Vaccinated Patients
Tumor progression is generally associated with both central and peripheral tolerance mechanisms to deplete or inactivate the relevant T cell repertoire, generating an immunosuppressive tumor microenvironment (TME) allowing tumor cells to evade immune attack [66]. Some of the mechanisms elaborated by tumors that have been observed in ovarian cancer include downregulation of class I MHC molecules [135], upregulation of surface molecules that induce T cell anergy of exhaustion (i.e., PD-L1 [136]), releasing immunosuppressive molecules such as IDO [12], or Treg recruitment to the TME [9]. Other cells in the TME can also release factors implicated in immunosuppression, such as VEGF release by tumor vascular cells [137,138]; or myeloid-derived suppressor cells (MDSC) recruited into the TME, which may release T cell inhibitors such as arginase and nitrous oxide synthase [139]. Additionally, it has recently been identified that the critical soluble mediators of type-1 immune effector cells, IFNγ and TNFα, synergize in the induction of COX-2, the key enzyme in PGE2 synthesis, implicated in hyperactivation of MDSC within the TME of ovarian cancer patients. Interestingly, this negative feedback limiting type-1 responses could be eliminated by COX-2 blockade, allowing amplification of type-1 immunity in the TME [140].
Consistent with reduced immunocompetence associated with tumor progression, in a review of 54 trials using DC vaccination including 967 patients with melanoma, it was observed that the objective response rate did not differ significantly between stages III and IV, but the clinical response differed significantly between the two groups (p = 0.03), and PD cases differed significantly between stages II (18.8%) and IV (52.6%) and between stages III (23.1%) and IV (both p = 0.0001) [125]. Therefore, efficacy of vaccination strategies and clinical benefit could be improved by selecting patients with either minimal burden disease, or with NED (no evidence of disease) after debulking strategies, and with good performance status, whenever possible.
Additionally, most patients enrolled in clinical trials involving cancer vaccines are elderly patients with already significantly compromised immune systems, due to immunosenescence [141]. This fact could contribute to the decreased ability of the elderly to control infectious diseases, their generally poor response to vaccination, as well as to the increased incidence of cancer with age. We as well as others have demonstrated that patients with advanced ovarian cancer exhibited a dampened T-cell response to the diphtheria carrier protein CRM197, a potent xeno-neoantigen of Prevnar™, which was given to monitor immune responsiveness in an autologous whole tumor lysate vaccine protocol [26,142]. Interestingly, myeloma patients have previously been shown to exhibit robust T-cell responses to CRM197, suggesting that ovarian cancer patients may be characterized by a profound level of systemic immunosuppression. Supplementary interventions might be required to boost T cell immunity. For instance, implanting genetically engineered stromal cells in the thymus to secrete IL-7 (a T cell survival factor) has been explored in the mouse [143]. Nutritional interventions might also be useful, such as supplements of either vitamin E [144], or conjugated linoleic acid [145], as well as controlling cholesterol levels [146], which could all improve T cell function in cancer patients.
4. Immunomodulatory and Combinatorial Strategies in Ovarian Cancer
One way to improve vaccine efficacy would be combining them with other immunomodulatory agents aiming to obtain a synergistic effect [147,148,149]. In this respect, immune checkpoints are mechanisms established to maintain self-tolerance (therefore preventing autoimmunity), and to protect tissue from damage after immune activation in response to pathogens. Checkpoint molecules include CTLA-4 (Cytotoxic T Lymphocyte Antigen-4), PD-1 (Programmed Death-1), LAG-3 (Lymphocyte Activation Gene-3), TIM-3 (T cell Immunoglobulin and Mucin protein-3), and several others (reviewed elsewhere; [150]). They have been found to modulate T cell responses to self-proteins, but also to chronic infections and tumor antigens, with CTLA-4 being the first shown to augment antitumor immune responses [151]. Following their success in other types of immunogenic tumors [152], immunomodulatory agents are currently being tested in ovarian cancer. For instance, Hodi et al. reported that periodic infusions of anti-CTLA-4 antibodies after vaccination with irradiated, autologous tumor cells engineered to secrete GM-CSF (GVAX) demonstrated an objective response with durable remission for four years in one patient with ovarian cancer [153]. To clarify the role of anti-CTLA-4 as monotherapy in ovarian cancer, a new Phase II clinical trial is being conducted in the USA (NCT01611558). Other immunomodulatory agents, such as antibodies blocking PD-1/PD-L1 pathway (from different sources [154]) are currently tested; for instance, a Phase I study is being conducted to test an anti-PD-L1 moAb as monotherapy in patients with metastatic or advanced solid tumors, including ovarian cancer (NCT01772004). However, although therapies blocking the immune checkpoints show significant clinical efficacy in advanced tumors [155], attributed to potent activation of T cells, in general terms monotherapy with a single moAb yields a low rate of objective responses [156]. For this reason, the combination of vaccination with immune checkpoint blocking agents is also worth testing in clinical trials, as tumors employ both PD-1 and CTLA-4 pathways to depress the immune system. CTLA-4 and PD-1 are both coinhibitory molecules that belong to the same family of molecules, yet there is evidence suggesting that they use distinct non-redundant mechanisms to inhibit T-cell activation [157]. In preclinical models using mice with pre-implanted B16 melanomas, it has been shown that concomitant blockade of both pathways can modulate Treg functions and enhance antitumor responses, as compared to single immune checkpoint blockade [158]. In this direction, clinical trials are currently being performed to test double immune checkpoint blockade, either specifically in ovarian cancer patients (NCT02498600), or in patients with solid tumors, including patients with ovarian cancer (NCT01975831).
Interestingly, preclinical studies in ovarian cancer mouse models have demonstrated a consistently greater anti-tumor effect when checkpoint blockade and vaccines are used in combination, compared with either alone. In an ID8 ovarian cancer tumor model it was demonstrated that PD-L1 blockade can restore antitumor immunity, and synergize with whole tumor antigen vaccine (GVAX) to produce tumor rejection. Remarkably, vaccine alone was completely ineffective, while PD-L1 antibody monotherapy produced half the cure rate than GVAX plus PD-L1 blockade combination [159]. Blockade of both PD-1 and CTLA-4 in those mice resulted in reversal of CD8+ TIL dysfunction and led to tumor rejection in two-thirds of mice. Double blockade was associated with increased proliferation of antigen-specific effector CD8+ and CD4+ T cells, antigen-specific cytokine release, inhibition of suppressive functions of Tregs, and upregulation of key signaling molecules critical for T cell function [160]. Therefore, these encouraging preclinical results warrant the initiation of clinical trials trying the combination of vaccines with checkpoint inhibitors in ovarian cancer.
The IDO pathway is another regulator of the immune response in the tumor microenvironment. Indoleamine 2,3-dioxygenase (IDO) is a tryptophan-catabolizing enzyme that induces immune tolerance, by depleting tryptophan locally and producing toxic tryptophan catabolites, such as kyneurine, inhibiting proliferation of T-cells (both CD4+ and CD8+), and natural killer (NK) cells [161,162]. There is evidence showing that ovarian cancer patients with elevated IDO expression show significant impairment both in OS and PFS as compared to patients with low or no IDO expression [12]. For this reason, one IDO inhibitor, epacadostat, is currently being tested in ovarian cancer in different clinical trials, either alone as neoadjuvant treatment (NCT02042430), or in combination with checkpoint inhibitors (NCT02327078, NCT02178722) aiming to obtain a synergistic effect. One phase I/II trial (NCT02575807) is currently testing CRS-207, a recombinant Listeria-based cancer vaccine containing a live-attenuated strain of the facultative intracellular bacterium Listeria monocytogenes (Lm) expressing human mesothelin, in combination with epacadostat in adults with platinum-resistant ovarian cancer.
