
The Gastrointestinal Barrier—Mechanisms of Barrier Dysfunction in Liver Cirrhosis and Spontaneous Bacterial Peritonitis
 , , ,
, , ,
Abstract
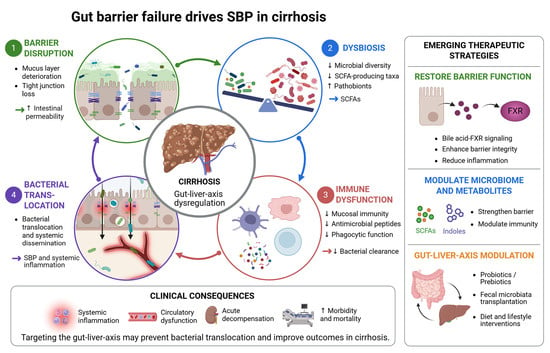
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Organisation of the Gastrointestinal Barrier
2.1. Epithelial Integrity and Tight Junctions
2.2. The Mucus Layer as a Protective Interface
3. Immune Homeostasis and Immune Dysregulation
4. Mechanisms of Barrier Failure in Liver Cirrhosis
4.1. Hemodynamic and Structural Alterations: The Impact of Portal Hypertension
4.2. Epithelial Barrier Disruption and Tight Junction Remodelling
4.3. Mucus Layer Deterioration and Loss of Spatial Segregation
4.4. Microbial Dysbiosis and Metabolic Perturbations
4.5. Immune Dysregulation and Failure of Mucosal Defence
4.6. Gut–Vascular Barrier Dysfunction and Systemic Dissemination
4.7. Integration of Barrier Failure and Link to SBP
5. Bacterial Translocation and SBP
5.1. Routes and Mechanisms of Bacterial Translocation
5.2. Drivers of Increased Bacterial Translocation in Cirrhosis
5.3. Spontaneous Bacterial Peritonitis: Pathogenesis
5.4. Systemic Consequences and Disease Progression
5.5. Clinical Implications and Risk Stratification
6. The Microbiome and Dysbiosis in Cirrhosis
7. Therapeutic Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ginès, P.; Krag, A.; Abraldes, J.G.; Solà, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Kamath, P.S.; Reddy, K.R. The Evolving Challenge of Infections in Cirrhosis. N. Engl. J. Med. 2021, 384, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; O’Leary, J.G.; Lai, J.C.; Wong, F.; Long, M.D.; Wong, R.J.; Kamath, P.S. Acute-on-Chronic Liver Failure Clinical Guidelines. Am. J. Gastroenterol. 2022, 117, 225–252. [Google Scholar] [CrossRef]
- Piano, S.; Bunchorntavakul, C.; Marciano, S.; Rajender Reddy, K. Infections in cirrhosis. Lancet Gastroenterol. Hepatol. 2024, 9, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140. [Google Scholar] [CrossRef]
- McGettigan, B.; Hernandez-Tejero, M.; Malhi, H.; Shah, V. Immune Dysfunction and Infection Risk in Advanced Liver Disease. Gastroenterology 2025, 168, 1085–1100. [Google Scholar] [CrossRef]
- Albillos, A.; Lario, M.; Álvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef]
- Simbrunner, B.; Mandorfer, M.; Trauner, M.; Reiberger, T. Gut-liver axis signaling in portal hypertension. World J. Gastroenterol. 2019, 25, 5897–5917. [Google Scholar] [CrossRef]
- Haderer, M.; Neubert, P.; Rinner, E.; Scholtis, A.; Broncy, L.; Gschwendtner, H.; Kandulski, A.; Pavel, V.; Mehrl, A.; Brochhausen, C.; et al. Novel pathomechanism for spontaneous bacterial peritonitis: Disruption of cell junctions by cellular and bacterial proteases. Gut 2022, 71, 580–592. [Google Scholar] [CrossRef]
- Smets, L.; Viola, M.F.; Boesch, M.; Raman, J.; van Melkebeke, L.; Nobis, M.; Flint, E.; Pajk, N.; Brescia, P.; Silvestri, A.; et al. Intestinal blood vessel-associated macrophages and gut-vascular barrier dysfunction in cirrhosis. Gut 2025. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Biggins, S.W.; Angeli, P.; Garcia-Tsao, G.; Ginès, P.; Ling, S.C.; Nadim, M.K.; Wong, F.; Kim, W.R. Diagnosis, Evaluation, and Management of Ascites, Spontaneous Bacterial Peritonitis and Hepatorenal Syndrome: 2021 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2021, 74, 1014–1048. [Google Scholar] [CrossRef]
- Pellegrini, C.; Fornai, M.; D’Antongiovanni, V.; Antonioli, L.; Bernardini, N.; Derkinderen, P. The intestinal barrier in disorders of the central nervous system. Lancet Gastroenterol. Hepatol. 2023, 8, 66–80. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Santacroce, G.; Rossi, C.M.; Broglio, G.; Lenti, M.V. Role of mucosal immunity and epithelial-vascular barrier in modulating gut homeostasis. Intern. Emerg. Med. 2023, 18, 1635–1646. [Google Scholar] [CrossRef]
- Neurath, M.F.; Artis, D.; Becker, C. The intestinal barrier: A pivotal role in health, inflammation, and cancer. Lancet Gastroenterol. Hepatol. 2025, 10, 573–592. [Google Scholar] [CrossRef]
- Breugelmans, T.; Oosterlinck, B.; Arras, W.; Ceuleers, H.; de Man, J.; Hold, G.L.; de Winter, B.Y.; Smet, A. The role of mucins in gastrointestinal barrier function during health and disease. Lancet Gastroenterol. Hepatol. 2022, 7, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Pelaseyed, T.; Bergström, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.H.; Schütte, A.; van der Post, S.; Svensson, F.; Rodríguez-Piñeiro, A.M.; Nyström, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Iftekhar, A.; Sigal, M. Defence and adaptation mechanisms of the intestinal epithelium upon infection. Int. J. Med. Microbiol. 2021, 311, 151486. [Google Scholar] [CrossRef]
- Chang, L.; Liu, Y.; Li, H.; Yan, J.; Wu, W.; Chen, N.; Ma, C.; Zhao, X.; Chen, J.; Zhang, J. Gut microbiome and its metabolites in liver cirrhosis: Mechanisms and clinical implications. Front. Cell. Infect. Microbiol. 2025, 15, 1717696. [Google Scholar] [CrossRef]
- Kuo, W.-T.; Odenwald, M.A.; Turner, J.R.; Zuo, L. Tight junction proteins occludin and ZO-1 as regulators of epithelial proliferation and survival. Ann. N. Y. Acad. Sci. 2022, 1514, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T. Regulation of intestinal epithelial permeability by tight junctions. Cell. Mol. Life Sci. 2013, 70, 631–659. [Google Scholar] [CrossRef]
- Zhang, K.; Hornef, M.W.; Dupont, A. The intestinal epithelium as guardian of gut barrier integrity. Cell. Microbiol. 2015, 17, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Guerrieri, F.; Di Gregorio, V.; Levrero, M.; Gagliardi, A.; Santangelo, F.; Sobolev, A.P.; Circi, S.; Giannelli, V.; Mannina, L.; et al. Combining amplicon sequencing and metabolomics in cirrhotic patients highlights distinctive microbiota features involved in bacterial translocation, systemic inflammation and hepatic encephalopathy. Sci. Rep. 2018, 8, 8210. [Google Scholar] [CrossRef]
- Horowitz, A.; Chanez-Paredes, S.D.; Haest, X.; Turner, J.R. Paracellular permeability and tight junction regulation in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 417–432. [Google Scholar] [CrossRef]
- Günzel, D.; Yu, A.S.L. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef]
- Le, S.; Weber, C.R.; Raleigh, D.R.; Yu, D.; Turner, J.R. Tight junction pore and leak pathways: A dynamic duo. Annu. Rev. Physiol. 2011, 73, 283–309. [Google Scholar] [CrossRef]
- Jin, Y.; Blikslager, A.T. The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases. Int. J. Mol. Sci. 2020, 21, 3550. [Google Scholar] [CrossRef]
- Lynn, K.S.; Peterson, R.J.; Koval, M. Ruffles and spikes: Control of tight junction morphology and permeability by claudins. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183339. [Google Scholar] [CrossRef] [PubMed]
- Le, S. Tight junctions on the move: Molecular mechanisms for epithelial barrier regulation. Ann. N. Y. Acad. Sci. 2012, 1258, 9–18. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Tsamandas, A.C.; Tsiaoussis, G.I.; Karatza, E.; Triantos, C.; Vagianos, C.E.; Spiliopoulou, I.; Kaltezioti, V.; Charonis, A.; Nikolopoulou, V.N.; et al. Altered intestinal tight junctions’ expression in patients with liver cirrhosis: A pathogenetic mechanism of intestinal hyperpermeability. Eur. J. Clin. Investig. 2012, 42, 439–446. [Google Scholar] [CrossRef]
- Muñoz, L.; Borrero, M.-J.; Úbeda, M.; Conde, E.; Del Campo, R.; Rodríguez-Serrano, M.; Lario, M.; Sánchez-Díaz, A.-M.; Pastor, O.; Díaz, D.; et al. Intestinal Immune Dysregulation Driven by Dysbiosis Promotes Barrier Disruption and Bacterial Translocation in Rats With Cirrhosis. Hepatology 2019, 70, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Du Plessis, J.; Vanheel, H.; Janssen, C.E.I.; Roos, L.; Slavik, T.; Stivaktas, P.I.; Nieuwoudt, M.; van Wyk, S.G.; Vieira, W.; Pretorius, E.; et al. Activated intestinal macrophages in patients with cirrhosis release NO and IL-6 that may disrupt intestinal barrier function. J. Hepatol. 2013, 58, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Sjövall, H.; Hansson, G.C. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, F.R.; Eguileor, A.; Llorente, C. Goblet cells: Guardians of gut immunity and their role in gastrointestinal diseases. eGastroenterology 2024, 2, e100098. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. The role of the mucosal barrier system in maintaining gut symbiosis to prevent intestinal inflammation. Semin. Immunopathol. 2024, 47, 2. [Google Scholar] [CrossRef]
- Cheng, H.; Li, H.; Li, Z.; Wang, Y.; Liu, L.; Wang, J.; Ma, X.; Tan, B. The role of glycosylated mucins in maintaining intestinal homeostasis and gut health. Anim. Nutr. (Zhongguo Xu Mu Shou Yi Xue Hui) 2025, 21, 439–446. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Larsson, J.M.H.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 4659–4665. [Google Scholar] [CrossRef]
- Bergstrom, K.; Xia, L. The barrier and beyond: Roles of intestinal mucus and mucin-type O-glycosylation in resistance and tolerance defense strategies guiding host-microbe symbiosis. Gut Microbes 2022, 14, 2052699. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef]
- Hockenberry, A.; Slack, E.; Stadtmueller, B.M. License to Clump: Secretory IgA Structure-Function Relationships Across Scales. Annu. Rev. Microbiol. 2023, 77, 645–668. [Google Scholar] [CrossRef]
- Mantis, N.J.; Rol, N.; Corthésy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef]
- Song, G.; Xie, Y.; Yi, L.; Cheng, W.; Jia, H.; Shi, W.; Liu, Q.; Fang, L.; Xue, S.; Liu, D.; et al. Bile acids affect intestinal barrier function through FXR and TGR5. Front. Med. 2025, 12, 1607899. [Google Scholar] [CrossRef]
- Simbrunner, B.; Hofer, B.S.; Schwabl, P.; Zinober, K.; Petrenko, O.; Fuchs, C.; Semmler, G.; Marculescu, R.; Mandorfer, M.; Datz, C.; et al. FXR-FGF19 signaling in the gut-liver axis is dysregulated in patients with cirrhosis and correlates with impaired intestinal defence. Hepatol. Int. 2024, 18, 929–942. [Google Scholar] [CrossRef]
- Costa, D.; Trebicka, J.; Ripoll, C.; Moreau, R.; Jalan, R.; Reiberger, T. Interaction of inflammation and portal hypertension in cirrhosis progression. Nat. Rev. Gastroenterol. Hepatol. 2025, 22, 846–865. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Simbrunner, B.; Baumgartner, M.; Campbell, C.; Reiberger, T.; Trauner, M. Bile acid metabolism and signalling in liver disease. J. Hepatol. 2025, 82, 134–153. [Google Scholar] [CrossRef]