Other combinations of immunomodulatory therapies with vaccines in patients with advanced ovarian cancer include the use of Ontak (denileukin diftitox, a cytotoxic recombinant protein consisting of IL-2 protein sequences fused to diphtheria toxin) aiming to deplete CD4+CD25+ immunoregulatory T-cells (Treg) in combination with a DC based vaccine, which is currently being investigated in a Phase II study (NCT00703105). Additionally, combinatorial strategies with DC vaccination and other interventions such as radiation, chemotherapy, hyperthermia, T-cell transfer, and antibody therapy might produce important breakthroughs in treating patients with ovarian cancer. There is growing evidence that radiation therapy targeted to the tumor can convert it into an in situ tumor vaccine by inducing release of antigens during cancer cell death in association with pro-inflammatory signals that trigger the innate immune system to activate tumor-specific T cells [163]. Radiation also affects the tumor microenvironment allowing increased infiltration by activated T cells and overcoming some of the mechanisms of tumor immunosuppression. Recently, in a pilot clinical trial including patients with metastatic solid tumors, the combination of radiotherapy with GM-CSF produced objective abscopal responses (radiotherapy-induced immune-mediated tumor regression at sites distant to the irradiated field) in some patients (26.8%) [164]. Additionally, it has been observed that including TGFβ blockade and anti-PD-1 antibodies extended survival achieved with radiation [165]. Further strategies using combinations of radiotherapy and immunotherapy are therefore currently being explored [166,167]. In ovarian cancer, although radiation is not widely accepted as a routine treatment modality in the initial treatment in EOC patients, it can be considered in higher-risk stage I and II disease and in stage III disease where small-volume residual disease is present after surgery.
The potential of chemotherapy combination with DC vaccination has also been explored. In a pilot clinical study including seven stage III colon cancer patients receiving standard adjuvant oxaliplatin/capecitabine chemotherapy and vaccinated at the same time with keyhole limpet hemocyanin (KLH) and carcinoembryonic antigen (CEA)-peptide pulsed DCs, an enhanced non-specific T-cell reactivity upon oxaliplatin administration was observed, results that support further testing of the combined use of tumor vaccination with oxaliplatin-based chemotherapy [168]. Importantly, platinum-based chemotherapy is the mainstay of treatment in EOC, and therefore chemotherapy should be considered in combination strategies to improve vaccine efficacy in ovarian cancer treatment.
Other immunomodulatory agents could also enhance antitumor responses in DC-based immunotherapy. It was recently described that the infiltration of T cells into the tumor endothelial barrier was mediated by the death mediator Fas ligand (FasL/CD95L) in the tumor vasculature of human and mouse solid tumors [169] and it was demonstrated that tumor-derived VEGF-A, IL-10 and prostaglandin E (PGE) cooperatively induced FasL expression in endothelial cells, allowing them to kill effector CD8+ T cells but not Treg cells, which express higher levels of c-FLIP. Dual inhibition of VEGF and PGE with anti-VEGF and aspirin, demonstrated a significant effect in tumor regression. These observations led us to conclude that modulating the tumor endothelial barrier with aspirin and bevacizumab is a promising approach to combine with vaccinations. Thereby, we conducted a clinical trial for subjects with recurrent ovarian cancer using OCDC (DC loaded with oxidized tumor lysate) administered intranodally alone, or in combination with i.v. bevacizumab and cyclophosphamide (aiming to reducing Tregs), or in combination with i.v. bevacizumab, cyclophosphamide and aspirin (NCT01132014) and the results are currently being analyzed.
5. Concluding Remarks
At present, a plethora of different options is open to discovery in the treatment of ovarian cancer. This represents a great opportunity to improve patients’ condition, and we can be reasonably optimistic that some of those options will surely translate into clinical benefit for patients. However, new opportunities come always together with new challenges, which we will have to confront and overcome. Among these, the first obvious challenge is to choose the options with more chances to yield clinical benefit, which requires a sound understanding of deep biological processes engaged in immune response to tumor development, and this implicates continuous investment in fundamental research. DC vaccines are one of the most promising options, due to a currently acceptable objective response rate, comparable to that obtained using standard of care approaches or checkpoint inhibitors, which has been demonstrated in different indications [85], coupled to a good safety profile (rarely presenting immune-related toxicities [60]), and associated with preserved quality of life of cancer patients [61]. Nevertheless, as we have previously discussed, results could be further improved by carefully selecting the right antigen to target, the right DC subtype, and the adequate adjuvant and delivery route. In addition, choosing the right patient population to vaccinate and improving patients’ immunocompetent status could also contribute to increased clinical benefit (see Figure 1).
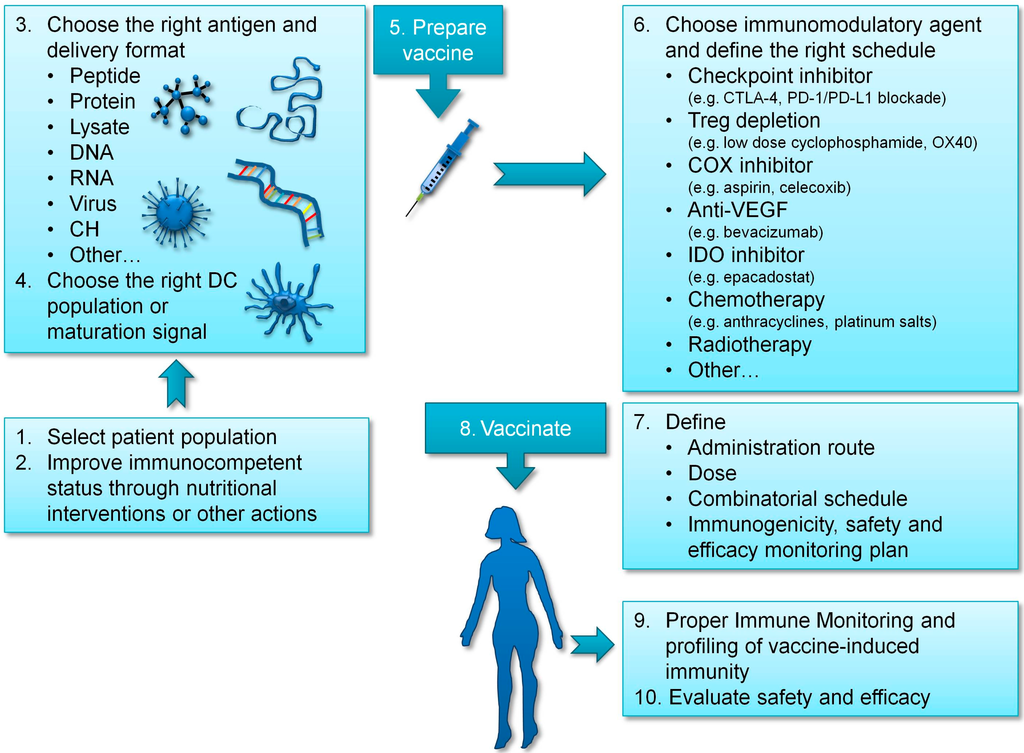
Figure 1.
Summary of required considerations to implement a successful vaccination strategy in ovarian cancer.
Finally, using combinatorial approaches aiming to obtain synergistic effects could lead to major breakthroughs in treating patients with ovarian cancer. However, it should be taken into account that some options will be limited by technical capacities, being restricted to centers with the right infrastructure and finances to successfully develop these strategies. This will undoubtedly generate deep discussions regarding patients’ access to treatment. From the clinical point of view, although combination of DC vaccines with other agents can provide long-term benefit to incurable patients, this has to be tempered by the observation that patients may present severe (even lethal in some cases) auto-inflammatory events, mostly in the lungs and in the gastrointestinal tract, that need to be early recognized by oncologists to allow effective management, implicating close monitoring of patients during and after treatment, as it is the case for checkpoint inhibitors [170]. Furthermore, it has to be noted that clinical trials design in immuno-oncology needs to be carefully planned, as safety and efficacy concerns differ substantially from those evaluated in clinical trials with cytotoxic agents [171]: immuno-oncology agents present unique kinetics, compared with traditional targeted or chemotherapeutic agents [172], and their combination require a rational design of synergistic combination strategies [173]. Finally, a detailed strategy for early detection and management of toxicity as well as planned demonstration of predicted additive or synergistic benefit are also required.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BC | breast cancer |
| CEA | carcinoembryonic antigen |
| CRC | colorectal cancer |
| CTLA-4 | cytotoxic T-lymphocyte-associated antigen 4 |
| DC | dendritic cells |
| EOC | epithelial ovarian cancer |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| i.p. | intraperitoneal |
| IDO1 | indoleamine 2,3-dioxygenase |
| IFN | interferon |
| KLH | keyhole limpet hemocyanin |
| LC | lung cancer |
| MDSC | myeloid derived suppressor cells |
| MEL | melanoma |
| MES | mesothelioma |
| MHC | major histocompatibility complex |
| moAb | monoclonal antibody |
| mOC | metastatic ovarian cancer |
| NED | no evidence of disease |
| NSCLC | non-small cell lung cancer |
| OC | ovarian cancer |
| OCDC | DC loaded with oxidized tumor lysate |
| PCRC | pancreatic cancer |
| PC | prostate cancer |
| PD-(L)1 | programmed death-(ligand)1 |
| PGE | prostaglandine E |
| PPV | personalized peptide vaccine |
| ROC | recurrent ovarian cancer |
| SAR | sarcoma |
| shRNA | short hairpin RNA |
| TAA | tumor-associated antigen |
| TGF | transforming growth factor |
| TIL | tumor infiltrating lymphocytes |
| TME | tumor microenvironment |
| Tregs | regulatory T cells |
| VEGF(R) | vascular endothelial growth factor (receptor) |
References
- Ovarian Cancer: Statistics. Available online: http://www.cancer.net/cancer-types/ovarian-cancer/statistics (accessed on 22 January 2016).
- IARC. GLOBOCAN 2012. Incidence, Mortality and Prevalence Worldwide (Ovary). Available online: http://globocan.iarc.fr/old/burden.asp?selection_pop=224900&Text-p=World&selection_cancer=22182&Text-c=Ovary&pYear=8&type=0&window=1&submit=%C2%A0Execute (accessed 18 February 2016).
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral t cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Schlienger, K.; Chu, C.S.; Woo, E.Y.; Rivers, P.M.; Toll, A.J.; Hudson, B.; Maus, M.V.; Riley, J.L.; Choi, Y.; Coukos, G.; et al. TRANCE- and CD40 ligand-matured dendritic cells reveal MHC class I-restricted T cells specific for autologous tumor in late-stage ovarian cancer patients. Clin. Cancer Res. 2003, 9, 1517–1527. [Google Scholar] [PubMed]
- Goodell, V.; Salazar, L.G.; Urban, N.; Drescher, C.W.; Gray, H.; Swensen, R.E.; McIntosh, M.W.; Disis, M.L. Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J. Clin. Oncol. 2006, 24, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Hermonat, P.L.; Ravaggi, A.; Bellone, S.; Roman, J.J.; Smith, C.V.; Pecorelli, S.; Radominska-Pandya, A.; Cannon, M.J.; Parham, G.P. Phenotypic and functional analysis of tumor-infiltrating lymphocytes compared with tumor-associated lymphocytes from ascitic fluid and peripheral blood lymphocytes in patients with advanced ovarian cancer. Gynecol. Obstet. Investig. 2001, 51, 254–261. [Google Scholar] [CrossRef]
- Gnjatic, S.; Ritter, E.; Buchler, M.W.; Giese, N.A.; Brors, B.; Frei, C.; Murray, A.; Halama, N.; Zornig, I.; Chen, Y.T.; et al. Seromic profiling of ovarian and pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 5088–5093. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory t cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Wolf, A.M.; Rumpold, H.; Fiegl, H.; Zeimet, A.G.; Muller-Holzner, E.; Deibl, M.; Gastl, G.; Gunsilius, E.; Marth, C. The expression of the regulatory T cell-specific forkhead box transcription factor foxp3 is associated with poor prognosis in ovarian cancer. Clin. Cancer Res. 2005, 11, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Inaba, T.; Ino, K.; Kajiyama, H.; Yamamoto, E.; Shibata, K.; Nawa, A.; Nagasaka, T.; Akimoto, H.; Takikawa, O.; Kikkawa, F. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol. Oncol. 2009, 115, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Sisirak, V.; Meeus, P.; Gobert, M.; Treilleux, I.; Bajard, A.; Combes, J.D.; Faget, J.; Mithieux, F.; Cassignol, A.; et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011, 71, 5423–5434. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41 (Suppl. 5), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; van Netten, J.P.; van Netten, C. Dr william coley and tumour regression: A place in history or in the future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar] [PubMed]
- D’Amelio, E.; Salemi, S.; D’Amelio, R. Anti-infectious human vaccination in historical perspective. Int. Rev. Immunol. 2015, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Lowy, D.R. Virus infection and human cancer: An overview. Recent Res. Cancer 2014, 193, 1–10. [Google Scholar]