- Verbeke, L.; Farre, R.; Verbinnen, B.; Covens, K.; Vanuytsel, T.; Verhaegen, J.; Komuta, M.; Roskams, T.; Chatterjee, S.; Annaert, P.; et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am. J. Pathol. 2015, 185, 409–419. [Google Scholar] [CrossRef]
- Wu, Z.; Zhou, H.; Liu, D.; Deng, F. Alterations in the gut microbiota and the efficacy of adjuvant probiotic therapy in liver cirrhosis. Front. Cell. Infect. Microbiol. 2023, 13, 1218552. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Li, C.; Sun, T.; Luo, X.; Jiang, B.; Liu, M.; Wang, Q.; Li, T.; Cao, J.; et al. Associations between changes in the gut microbiota and liver cirrhosis: A systematic review and meta-analysis. BMC Gastroenterol. 2025, 25, 16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, C.; Zuo, S.; Cao, K.; Li, H. Integrative analysis of the gut microbiota and faecal and serum short-chain fatty acids and tryptophan metabolites in patients with cirrhosis and hepatic encephalopathy. J. Transl. Med. 2023, 21, 395. [Google Scholar] [CrossRef]
- Tranah, T.H.; Edwards, L.A.; Schnabl, B.; Shawcross, D.L. Targeting the gut-liver-immune axis to treat cirrhosis. Gut 2021, 70, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; He, C.; Xia, Y.; Ocansey, D.K.W.; Mao, F. Intestinal mucus barrier: A potential therapeutic target for IBD. Autoimmun. Rev. 2025, 24, 103717. [Google Scholar] [CrossRef]
- Tingler, A.M.; Engevik, M.A. Breaking down barriers: Is intestinal mucus degradation by Akkermansia muciniphila beneficial or harmful? Infect. Immun. 2025, 93, e0050324. [Google Scholar] [CrossRef]
- Almeqdadi, M.; Gordon, F.D. Farnesoid X Receptor Agonists: A Promising Therapeutic Strategy for Gastrointestinal Diseases. Gastro Hep Adv. 2024, 3, 344–352. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Ng, S.C.; Schnabl, B. Promises of microbiome-based therapies. J. Hepatol. 2022, 76, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Bloom, P.P.; Tapper, E.B. Lactulose in cirrhosis: Current understanding of efficacy, mechanism, and practical considerations. Hepatol. Commun. 2023, 7, e0295. [Google Scholar] [CrossRef]
- Rodríguez-Negrete, E.V.; Gálvez-Martínez, M.; Sánchez-Reyes, K.; Fajardo-Felix, C.F.; Pérez-Reséndiz, K.E.; Madrigal-Santillán, E.O.; Morales-González, Á.; Morales-González, J.A. Liver Cirrhosis: The Immunocompromised State. J. Clin. Med. 2024, 13, 5582. [Google Scholar] [CrossRef]
- Albillos, A.; Martin-Mateos, R.; van der Merwe, S.; Wiest, R.; Jalan, R.; Álvarez-Mon, M. Cirrhosis-associated immune dysfunction. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 112–134. [Google Scholar] [CrossRef]
- Yu, S.; Gao, N. Compartmentalizing intestinal epithelial cell toll-like receptors for immune surveillance. Cell. Mol. Life Sci. 2015, 72, 3343–3353. [Google Scholar] [CrossRef]
- Owen, A.M.; Luan, L.; Burelbach, K.R.; McBride, M.A.; Stothers, C.L.; Boykin, O.A.; Sivanesam, K.; Schaedel, J.F.; Patil, T.K.; Wang, J.; et al. MyD88-dependent signaling drives toll-like receptor-induced trained immunity in macrophages. Front. Immunol. 2022, 13, 1044662. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, Y.; Chu, Y.; Liu, J.; Cui, L.; Zhang, D. Toll-Like Receptors Recognize Intestinal Microbes in Liver Cirrhosis. Front. Immunol. 2021, 12, 608498. [Google Scholar] [CrossRef]
- Zhou, Y.; Yu, S.; Zhang, W. NOD-like Receptor Signaling Pathway in Gastrointestinal Inflammatory Diseases and Cancers. Int. J. Mol. Sci. 2023, 24, 4511. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Lu, Q.; Hitch, T.C.A.; Zhou, J.Y.; Dwidar, M.; Sangwan, N.; Lawrence, D.; Nolan, L.S.; Espenschied, S.T.; Newhall, K.P.; Han, Y.; et al. A host-adapted auxotrophic gut symbiont induces mucosal immunodeficiency. Science 2024, 385, eadk2536. [Google Scholar] [CrossRef]
- Hackstein, C.-P.; Spitzer, J.; Symeonidis, K.; Horvatic, H.; Bedke, T.; Steglich, B.; Klein, S.; Assmus, L.M.; Odainic, A.; Szlapa, J.; et al. Interferon-induced IL-10 drives systemic T-cell dysfunction during chronic liver injury. J. Hepatol. 2023, 79, 150–166. [Google Scholar] [CrossRef]
- van der Merwe, S.; Chokshi, S.; Bernsmeier, C.; Albillos, A. The multifactorial mechanisms of bacterial infection in decompensated cirrhosis. J. Hepatol. 2021, 75, S82–S100. [Google Scholar] [CrossRef]
- Ohlendorf, V.; Buttler, L.; Maasoumy, B. Leberzirrhose-assoziierte Immundysfunktion (CAID). Dtsch. Med. Wochenschr. 2026, 151, 156–162. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Rimola, A.; Soto, R.; Bory, F.; Arroyo, V.; Piera, C.; Rodes, J. Reticuloendothelial system phagocytic activity in cirrhosis and its relation to bacterial infections and prognosis. Hepatology 1984, 4, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Seitz, R.; Müller, M.; Gülow, K. Extracellular Redox Balance as a Determinant of Immune Regulation and Tissue Inflammation. Antioxidants 2026, 15, 280. [Google Scholar] [CrossRef]