- Szarewski, A.; Poppe, W.A.; Skinner, S.R.; Wheeler, C.M.; Paavonen, J.; Naud, P.; Salmeron, J.; Chow, S.N.; Apter, D.; Kitchener, H.; et al. Efficacy of the human papillomavirus (hpv)-16/18 as04-adjuvanted vaccine in women aged 15-25 years with and without serological evidence of previous exposure to hpv-16/18. Int. J. Cancer 2012, 131, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.-Y. Therapeutic cancer vaccines: Past, present and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [PubMed]
- Seya, T.; Shime, H.; Takeda, Y.; Tatematsu, M.; Takashima, K.; Matsumoto, M. Adjuvant for vaccine immunotherapy of cancer - focusing on toll-like receptor 2 and 3 agonists for safely enhancing antitumor immunity. Cancer Sci. 2015, 106, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.-L.; Coukos, G.; Kandalaft, L.E. Whole tumor antigen vaccines: Where are we? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [PubMed]
- Guidance Development Review Committee; Working Group for Clinical Studies of Cancer Immunotherapy; Working Group for Effector Cell Therapy; Working Group for CMC/Non-clinical Studies; Working Group for Cancer Vaccines and Adjuvants; Working Group for Anti-immune Checkpoint Therapy and Comprehensive Cancer Immunotherapy; Biostatistics Subcommittee. 2015 guidance on cancer immunotherapy development in early-phase clinical studies. Cancer Sci. 2015, 106, 1761–1771. [Google Scholar]
- Bapsy, P.P.; Sharan, B.; Kumar, C.; Das, R.P.; Rangarajan, B.; Jain, M.; Suresh Attili, V.S.; Subramanian, S.; Aggarwal, S.; Srivastava, M.; et al. Open-label, multi-center, non-randomized, single-arm study to evaluate the safety and efficacy of dendritic cell immunotherapy in patients with refractory solid malignancies, on supportive care. Cytotherapy 2014, 16, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.-L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Nisenbaum, H.L.; Levine, B.L.; et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Powell, D.J.; Chiang, C.L.; Tanyi, J.; Kim, S.; Bosch, M.; Montone, K.; Mick, R.; Levine, B.L.; Torigian, D.A.; et al. Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated t cells in recurrent ovarian cancer. Oncoimmunology 2013, 2, e22664. [Google Scholar] [CrossRef] [PubMed]
- Hernando, J.; Park, T.-W.; Kübler, K.; Offergeld, R.; Schlebusch, H.; Bauknecht, T. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: Clinical and immunological evaluation of a phase i trial. Cancer Immunol. Immunother. 2002, 51, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Loveland, B.E.; Zhao, A.; White, S.; Gan, H.; Hamilton, K.; Xing, P.X.; Pietersz, G.A.; Apostolopoulos, V.; Vaughan, H.; Karanikas, V.; et al. Mannan-MUC1–pulsed dendritic cell immunotherapy: A phase I trial in patients with adenocarcinoma. Clin. Cancer Res. 2006, 12, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Peethambaram, P.P.; Melisko, M.E.; Rinn, K.J.; Alberts, S.R.; Provost, N.M.; Jones, L.A.; Sims, R.B.; Lin, L.R.C.; Frohlich, M.W.; Park, J.W. A phase I trial of immunotherapy with lapuleucel-T (APC8024) in patients with refractory metastatic tumors that express her-2/neu. Clin. Cancer Res. 2009, 15, 5937–5944. [Google Scholar] [CrossRef] [PubMed]
- Brossart, P.; Wirths, S.; Stuhler, G.; Reichardt, V.L.; Kanz, L.; Brugger, W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood 2000, 96, 3102–3108. [Google Scholar] [PubMed]
- Chu, C.; Boyer, J.; Schullery, D.; Gimotty, P.; Gamerman, V.; Bender, J.; Levine, B.; Coukos, G.; Rubin, S.; Morgan, M.; et al. Phase I/II randomized trial of dendritic cell vaccination with or without cyclophosphamide for consolidation therapy of advanced ovarian cancer in first or second remission. Cancer Immunol. Immunother. 2012, 61, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Chiba, A.; Izawa, H.; Yanagida, E.; Okamoto, M.; Shimodaira, S.; Yonemitsu, Y.; Shibamoto, Y.; Suzuki, N.; Nagaya, M. The feasibility and clinical effects of dendritic cell-based immunotherapy targeting synthesized peptides for recurrent ovarian cancer. J. Ovarian Res. 2014, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.; Barve, M.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Lazar, M.; Lifshitz, S.; Magee, M.; Oh, J.; Mill, S.W.; et al. Phase I trial of “bi-shRNAifurin/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol. Ther. 2012, 20, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.H.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A live-attenuated listeria vaccine (ANZ-100) and a live-attenuated listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clin. Cancer Res. 2012, 18, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, N.; Mochizuki, K.; Harada, M.; Sukehiro, A.; Kawano, K.; Yamada, A.; Ushijima, K.; Sugiyama, T.; Nishida, T.; Yamana, H.; et al. Vaccination with predesignated or evidence-based peptides for patients with recurrent gynecologic cancers. J. Immunother. 2004, 27, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Secord, A.A.; Blackwell, K.; Hobeika, A.C.; Sinnathamby, G.; Osada, T.; Hafner, J.; Philip, M.; Clay, T.M.; Lyerly, H.K.; et al. MHC class I–presented tumor antigens identified in ovarian cancer by immunoproteomic analysis are targets for T-cell responses against breast and ovarian cancer. Clin. Cancer Res. 2011, 17, 3408–3419. [Google Scholar] [CrossRef] [PubMed]
- Chianese-Bullock, K.A.; Irvin, W.P., Jr.; Petroni, G.R.; Murphy, C.; Smolkin, M.; Olson, W.C.; Coleman, E.; Boerner, S.A.; Nail, C.J.; Neese, P.Y.; et al. A multipeptide vaccine is safe and elicits T-cell responses in participants with advanced stage ovarian cancer. J. Immunother. 2008, 31, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Schiffman, K.; Cheever, M.A.; Disis, M.L. Immunization of cancer patients with a HER-2/neu, HLA-A2 peptide, p369–377, results in short-lived peptide-specific immunity. Clin. Cancer Res. 2002, 8, 1014–1018. [Google Scholar] [PubMed]