- Rooney, M.; Duduskar, S.N.; Ghait, M.; Reißing, J.; Stengel, S.; Reuken, P.A.; Quickert, S.; Zipprich, A.; Bauer, M.; Russo, A.J.; et al. Type-I interferon shapes peritoneal immunity in cirrhosis and drives caspase-5-mediated progranulin release upon infection. J. Hepatol. 2024, 81, 971–982. [Google Scholar] [CrossRef]
- Angeli, P.; Bernardi, M.; Villanueva, C.; Francoz, C.; Mookerjee, R.P.; Trebicka, J.; Krag, A.; Laleman, W.; Gines, P. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Zocco, M.A.; Cerrito, L.; Gasbarrini, A.; Pompili, M. Bacterial translocation in patients with liver cirrhosis: Physiology, clinical consequences, and practical implications. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 641–656. [Google Scholar] [CrossRef]
- Tsiaoussis, G.I.; Papaioannou, E.C.; Kourea, E.P.; Assimakopoulos, S.F.; Theocharis, G.I.; Petropoulos, M.; Theopistos, V.I.; Diamantopoulou, G.G.; Lygerou, Z.; Spiliopoulou, I.; et al. Expression of α-Defensins, CD20+ B-lymphocytes, and Intraepithelial CD3+ T-lymphocytes in the Intestinal Mucosa of Patients with Liver Cirrhosis: Emerging Mediators of Intestinal Barrier Function. Dig. Dis. Sci. 2018, 63, 2582–2592. [Google Scholar] [CrossRef]
- Pijls, K.E.; Jonkers, D.M.A.E.; Elamin, E.E.; Masclee, A.A.M.; Koek, G.H. Intestinal epithelial barrier function in liver cirrhosis: An extensive review of the literature. Liver Int. 2013, 33, 1457–1469. [Google Scholar] [CrossRef]
- Fondevila, M.F.; Kreimeyer, H.; Hsu, C.L.; Tamargo-Azpilicueta, J.; Le Day, Z.; Gritsenko, M.; Attah, K.; Cabré, N.; Harberts, A.; Tonetti, F.R.; et al. Macrophage-derived cathepsin B disrupts intestinal tight junctions through occludin degradation and promotes alcohol-associated liver disease. J. Hepatol. 2026. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Devaux, C.A.; Mezouar, S.; Mege, J.-L. The E-Cadherin Cleavage Associated to Pathogenic Bacteria Infections Can Favor Bacterial Invasion and Transmigration, Dysregulation of the Immune Response and Cancer Induction in Humans. Front. Microbiol. 2019, 10, 2598. [Google Scholar] [CrossRef]
- Huber, P. Targeting of the apical junctional complex by bacterial pathogens. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183237. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.P.; Mileto, S.J.; Lyras, D. Impact of enteric bacterial infections at and beyond the epithelial barrier. Nat. Rev. Microbiol. 2023, 21, 260–274. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, X.; Wang, H.; Hou, L.; Zheng, R.; Wang, Y.; Jiang, W.; Tang, W. Investigating intestinal farnesoid X receptor functions at the intestinal mucosal barrier and in the intestinal microbiota in a biliary obstruction mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 2025, 329, G313–G327. [Google Scholar] [CrossRef]
- Distrutti, E.; Santucci, L.; Cipriani, S.; Renga, B.; Schiaroli, E.; Ricci, P.; Donini, A.; Fiorucci, S. Bile acid activated receptors are targets for regulation of integrity of gastrointestinal mucosa. J. Gastroenterol. 2015, 50, 707–719. [Google Scholar] [CrossRef]
- Jin, M.; Kalainy, S.; Baskota, N.; Chiang, D.; Deehan, E.C.; McDougall, C.; Tandon, P.; Martínez, I.; Cervera, C.; Walter, J.; et al. Faecal microbiota from patients with cirrhosis has a low capacity to ferment non-digestible carbohydrates into short-chain fatty acids. Liver Int. 2019, 39, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Haidar, G.; Singh, N. The Evolving Challenge of Infections in Cirrhosis. N. Engl. J. Med. 2021, 385, 1150–1152. [Google Scholar] [CrossRef]
- Solé, C.; Guilly, S.; Da Silva, K.; Llopis, M.; Le-Chatelier, E.; Huelin, P.; Carol, M.; Moreira, R.; Fabrellas, N.; de Prada, G.; et al. Alterations in Gut Microbiome in Cirrhosis as Assessed by Quantitative Metagenomics: Relationship With Acute-on-Chronic Liver Failure and Prognosis. Gastroenterology 2021, 160, 206–218.e13. [Google Scholar] [CrossRef]
- Ma, C.; Yang, J.; Fu, X.-N.; Luo, J.-Y.; Liu, P.; Zeng, X.-L.; Li, X.-Y.; Zhang, S.-L.; Zheng, S. Microbial characteristics of gut microbiome dysbiosis in patients with chronic liver disease. World J. Hepatol. 2025, 17, 106124. [Google Scholar] [CrossRef]
- Juanola, O.; Ferrusquía-Acosta, J.; García-Villalba, R.; Zapater, P.; Magaz, M.; Marín, A.; Olivas, P.; Baiges, A.; Bellot, P.; Turon, F.; et al. Circulating levels of butyrate are inversely related to portal hypertension, endotoxemia, and systemic inflammation in patients with cirrhosis. FASEB J. 2019, 33, 11595–11605. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zheng, S.; Zhao, M.; Shi, F.; Zheng, L.; Wang, H. The regulatory role of bile acid microbiota in the progression of liver cirrhosis. Front. Pharmacol. 2023, 14, 1214685. [Google Scholar] [CrossRef]
- Simbrunner, B.; Trauner, M.; Reiberger, T. Review article: Therapeutic aspects of bile acid signalling in the gut-liver axis. Aliment. Pharmacol. Ther. 2021, 54, 1243–1262. [Google Scholar] [CrossRef]
- Pose, E.; Coll, M.; Martínez-Sánchez, C.; Zeng, Z.; Surewaard, B.G.J.; Català, C.; Velasco-de Andrés, M.; Lozano, J.J.; Ariño, S.; Fuster, D.; et al. Programmed Death Ligand 1 Is Overexpressed in Liver Macrophages in Chronic Liver Diseases, and Its Blockade Improves the Antibacterial Activity Against Infections. Hepatology 2021, 74, 296–311. [Google Scholar] [CrossRef] [PubMed]
- Akalin, H.E.; Laleli, Y.; Telatar, H. Bactericidal and opsonic activity of ascitic fluid from cirrhotic and noncirrhotic patients. J. Infect. Dis. 1983, 147, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Runyon, B.A.; Morrissey, R.L.; Hoefs, J.C.; Wyle, F.A. Opsonic activity of human ascitic fluid: A potentially important protective mechanism against spontaneous bacterial peritonitis. Hepatology 1985, 5, 634–637. [Google Scholar] [CrossRef]
- Taylor, N.J.; Manakkat Vijay, G.K.; Abeles, R.D.; Auzinger, G.; Bernal, W.; Ma, Y.; Wendon, J.A.; Shawcross, D.L. The severity of circulating neutrophil dysfunction in patients with cirrhosis is associated with 90-day and 1-year mortality. Aliment. Pharmacol. Ther. 2014, 40, 705–715. [Google Scholar] [CrossRef]
- Rajkovic, I.A.; Williams, R. Abnormalities of neutrophil phagocytosis, intracellular killing and metabolic activity in alcoholic cirrhosis and hepatitis. Hepatology 1986, 6, 252–262. [Google Scholar] [CrossRef]
- Moreau, R.; Périanin, A.; Arroyo, V. Review of Defective NADPH Oxidase Activity and Myeloperoxidase Release in Neutrophils From Patients With Cirrhosis. Front. Immunol. 2019, 10, 1044. [Google Scholar] [CrossRef]
- Haedge, F.; Reuken, P.A.; Reißing, J.; Große, K.; Frissen, M.; El-Hassani, M.; Aschenbach, R.; Teichgräber, U.; Stallmach, A.; Bruns, T. Surrogate Markers of Intestinal Permeability, Bacterial Translocation and Gut-Vascular Barrier Damage Across Stages of Cirrhosis. Liver Int. 2025, 45, e70119. [Google Scholar] [CrossRef] [PubMed]
- Nie, G.; Zhang, H.; Xie, D.; Yan, J.; Li, X. Liver cirrhosis and complications from the perspective of dysbiosis. Front. Med. 2023, 10, 1320015. [Google Scholar] [CrossRef]
- Guan, H.; Zhang, X.; Kuang, M.; Yu, J. The gut-liver axis in immune remodeling of hepatic cirrhosis. Front. Immunol. 2022, 13, 946628. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, Y.; Chen, C.; Song, M.; Hylemon, P.B. Gut microbiome and bile acid metabolism in liver disease: Mechanisms, clinical implications, and therapeutic opportunities. Pharmacol. Rev. 2026, 78, 100120. [Google Scholar] [CrossRef] [PubMed]
- Bellot, P.; Francés, R.; Such, J. Pathological bacterial translocation in cirrhosis: Pathophysiology, diagnosis and clinical implications. Liver Int. 2013, 33, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tai, Y.; Tang, S.; Zhao, C.; Tong, H.; Gao, J.; Tang, C. Compromised Ileal Mucus Barrier Due to Impaired Epithelial Homeostasis Caused by Notch1 Signaling in Cirrhotic Rats. Dig. Dis. Sci. 2021, 66, 131–142. [Google Scholar] [CrossRef]
- Shi, H.; Lv, L.; Cao, H.; Lu, H.; Zhou, N.; Yang, J.; Jiang, H.; Dong, H.; Hu, X.; Yu, W.; et al. Bacterial translocation aggravates CCl4-induced liver cirrhosis by regulating CD4+ T cells in rats. Sci. Rep. 2017, 7, 40516. [Google Scholar] [CrossRef]
- Bernsmeier, C.; van der Merwe, S.; Périanin, A. Innate immune cells in cirrhosis. J. Hepatol. 2020, 73, 186–201. [Google Scholar] [CrossRef]
- Ginès, P.; Cárdenas, A.; Arroyo, V.; Rodés, J. Management of cirrhosis and ascites. N. Engl. J. Med. 2004, 350, 1646–1654. [Google Scholar] [CrossRef]
- Ibidapo-Obe, O.; Rooney, M.D.; Bruns, T. Peritoneal immunity in decompensated cirrhosis. Expert Rev. Clin. Immunol. 2025, 21, 1355–1371. [Google Scholar] [CrossRef]
- Nieto, J.C.; Perea, L.; Soriano, G.; Zamora, C.; Cantó, E.; Medina, A.; Poca, M.; Sanchez, E.; Roman, E.; Julià, G.; et al. Ascitic fluid regulates the local innate immune response of patients with cirrhosis. J. Leukoc. Biol. 2018, 104, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Soriano, G.; Castellote, J.; Alvarez, C.; Girbau, A.; Gordillo, J.; Baliellas, C.; Casas, M.; Pons, C.; Román, E.M.; Maisterra, S.; et al. Secondary bacterial peritonitis in cirrhosis: A retrospective study of clinical and analytical characteristics, diagnosis and management. J. Hepatol. 2010, 52, 39–44. [Google Scholar] [CrossRef]
- Miller, J.M.; Binnicker, M.J.; Campbell, S.; Carroll, K.C.; Chapin, K.C.; Gonzalez, M.D.; Harrington, A.; Jerris, R.C.; Kehl, S.C.; Leal, S.M.; et al. Guide to Utilization of the Microbiology Laboratory for Diagnosis of Infectious Diseases: 2024 Update by the Infectious Diseases Society of America (IDSA) and the American Society for Microbiology (ASM). Clin. Infect. Dis. 2024, ciae104. [Google Scholar] [CrossRef]
- Furey, C.; Zhou, S.; Park, J.H.; Foong, A.; Chowdhury, A.; Dawit, L.; Lee, V.; Vergara-Lluri, M.; She, R.; Kahn, J.; et al. Impact of Bacteria Types on the Clinical Outcomes of Spontaneous Bacterial Peritonitis. Dig. Dis. Sci. 2023, 68, 2140–2148. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X.-X.; Song, J.-W.; Zhang, X.-C.; Zhen, C.; Bi, J.-F.; Lu, F.-Y.; Chen, S.-M.; Dan Huo, D.; Zhao, P.; et al. Clinical features, microbial spectrum, and antibiotic susceptibility patterns of spontaneous bacterial peritonitis in cirrhotic patients. Dig. Liver Dis. 2023, 55, 1554–1561. [Google Scholar] [CrossRef]