- Disis, M.L.; Gooley, T.A.; Rinn, K.; Davis, D.; Piepkorn, M.; Cheever, M.A.; Knutson, K.L.; Schiffman, K. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide–based vaccines. J. Clin. Oncol. 2002, 20, 2624–2632. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Rinn, K.; Knutson, K.L.; Davis, D.; Caron, D.; dela Rosa, C.; Schiffman, K. Flt3 ligand as a vaccine adjuvant in association with HER-2/neu peptide-based vaccines in patients with HER-2/neu–overexpressing cancers. Blood 2002, 99, 2845–2850. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Goodell, V.; Schiffman, K.; Knutson, K.L. Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients. J. Clin. Immunol. 2004, 24, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Schiffman, K.; Guthrie, K.; Salazar, L.G.; Knutson, K.L.; Goodell, V.; dela Rosa, C.; Cheever, M.A. Effect of dose on immune response in patients vaccinated with an HER-2/neu intracellular domain protein—based vaccine. J. Clin. Oncol. 2004, 22, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Qian, F.; Matsuzaki, J.; Mhawech-Fauceglia, P.; Andrews, C.; Hoffman, E.W.; Pan, L.; Ritter, G.; Villella, J.; Thomas, B.; et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12837–12842. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, C.S.M.; Gnjatic, S.; Sabbatini, P.; Aghajanian, C.; Hensley, M.L.; Spriggs, D.R.; Iasonos, A.; Lee, H.; Dupont, B.; Pezzulli, S.; et al. Safety and immunogenicity study of NY-ESO-1b peptide and montanide ISA-51 vaccination of patients with epithelial ovarian cancer in high-risk first remission. Clin. Cancer Res. 2008, 14, 2740–2748. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.; Tsuji, T.; Ferran, L.; Ritter, E.; Sedrak, C.; Tuballes, K.; Jungbluth, A.A.; Ritter, G.; Aghajanian, C.; Bell-McGuinn, K.; et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin. Cancer Res. 2012, 18, 6497–6508. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Matsuzaki, J.; James, S.R.; Mhawech-Fauceglia, P.; Tsuji, T.; Miller, A.; Zhang, W.; Akers, S.N.; Griffiths, E.A.; Miliotto, A.; et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol. Res. 2014, 2, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Ashtar, E.; Czystowska, M.; Szajnik, M.E.; Wieckowski, E.; Bernstein, S.; Herrin, V.E.; Shams, M.A.; Steinberg, S.M.; Merino, M.; et al. A gynecologic oncology group phase ii trial of two p53 peptide vaccine approaches: Subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol. Immunother. 2012, 61, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Lambeck, A.J.A.; Gooden, M.J.M.; Hoogeboom, B.-N.; Wolf, R.; Hamming, I.E.; Hepkema, B.G.; Willemse, P.H.B.; Molmans, B.H.W.; Hollema, H.; et al. Immunization with a p53 synthetic long peptide vaccine induces p53-specific immune responses in ovarian cancer patients, a phase II trial. Int. J. Cancer 2009, 125, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Vermeij, R.; Hoogeboom, B.-N.; Schulze, U.R.; Wolf, R.; Hamming, I.E.; van der Zee, A.G.; Melief, K.J.; van der Burg, S.H.; Daemen, T.; et al. Long-term clinical and immunological effects of p53-slp® vaccine in patients with ovarian cancer. Int. J. Cancer 2012, 130, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, R.; Leffers, N.; Hoogeboom, B.-N.; Hamming, I.L.E.; Wolf, R.; Reyners, A.K.L.; Molmans, B.H.W.; Hollema, H.; Bart, J.; Drijfhout, J.W.; et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: A single-arm phase II study. Int. J. Cancer 2012, 131, E670–E680. [Google Scholar] [CrossRef] [PubMed]
- Kawano, K.; Tsuda, N.; Matsueda, S.; Sasada, T.; Watanabe, N.; Ushijima, K.; Yamaguchi, T.; Yokomine, M.; Itoh, K.; Yamada, A.; et al. Feasibility study of personalized peptide vaccination for recurrent ovarian cancer patients. Immunopharmacol. Immunotoxicol. 2014, 36, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.S.; Vadhan-Raj, S.; Butts, C.; Savary, C.; Melichar, B.; Verschraegen, C.; Kavanagh, J.J.; Hicks, M.E.; Levy, L.B.; Folloder, J.K.; et al. Pilot study of Flt3 ligand comparing intraperitoneal with subcutaneous routes on hematologic and immunologic responses in patients with peritoneal carcinomatosis and mesotheliomas. Clin. Cancer Res. 2003, 9, 5228–5237. [Google Scholar] [PubMed]
- Gulley, J.L.; Arlen, P.M.; Tsang, K.-Y.; Yokokawa, J.; Palena, C.; Poole, D.J.; Remondo, C.; Cereda, V.; Jones, J.L.; Pazdur, M.P.; et al. A pilot study to evaluate the safety and clinical outcomes of vaccination with recombinant CEA-MUC-1-TRICOM (PANVAC) poxviral-based vaccines in patients with metastatic carcinoma. Clin. Cancer Res. 2008, 14, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Jäger, E.; Karbach, J.; Gnjatic, S.; Neumann, A.; Bender, A.; Valmori, D.; Ayyoub, M.; Ritter, E.; Ritter, G.; Jäger, D.; et al. Recombinant vaccinia/fowlpox NY-ESO-1 vaccines induce both humoral and cellular NY-ESO-1-specific immune responses in cancer patients. Proc. Natl. Acad. Sci. USA 2006, 103, 14453–14458. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Matsuzaki, J.; Karbach, J.; Neumann, A.; Mhawech-Fauceglia, P.; Miller, A.; Beck, A.; Morrison, C.D.; Ritter, G.; Godoy, H.; et al. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proc. Natl. Acad. Sci. USA 2012, 109, 5797–5802. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, L.A.; Oparin, D.V.; Gooley, T.; Sandmaier, B.M. The role of cancer vaccines following autologous stem cell rescue in breast and ovarian cancer patients: Experience with the STn-KLH vaccine (theratope®). Clin. Breast Cancer 2003, 3 (Suppl. 4), S144–S151. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.J.; Kudryashov, V.; Ragupathi, G.; Danishefsky, S.J.; Livingston, P.O.; Bornmann, W.; Spassova, M.; Zatorski, A.; Spriggs, D.; Aghajanian, C.; et al. Immunization of ovarian cancer patients with a synthetic lewis(y)-protein conjugate vaccine: A phase I trial. Int. J. Cancer 2000, 87, 79–85. [Google Scholar] [CrossRef]
- EMBL-EBI. IPD-IMGT/HLA. 2016 [The IPD-IMGT/HLA Database allows you to retrieve information upon a specific HLA allele as named in the WHO Nomenclature Committee Reports]. Available online: http://www.ebi.ac.uk/ipd/imgt/hla/allele.html (accessed on 25 April 2016).