- Blanchard, F.; Henry, B.; Vijayaratnam, S.; Canouï, E.; Moura, A.; Thouvenot, P.; Bracq-Dieye, H.; Tessaud-Rita, N.; Valès, G.; Diakité, A.; et al. Listeria monocytogenes-associated spontaneous bacterial peritonitis in France: A nationwide observational study of 208 cases. Lancet Infect. Dis. 2024, 24, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Schütte, S.L.; Tergast, T.L.; Maasoumy, B.; Braun, L.; Esche, U.V.d.; Häcker, G.; Mücke, M.M.; Reincke, M.; Schultheiss, M.; et al. Altered Pathogen Spectrum of Spontaneous Bacterial Peritonitis in Patients Treated With Proton Pump Inhibitors. Aliment. Pharmacol. Ther. 2026. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, A.; Papadopoulos, N.; Eliopoulos, D.G.; Alexaki, A.; Tsiriga, A.; Toutouza, M.; Pectasides, D. Increasing frequency of gram-positive cocci and gram-negative multidrug-resistant bacteria in spontaneous bacterial peritonitis. Liver Int. 2013, 33, 975–981. [Google Scholar] [CrossRef]
- Li, H.; Wieser, A.; Zhang, J.; Liss, I.; Markwardt, D.; Hornung, R.; Neumann-Cip, A.C.; Mayerle, J.; Gerbes, A.; Steib, C.J. Patients with cirrhosis and SBP: Increase in multidrug-resistant organisms and complications. Eur. J. Clin. Investig. 2020, 50, e13198. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.-H.; Wang, C.-Y.; Tsai, C.-C.; Lee, H.-F. Short and long-term mortality of spontaneous bacterial peritonitis in cirrhotic patients. Medicine 2024, 103, e40851. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-del-Arbol, L.; Urman, J.; Fernández, J.; González, M.; Navasa, M.; Monescillo, A.; Albillos, A.; Jiménez, W.; Arroyo, V. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology 2003, 38, 1210–1218. [Google Scholar] [CrossRef]
- Simbrunner, B.; Caparrós, E.; Neuwirth, T.; Schwabl, P.; Königshofer, P.; Bauer, D.; Marculescu, R.; Trauner, M.; Scheiner, B.; Stary, G.; et al. Bacterial translocation occurs early in cirrhosis and triggers a selective inflammatory response. Hepatol. Int. 2023, 17, 1045–1056. [Google Scholar] [CrossRef]
- Xu, H.B.; Wang, H.D.; Li, C.H.; Ye, S.; Dong, M.S.; Xia, Q.J.; Zhang, A.Q.; Pan, K.; Ge, X.L.; Dong, J.H. Proton pump inhibitor use and risk of spontaneous bacterial peritonitis in cirrhotic patients: A systematic review and meta-analysis. Genet. Mol. Res. 2015, 14, 7490–7501. [Google Scholar] [CrossRef]
- El-Azab, G. Proton Pump Inhibitors in Patients with Cirrhosis: Pharmacokinetics, Benefits and Drawbacks. Curr. Gastroenterol. Rep. 2024, 26, 323–334. [Google Scholar] [CrossRef]
- Philips, C.A.; Augustine, P. Gut Barrier and Microbiota in Cirrhosis. J. Clin. Exp. Hepatol. 2022, 12, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Chopyk, D.M.; Grakoui, A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology 2020, 159, 849–863. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Qian, S.; Su, Z.; Lin, J.; Hou, Q.; Wang, X.; Li, Y.; Wang, J.; Huang, C.; Wang, Z.; Cubero, F.J.; et al. Inhibition of Farnesoid-x-receptor signaling during abdominal sepsis by dysbiosis exacerbates gut barrier dysfunction. Cell Commun. Signal. 2025, 23, 236. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Jakab, S.S.; Jesudian, A.B.; Rahimi, R.S.; Duarte-Rojo, A.; Chen, P.-H.; Wong, R.J.; Tapper, E.B.; Tandon, P. ACG Clinical Guideline: Hepatic Encephalopathy. Am. J. Gastroenterol. 2026, 121, 588–618. [Google Scholar] [CrossRef]
- Thévenot, T.; Elkrief, L.; Bureau, C.; Bardou-Jacquet, E.; Rosa, I.; Nguyen-Khac, E.; Oberti, F.; Pitta, A.; Mallet, M.; Lebossé, F.; et al. Effect of rifaximin in patients with severe cirrhosis and ascites: A randomized double-blind placebo-controlled trial. J. Hepatol. 2025, 83, 1320–1327. [Google Scholar] [CrossRef]
- Caraceni, P.; Vargas, V.; Solà, E.; Alessandria, C.; de Wit, K.; Trebicka, J.; Angeli, P.; Mookerjee, R.P.; Durand, F.; Pose, E.; et al. The Use of Rifaximin in Patients With Cirrhosis. Hepatology 2021, 74, 1660–1673. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yu, Q.; Peng, H.; Zhen, Z. Alterations of gut microbiome and effects of probiotic therapy in patients with liver cirrhosis: A systematic review and meta-analysis. Medicine 2022, 101, e32335. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Fagan, A.; Gavis, E.A.; Sterling, R.K.; Gallagher, M.L.; Lee, H.; Matherly, S.C.; Siddiqui, M.S.; Bartels, A.; Mousel, T.; et al. Microbiota transplant for hepatic encephalopathy in cirrhosis: The THEMATIC trial. J. Hepatol. 2025, 83, 81–91. [Google Scholar] [CrossRef]
- Lombardi, M.; Troisi, J.; Motta, B.M.; Torre, P.; Masarone, M.; Persico, M. Gut-Liver Axis Dysregulation in Portal Hypertension: Emerging Frontiers. Nutrients 2024, 16, 1025. [Google Scholar] [CrossRef]
- Anand, S.; Mande, S.S. Host-microbiome interactions: Gut-Liver axis and its connection with other organs. NPJ Biofilms Microbiomes 2022, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Abraldes, J.G.; Rich, N.E.; Wong, V.W.-S. AGA Clinical Practice Update on the Use of Vasoactive Drugs and Intravenous Albumin in Cirrhosis: Expert Review. Gastroenterology 2024, 166, 202–210. [Google Scholar] [CrossRef]
- Silvey, S.; Patel, N.; Tsai, S.Y.; Nadeem, M.; Sterling, R.K.; Markley, J.D.; French, E.; O’Leary, J.G.; Bajaj, J.S. Higher Rate of Spontaneous Bacterial Peritonitis Recurrence With Secondary Spontaneous Bacterial Peritonitis Prophylaxis Compared With No Prophylaxis in 2 National Cirrhosis Cohorts. Am. J. Gastroenterol. 2025, 120, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Lontos, S.; Gow, P.J.; Vaughan, R.B.; Angus, P.W. Norfloxacin and trimethoprim-sulfamethoxazole therapy have similar efficacy in prevention of spontaneous bacterial peritonitis. J. Gastroenterol. Hepatol. 2008, 23, 252–255. [Google Scholar] [CrossRef]
- Alvarez, R.F.; de Mattos, A.A.; Corrêa, E.B.D.; Cotrim, H.P.; Nascimento, T.V.S.B. Trimethoprim-sulfamethoxazole versus norfloxacin in the prophylaxis of spontaneous bacterial peritonitis in cirrhosis. Arq. Gastroenterol. 2005, 42, 256–262. [Google Scholar] [CrossRef]
- Iyer, U.; Jara-Tantoco, M.N.; Delgado, A. P-163. Trimethoprim-Sulfamethoxazole is Associated with Decreased Mortality Compared to Fluoroquinolones for Secondary Prophylaxis of Spontaneous Bacterial Peritonitis: A Global Network Analysis. Open Forum Infect. Dis. 2026, 13, ofaf695.387. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Rodriguez, M.P.; Fagan, A.; McGeorge, S.; Sterling, R.K.; Lee, H.; Luketic, V.; Fuchs, M.; Davis, B.C.; Sikaroodi, M.; et al. Impact of bacterial infections and spontaneous bacterial peritonitis prophylaxis on phage-bacterial dynamics in cirrhosis. Hepatology 2022, 76, 1723–1734. [Google Scholar] [CrossRef]
- Juncu, S.; Minea, H.; Lungu, A.; Jucan, A.; Avram, R.; Buzuleac, A.-M.; Cojocariu, C.; Diaconu, L.S.; Stanciu, C.; Trifan, A.; et al. Fluoroquinolones for the Prophylaxis of Spontaneous Bacterial Peritonitis in Patients with Liver Cirrhosis: Are They Losing Ground? Life 2025, 15, 586. [Google Scholar] [CrossRef]
- Salehi, S.; Tranah, T.H.; Lim, S.; Heaton, N.; Heneghan, M.; Aluvihare, V.; Patel, V.C.; Shawcross, D.L. Rifaximin reduces the incidence of spontaneous bacterial peritonitis, variceal bleeding and all-cause admissions in patients on the liver transplant waiting list. Aliment. Pharmacol. Ther. 2019, 50, 435–441. [Google Scholar] [CrossRef]
- Fernández, J.; Prado, V.; Trebicka, J.; Amoros, A.; Gustot, T.; Wiest, R.; Deulofeu, C.; Garcia, E.; Acevedo, J.; Fuhrmann, V.; et al. Multidrug-resistant bacterial infections in patients with decompensated cirrhosis and with acute-on-chronic liver failure in Europe. J. Hepatol. 2019, 70, 398–411. [Google Scholar] [CrossRef]
- Liu, J.; Guevara, J.G.; Macnaughtan, J.; Jin, Y.; Clasen, F.; Kerbert, A.; Portlock, T.; González, J.M.; Habtesion, A.; Phillips, A.; et al. TOP-218 Yaq-001 positively impacts gut microbiome composition, virulence, antimicrobial resistance gene profile resulting in significant effects on ammonia, endotoxemia and inflammation in cirrhosis patients. J. Hepatol. 2025, 82, S132–S133. [Google Scholar] [CrossRef]
- Liu, J.; Macnaughtan, J.; Kerbert, A.J.C.; Portlock, T.; Martínez Gonzalez, J.; Jin, Y.; Clasen, F.; Habtesion, A.; Ji, H.; Jin, Q.; et al. Clinical, experimental and pathophysiological effects of Yaq-001: A non-absorbable, gut-restricted adsorbent in models and patients with cirrhosis. Gut 2024, 73, 1183–1198. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Fagan, A.; Gavis, E.A.; Kassam, Z.; Sikaroodi, M.; Gillevet, P.M. Long-term Outcomes of Fecal Microbiota Transplantation in Patients With Cirrhosis. Gastroenterology 2019, 156, 1921–1923.e3. [Google Scholar] [CrossRef]
- University of Chicago. The Microbiota Augmentation to Reestablish Commensal Organisms (MARCO) Trial. Available online: https://clinicaltrials.gov/study/NCT06871111 (accessed on 23 March 2026).
- Brödel, A.K.; Charpenay, L.H.; Galtier, M.; Fuche, F.J.; Terrasse, R.; Poquet, C.; Havránek, J.; Pignotti, S.; Krawczyk, A.; Arraou, M.; et al. In situ targeted base editing of bacteria in the mouse gut. Nature 2024, 632, 877–884. [Google Scholar] [CrossRef]
- Lam, K.N.; Spanogiannopoulos, P.; Soto-Perez, P.; Alexander, M.; Nalley, M.J.; Bisanz, J.E.; Nayak, R.R.; Weakley, A.M.; Yu, F.B.; Turnbaugh, P.J. Phage-delivered CRISPR-Cas9 for strain-specific depletion and genomic deletions in the gut microbiome. Cell Rep. 2021, 37, 109930. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Bhattacharjee, R.; Nandi, A.; Sinha, A.; Kar, S.; Manoharan, N.; Mitra, S.; Mojumdar, A.; Panda, P.K.; Patro, S.; et al. Phage delivered CRISPR-Cas system to combat multidrug-resistant pathogens in gut microbiome. Biomed. Pharmacother. 2022, 151, 113122. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Bloom, P.P.; Tapper, E.B.; Young, V.B.; Lok, A.S. Microbiome therapeutics for hepatic encephalopathy. J. Hepatol. 2021, 75, 1452–1464. [Google Scholar] [CrossRef]
- Zhou, Y.-L.; Pu, S.-T.; Xiao, J.-B.; Luo, J.; Xue, L. Meta-analysis of probiotics efficacy in the treatment of minimum hepatic encephalopathy. Liver Int. 2024, 44, 3164–3173. [Google Scholar] [CrossRef]
- Dalal, R.; McGee, R.G.; Riordan, S.M.; Webster, A.C. Probiotics for people with hepatic encephalopathy. Cochrane Database Syst. Rev. 2017, 2, CD008716. [Google Scholar] [CrossRef]
- Rose, E.C.; Odle, J.; Blikslager, A.T.; Ziegler, A.L. Probiotics, Prebiotics and Epithelial Tight Junctions: A Promising Approach to Modulate Intestinal Barrier Function. Int. J. Mol. Sci. 2021, 22, 6729. [Google Scholar] [CrossRef]