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Draube, A.; Klein-González, N.; Mattheus, S.; Brillant, C.; Hellmich, M.; Engert, A.; von Bergwelt-Baildon, M. Dendritic cell based tumor vaccination in prostate and renal cell cancer: A systematic review and meta-analysis. PLoS ONE 2011, 6, e18801. [Google Scholar] [CrossRef] [PubMed]
- Leonhartsberger, N.; Ramoner, R.; Falkensammer, C.; Rahm, A.; Gander, H.; Holtl, L.; Thurnher, M. Quality of life during dendritic cell vaccination against metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2012, 61, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Galon, J.; Sautès-Fridman, C.; Cremer, I.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014, 3, e27878. [Google Scholar] [CrossRef] [PubMed]
- Neller, M.A.; Lopez, J.A.; Schmidt, C.W. Antigens for cancer immunotherapy. Semin. Immunol. 2008, 20, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Small, E.J.; Fratesi, P.; Reese, D.M.; Strang, G.; Laus, R.; Peshwa, M.V.; Valone, F.H. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J. Clin. Oncol. 2000, 18, 3894–3903. [Google Scholar] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Bijker, M.S.; van den Eeden, S.J.; Franken, K.L.; Melief, C.J.; van der Burg, S.H.; Offringa, R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur. J. Immunol. 2008, 38, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 receptor in breast cancer: Pathophysiology, clinical use, and new advances in therapy. Chemother. Res. Prac. 2012, 2012, 743193. [Google Scholar] [CrossRef] [PubMed]
- Tuefferd, M.; Couturier, J.; Penault-Llorca, F.; Vincent-Salomon, A.; Broët, P.; Guastalla, J.-P.; Allouache, D.; Combe, M.; Weber, B.; Pujade-Lauraine, E.; et al. HER2 status in ovarian carcinomas: A multicenter gineco study of 320 patients. PLoS ONE 2007, 2, e1138. [Google Scholar] [CrossRef] [PubMed]
- Aurisicchio, L.; Ciliberto, G. Genetic cancer vaccines: Current status and perspectives. Exp. Opin. Biol. Ther. 2012, 12, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Kannagi, R.; Yin, J.; Miyazaki, K.; Izawa, M. Current relevance of incomplete synthesis and neo-synthesis for cancer-associated alteration of carbohydrate determinants—hakomori’s concepts revisited. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.I.; Toyota, M.; Kawashima, R.; Hagiwara, T.; Suzuki, H.; Imai, K.; Shinomura, Y.; Tokino, T.; Kannagi, R.; Dohi, T. DNA hypermethylation contributes to incomplete synthesis of carbohydrate determinants in gastrointestinal cancer. Gastroenterology 2008, 135, 142–151.e3. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Deng, G. Aberrant expression of carbohydrate antigens in cancer: The role of genetic and epigenetic regulation. Gastroenterology 2008, 135, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Dall’Olio, F.; Malagolini, N.; Trinchera, M.; Chiricolo, M. Mechanisms of cancer-associated glycosylation changes. Front. Biosci. 2012, 17, 670–699. [Google Scholar] [CrossRef]
- Avci, F.Y.; Li, X.; Tsuji, M.; Kasper, D.L. Carbohydrates and t cells: A sweet twosome. Semin. Immunol. 2013, 25, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S. Tumor-associated carbohydrate antigens defining tumor malignancy: Basis for development of anti-cancer vaccines. Adv. Exp. Med. Biol. 2001, 491, 369–402. [Google Scholar] [PubMed]
- Dalziel, M.; Crispin, M.; Scanlan, C.N.; Zitzmann, N.; Dwek, R.A. Emerging principles for the therapeutic exploitation of glycosylation. Science 2014, 343, 1235681. [Google Scholar] [CrossRef] [PubMed]
- Federici, M.F.; Kudryashov, V.; Saigo, P.E.; Finstad, C.L.; Lloyd, K.O. Selection of carbohydrate antigens in human epithelial ovarian cancers as targets for immunotherapy: Serous and mucinous tumors exhibit distinctive patterns of expression. Int. J. Cancer 1999, 81, 193–198. [Google Scholar] [CrossRef]
- Kinney, A.Y.; Sahin, A.; Vernon, S.W.; Frankowski, R.F.; Annegers, J.F.; Hortobagyi, G.N.; Buzdar, A.U.; Frye, D.K.; Dhingra, K. The prognostic significance of sialyl-tn antigen in women treated with breast carcinoma treated with adjuvant chemotherapy. Cancer 1997, 80, 2240–2249. [Google Scholar] [CrossRef]
- Kobayashi, H.; Terao, T.; Kawashima, Y. Serum sialyl Tn as an independent predictor of poor prognosis in patients with epithelial ovarian cancer. J. Clin. Oncol. 1992, 10, 95–101. [Google Scholar] [PubMed]
- Miles, D.; Roché, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Phase III multicenter clinical trial of the sialyl-Tn (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011, 16, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cordon-Cardo, C.; Zhang, H.S.; Reuter, V.E.; Adluri, S.; Hamilton, W.B.; Lloyd, K.O.; Livingston, P.O. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int. J. Cancer 1997, 73, 42–49. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.L.; Haynes, L.; Parker, C.; Iversen, P. Interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J. Natl. Cancer Inst. 2012, 104, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Ophir, E.; Bobisse, S.; Coukos, G.; Harari, A.; Kandalaft, L.E. Personalized approaches to active immunotherapy in cancer. Biochim. Biophys. Acta 2016, 1865, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wolfel, C.; Huber, C.; Wolfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific t cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, P.; Merrick, A.E.; West, E.; O‘Donnell, D.; Selby, P.; Vile, R.; Melcher, A.A. Optimization of dendritic cell loading with tumor cell lysates for cancer immunotherapy. J. Immunother. 2008, 31, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.Y.; Yang, W.K.; Lee, H.C.; Hsu, D.M.; Lin, H.L.; Lin, S.Z.; Chen, C.C.; Harn, H.J.; Liu, C.L.; Lee, W.Y.; et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: A phase ii clinical trial. World Neurosurg. 2012, 77, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Reyes, D.; Salazar, L.; Espinoza, E.; Pereda, C.; Castellon, E.; Valdevenito, R.; Huidobro, C.; Ines Becker, M.; Lladser, A.; Lopez, M.N.; et al. Tumour cell lysate-loaded dendritic cell vaccine induces biochemical and memory immune response in castration-resistant prostate cancer patients. Br. J. Cancer 2013, 109, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Von Euw, E.M.; Barrio, M.M.; Furman, D.; Levy, E.M.; Bianchini, M.; Peguillet, I.; Lantz, O.; Vellice, A.; Kohan, A.; Chacón, M.; et al. A phase I clinical study of vaccination of melanoma patients with dendritic cells loaded with allogeneic apoptotic/necrotic melanoma cells. Analysis of toxicity and immune response to the vaccine and of IL-10 -1082 promoter genotype as predictor of disease progression. J. Transl. Med. 2008, 6, 6. [Google Scholar] [PubMed]