- Montagnese, S.; Rautou, P.E.; Romero-Gómez, M.; Larsen, F.S.; Shawcross, D.L.; Thabut, D.; Vilstrup, H.; Weissenborn, K. EASL Clinical Practice Guidelines on the management of hepatic encephalopathy. J. Hepatol. 2022, 77, 807–824. [Google Scholar] [CrossRef]
- Úbeda, M.; Lario, M.; Muñoz, L.; Borrero, M.-J.; Rodríguez-Serrano, M.; Sánchez-Díaz, A.-M.; Del Campo, R.; Lledó, L.; Pastor, Ó.; García-Bermejo, L.; et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057. [Google Scholar] [CrossRef]
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The microbiota in cirrhosis and its role in hepatic decompensation. J. Hepatol. 2021, 75, S67–S81. [Google Scholar] [CrossRef]
- Fiorucci, S.; Urbani, G.; Distrutti, E.; Biagioli, M. Obeticholic Acid and Other Farnesoid-X-Receptor (FXR) Agonists in the Treatment of Liver Disorders. Pharmaceuticals 2025, 18, 1424. [Google Scholar] [CrossRef]
- Ma, K.; Tang, D.; Yu, C.; Zhao, L. Progress in research on the roles of TGR5 receptor in liver diseases. Scand. J. Gastroenterol. 2021, 56, 717–726. [Google Scholar] [CrossRef]
- Canovai, E.; Farré, R.; Accarie, A.; Lauriola, M.; de Hertogh, G.; Vanuytsel, T.; Pirenne, J.; Ceulemans, L.J. INT-767-A Dual Farnesoid-X Receptor (FXR) and Takeda G Protein-Coupled Receptor-5 (TGR5) Agonist Improves Survival in Rats and Attenuates Intestinal Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2023, 24, 14881. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Passeri, D.; de Franco, F.; Ciaccioli, G.; Donadio, L.; Rizzo, G.; Orlandi, S.; Sadeghpour, B.; Wang, X.X.; Jiang, T.; et al. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol. Pharmacol. 2010, 78, 617–630. [Google Scholar] [CrossRef]
- Liu, W.; Hu, D.; Huo, H.; Zhang, W.; Adiliaghdam, F.; Morrison, S.; Ramirez, J.M.; Gul, S.S.; Hamarneh, S.R.; Hodin, R.A. Intestinal Alkaline Phosphatase Regulates Tight Junction Protein Levels. J. Am. Coll. Surg. 2016, 222, 1009–1017. [Google Scholar] [CrossRef]
- Liu, Y.; Cavallaro, P.M.; Kim, B.-M.; Liu, T.; Wang, H.; Kühn, F.; Adiliaghdam, F.; Liu, E.; Vasan, R.; Samarbafzadeh, E.; et al. A role for intestinal alkaline phosphatase in preventing liver fibrosis. Theranostics 2021, 11, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lv, H.; Zhang, H.; Li, H.; Wang, A.; Yu, S.; He, Q.; Chen, S.; Wang, J.; Ran, X.; et al. The protective role of intestinal alkaline phosphatase in inflammatory bowel disease-associated non-alcoholic fatty liver disease. Life Sci. 2026, 384, 124113. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Giovannini, M.; Di Micoli, V.; Grandi, E.; Borghi, C.; Cicero, A.F.G. Effect of Supplementation of a Butyrate-Based Formula in Individuals with Liver Steatosis and Metabolic Syndrome: A Randomized Double-Blind Placebo-Controlled Clinical Trial. Nutrients 2024, 16, 2454. [Google Scholar] [CrossRef]
- Abbasi, F.; Haghighat Lari, M.M.; Khosravi, G.R.; Mansouri, E.; Payandeh, N.; Milajerdi, A. A systematic review and meta-analysis of clinical trials on the effects of glutamine supplementation on gut permeability in adults. Amino Acids 2024, 56, 60. [Google Scholar] [CrossRef]
- Achamrah, N.; Déchelotte, P.; Coëffier, M. Glutamine and the regulation of intestinal permeability: From bench to bedside. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 86–91. [Google Scholar] [CrossRef]
- Cordes, F.; Demmig, C.; Bokemeyer, A.; Brückner, M.; Lenze, F.; Lenz, P.; Nowacki, T.; Tepasse, P.; Schmidt, H.H.; Schmidt, M.A.; et al. MicroRNA-320a Monitors Intestinal Disease Activity in Patients With Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2020, 11, e00134. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.-J.; Piurowski, V.; Rigot, B.; Croce, C.M. MicroRNAs in intestinal barrier function, inflammatory bowel disease and related cancers-their effects and therapeutic potentials. Curr. Opin. Pharmacol. 2017, 37, 142–150. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Olaru-Stavila, C.; Steinmann, S.M.; Mester, P.; Müller, M.; Tcaciuc, E.; Gülow, K. The Gastrointestinal Barrier—Mechanisms of Barrier Dysfunction in Liver Cirrhosis and Spontaneous Bacterial Peritonitis. Biomedicines 2026, 14, 1084. https://doi.org/10.3390/biomedicines14051084
Olaru-Stavila C, Steinmann SM, Mester P, Müller M, Tcaciuc E, Gülow K. The Gastrointestinal Barrier—Mechanisms of Barrier Dysfunction in Liver Cirrhosis and Spontaneous Bacterial Peritonitis. Biomedicines. 2026; 14(5):1084. https://doi.org/10.3390/biomedicines14051084
Chicago/Turabian StyleOlaru-Stavila, Catalina, Sara Martina Steinmann, Patricia Mester, Martina Müller, Eugen Tcaciuc, and Karsten Gülow. 2026. "The Gastrointestinal Barrier—Mechanisms of Barrier Dysfunction in Liver Cirrhosis and Spontaneous Bacterial Peritonitis" Biomedicines 14, no. 5: 1084. https://doi.org/10.3390/biomedicines14051084
APA StyleOlaru-Stavila, C., Steinmann, S. M., Mester, P., Müller, M., Tcaciuc, E., & Gülow, K. (2026). The Gastrointestinal Barrier—Mechanisms of Barrier Dysfunction in Liver Cirrhosis and Spontaneous Bacterial Peritonitis. Biomedicines, 14(5), 1084. https://doi.org/10.3390/biomedicines14051084