- Zappasodi, R.; Pupa, S.M.; Ghedini, G.C.; Bongarzone, I.; Magni, M.; Cabras, A.D.; Colombo, M.P.; Carlo-Stella, C.; Gianni, A.M.; Di Nicola, M. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010, 70, 9062–9072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-N.; Choi, I.-K.; Huang, J.-H.; Yoo, J.-Y.; Choi, K.-J.; Yun, C.-O. Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol. Ther. 2011, 19, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Woller, N.; Knocke, S.; Mundt, B.; Gürlevik, E.; Strüver, N.; Kloos, A.; Boozari, B.; Schache, P.; Manns, M.P.; Malek, N.P.; et al. Virus-induced tumor inflammation facilitates effective DC cancer immunotherapy in a Treg-dependent manner in mice. J. Clin. Invest. 2011, 121, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Kashiwagi, S.; Reeves, P.; Nezivar, J.; Yang, Y.; Arrifin, N.H.; Nguyen, M.; Jean-Mary, G.; Tong, X.; Uppal, P.; et al. A novel mycobacterial Hsp70-containing fusion protein targeting mesothelin augments antitumor immunity and prolongs survival in murine models of ovarian cancer and mesothelioma. J. Hematol. Oncol. 2014, 7, 15–15. [Google Scholar] [CrossRef] [PubMed]
- Darrasse-Jeze, G.; Deroubaix, S.; Mouquet, H.; Victora, G.D.; Eisenreich, T.; Yao, K.H.; Masilamani, R.F.; Dustin, M.L.; Rudensky, A.; Liu, K.; et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J. Exp. Med. 2009, 206, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Turley, S.; Mellman, I.; Inaba, K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000, 191, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Lefebvre, F.; Filali-Mouhim, A.; Cameron, M.J.; Goulet, J.P.; Haddad, E.K.; Breton, G.; Trumpfheller, C.; Pollak, S.; Shimeliovich, I.; et al. Synthetic double-stranded rna induces innate immune responses similar to a live viral vaccine in humans. J. Exp. Med. 2011, 208, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Wang, B.; Shivji, G.M.; Toto, P.; Amerio, P.; Tomai, M.A.; Miller, R.L.; Sauder, D.N. Imiquimod, a topical immune response modifier, induces migration of langerhans cells. J. Investig. Dermatol. 2000, 114, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; O’Neill, D.W.; Nonaka, D.; Hardin, E.; Chiriboga, L.; Siu, K.; Cruz, C.M.; Angiulli, A.; Angiulli, F.; Ritter, E.; et al. Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using toll-like receptor 7 agonist imiquimod as vaccine adjuvant. J. Immunol. 2008, 181, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Dietsch, G.N.; Matthews, M.A.; Yang, Y.; Ghanekar, S.; Inokuma, M.; Suni, M.; Maino, V.C.; Henderson, K.E.; Howbert, J.J.; et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin. Cancer Res. 2012, 18, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Northfelt, D.W.; Ramanathan, R.K.; Cohen, P.A.; Von Hoff, D.D.; Weiss, G.J.; Dietsch, G.N.; Manjarrez, K.L.; Randall, T.D.; Hershberg, R.M. A phase I dose-finding study of the novel toll-like receptor 8 agonist VTX-2337 in adult subjects with advanced solid tumors or lymphoma. Clin. Cancer Res. 2014, 20, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.-L.; Benencia, F.; Coukos, G. Whole tumor antigen vaccines. Sem. Immunol. 2010, 22, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Oh, S.; Gharagozlou, S.; Vedi, R.N.; Ericson, K.; Low, W.C.; Chen, W.; Ohlfest, J.R. In vivo vaccination with tumor cell lysate plus cpg oligodeoxynucleotides eradicates murine glioblastoma. J. Immunother. 2007, 30, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Sfondrini, L.; Rossini, A.; Besusso, D.; Merlo, A.; Tagliabue, E.; Menard, S.; Balsari, A. Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J. Immunol. 2006, 176, 6624–6630. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, M.M.; DeVeer, M.J.; Edling, A.; Oates, R.K.; Simons, B.; Lindner, D.; Williams, B.R. Synergistic activation of innate immunity by double-stranded RNA and CpG DNA promotes enhanced antitumor activity. Cancer Res. 2004, 64, 5850–5860. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Hu, J. Complexity of dendritic cell subsets and their function in the host immune system. Immunology 2011, 133, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to cpg oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Rothenfusser, S.; Hornung, V.; Jahrsdorfer, B.; Blackwell, S.; Ballas, Z.K.; Endres, S.; Krieg, A.M.; Hartmann, G. Identification of CpG oligonucleotide sequences with high induction of IFN-α/β in plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 2154–2163. [Google Scholar] [CrossRef]
- Gibson, S.J.; Lindh, J.M.; Riter, T.R.; Gleason, R.M.; Rogers, L.M.; Fuller, A.E.; Oesterich, J.L.; Gorden, K.B.; Qiu, X.; McKane, S.W.; et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell. Immunol. 2002, 218, 74–86. [Google Scholar] [CrossRef]
- King, J.W.; Taylor, E.M.; Crow, S.D.; White, M.C.; Todd, J.R.; Poe, M.B.; Conrad, S.A.; Gelder, F.B. Comparison of the immunogenicity of hepatitis b vaccine administered intradermally and intramuscularly. Rev. Infect. Dis. 1990, 12, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Tureci, O.; Sahin, U. Intranodal vaccination with naked antigen-encoding rna elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, N.; Torensma, R.; Tel, J. Deciphering the message broadcast by tumor-infiltrating dendritic cells. Am. J. Pathol. 2012, 181, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Hartmann, G.; Kishimoto, S.; Katoh, N. Dendritic cell vaccination in human melanoma: Relationships between clinical effects and vaccine parameters. Pigment Cell Melanoma Res. 2010, 23, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific t-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, F.; Schreibelt, G.; Skold, A.E.; Figdor, C.G.; De Vries, I.J. Paradigm shift in dendritic cell-based immunotherapy: From in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front. Immunol. 2014, 5, 165. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Lion, E.; Van den Bergh, J.; Van Acker, H.H.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F.; Berneman, Z.N. Interleukin-15 dendritic cells as vaccine candidates for cancer immunotherapy. Hum. Vaccines Immunother. 2013, 9, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Rossi, M.; Ratzinger, G.; de Cos, M.A.; Chung, D.J.; Panageas, K.S.; Wolchok, J.D.; Houghton, A.N.; Chapman, P.B.; Heller, G.; et al. Peptide-loaded langerhans cells, despite increased IL15 secretion and T-cell activation in vitro, elicit antitumor T-cell responses comparable to peptide-loaded monocyte-derived dendritic cells in vivo. Clin. Cancer Res. 2011, 17, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Lion, E.; Smits, E.L.J.M.; Berneman, Z.N.; Van Tendeloo, V.F.I. NK cells: Key to success of DC-based cancer vaccines? Oncologist 2012, 17, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis e Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.-L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Ollion, V.; Colletti, N.; Chelbi, R.; Montanana-Sanchis, F.; Liu, H.; Vu Manh, T.-P.; Sanchez, C.; Savoret, J.; Perrot, I.; et al. Human XCR1+ dendritic cells derived in vitro from CD34+ progenitors closely resemble blood dendritic cells, including their adjuvant responsiveness, contrary to monocyte-derived dendritic cells. J. Immunol. 2014, 193, 1622–1635. [Google Scholar] [CrossRef] [PubMed]
- Kooi, S.; Zhang, H.Z.; Patenia, R.; Edwards, C.L.; Platsoucas, C.D.; Freedman, R.S. HLA class I expression on human ovarian carcinoma cells correlates with T-cell infiltration in vivo and T-cell expansion in vitro in low concentrations of recombinant interleukin-2. Cell. Immunol. 1996, 174, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Maine, C.J.; Aziz, N.H.; Chatterjee, J.; Hayford, C.; Brewig, N.; Whilding, L.; George, A.J.; Ghaem-Maghami, S. Programmed death ligand-1 over-expression correlates with malignancy and contributes to immune regulation in ovarian cancer. Cancer Immunol. Immunother. 2014, 63, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Bouzin, C.; Brouet, A.; De Vriese, J.; Dewever, J.; Feron, O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: Identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O’Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Marigo, I.; Dolcetti, L.; Serafini, P.; Zanovello, P.; Bronte, V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 2008, 222, 162–179. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.L.; Obermajer, N.; Odunsi, K.; Edwards, R.P.; Kalinski, P. Synergistic COX2 induction by IFNγ and TNFα self-limits type-1 immunity in the human tumor microenvironment. Cancer Immunol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Derhovanessian, E.; Solana, R.; Larbi, A.; Pawelec, G. Immunity, ageing and cancer. Immun. Ageing 2008, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.S.; Boyer, J.D.; Jawad, A.; McDonald, K.; Rogers, W.T.; Prak, E.T.L.; Sullivan, K.E. Immunologic consequences of chemotherapy for ovarian cancer: Impaired responses to the influenza vaccine. Vaccine 2013, 31. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.A.; Brondstetter, T.I.; English, C.A.; Lee, H.E.; Virts, E.L.; Thoman, M.L. Il-7 gene therapy in aging restores early thymopoiesis without reversing involution. J. Immunol. 2004, 173, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Marko, M.G.; Ahmed, T.; Bunnell, S.C.; Wu, D.; Chung, H.; Huber, B.T.; Meydani, S.N. Age-associated decline in effective immune synapse formation of CD4+ T cells is reversed by vitamin e supplementation. J. Immunol. 2007, 178, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Albers, R.; van der Wielen, R.P.; Brink, E.J.; Hendriks, H.F.; Dorovska-Taran, V.N.; Mohede, I.C. Effects of cis-9, trans-11 and trans-10, cis-12 conjugated linoleic acid (CLA) isomers on immune function in healthy men. Eur. J. Clin. Nutr. 2003, 57, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Dupuis, G.; Fortin, C.; Douziech, N.; Larbi, A. T cell response in aging: Influence of cellular cholesterol modulation. Adv. Exp. Med. Biol. 2006, 584, 157–169. [Google Scholar] [PubMed]
- Kensler, T.W.; Spira, A.; Garber, J.E.; Szabo, E.; Lee, J.J.; Dong, Z.; Dannenberg, A.J.; Hait, W.N.; Blackburn, E.; Davidson, N.E.; et al. Transforming cancer prevention through precision medicine and immune-oncology. Cancer Prev. Res. 2016, 9, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Lollini, P.-L.; Cavallo, F.; Nanni, P.; Quaglino, E. The promise of preventive cancer vaccines. Vaccines 2015, 3, 467–489. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Lyerly, H.K. Checkpoint blockade in combination with cancer vaccines. Vaccine 2015, 33, 7377–7385. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Co-expression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Tamada, K. Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci. 2015, 106, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Butler, M.; Oble, D.A.; Seiden, M.V.; Haluska, F.G.; Kruse, A.; MacRae, S.; Nelson, M.; Canning, C.; Lowy, I.; et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Sharfman, W.H.; Drake, C.G.; Wollner, I.; Taube, J.M.; Anders, R.A.; Xu, H.; Yao, S.; Pons, A.; Chen, L.; et al. Durable cancer regression off-treatment and effective re-induction therapy with an anti-PD-1 antibody. Clin. Cancer Res. 2013, 19, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Terness, P.; Bauer, T.M.; Rose, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Chhabra, A.; Chachoua, A.; Adams, S.; Donach, M.; Fenton-Kerimian, M.; Friedman, K.; Ponzo, F.; Babb, J.S.; Goldberg, J.; et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: A proof-of-principle trial. Lancet Oncol. 2015, 16, 795–803. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. Tgfbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, M.; Kohrt, H.; Levy, R.; Jones, J.; Camphausen, K.; Dicker, A.; Demaria, S.; Formenti, S. Current clinical trials testing combinations of immunotherapy and radiation. Semin. Radiat. Oncol. 2015, 25, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Pilones, K.A.; Wennerberg, E.; Formenti, S.C.; Demaria, S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine 2015, 33, 7415–7422. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; de Vries, I.J.; Aarntzen, E.A.; de Boer, A.; Scharenborg, N.M.; van de Rakt, M.; van Spronsen, D.J.; Preijers, F.W.; Figdor, C.G.; Adema, G.J.; et al. A pilot study on the immunogenicity of dendritic cell vaccination during adjuvant oxaliplatin/capecitabine chemotherapy in colon cancer patients. Br. J. Cancer 2010, 103, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium fasl establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Dranitsaris, G.; Cohen, R.B.; Acton, G.; Keltner, L.; Price, M.; Amir, E.; Podack, E.R.; Schreiber, T.H. Statistical considerations in clinical trial design of immunotherapeutic cancer agents. J. Immunother. 2015, 38, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Perez Gracia, J.L.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef] [PubMed]
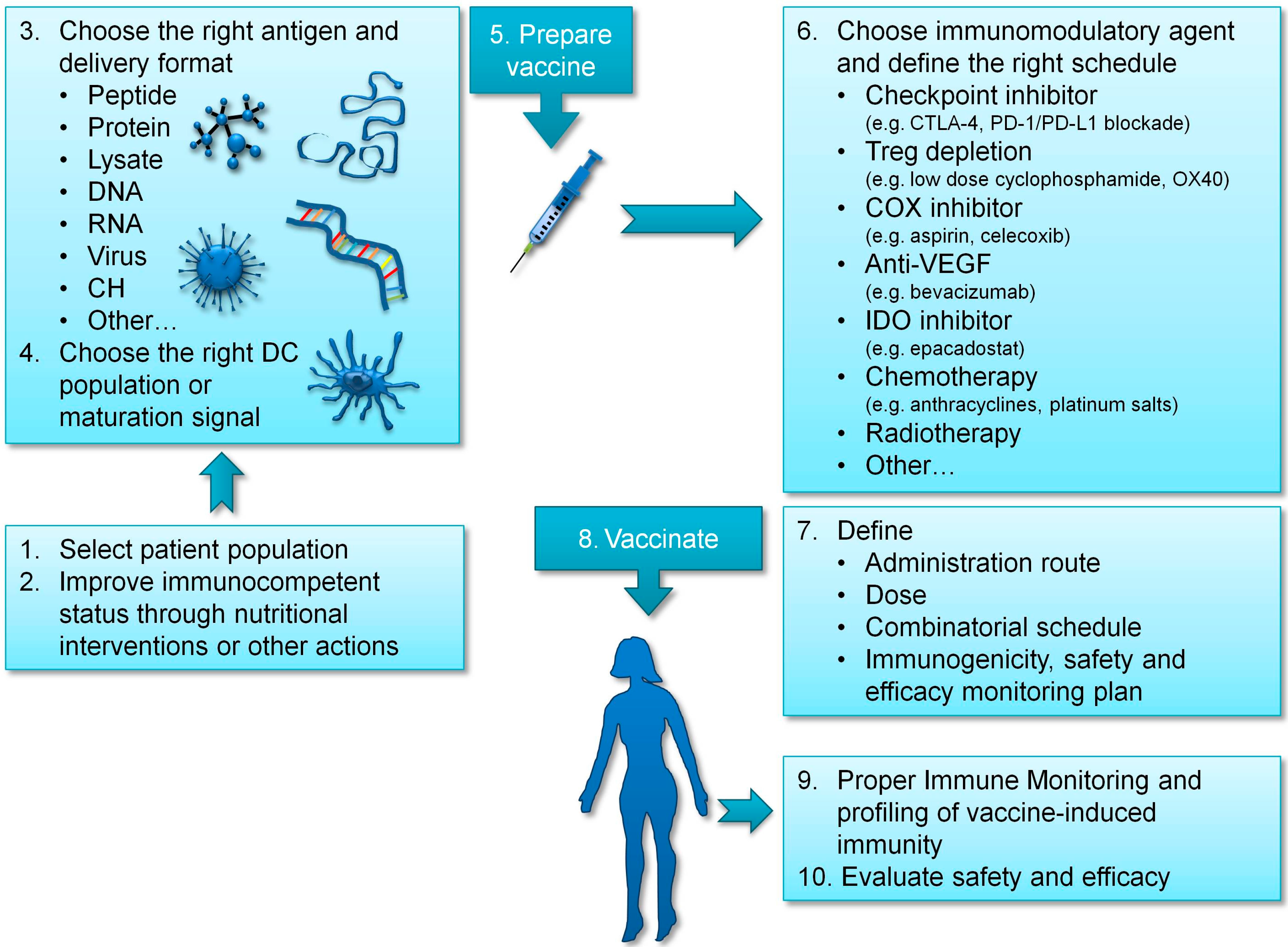
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).