Abstract
Sleep is a vital biological function governed by neuronal networks in the brainstem, hypothalamus, and thalamus. Disruptions in these circuits contribute to the sleep disturbances observed in neurodegenerative disorders, including Parkinson’s disease, epilepsy, Huntington’s disease, and Alzheimer’s disease. Oxidative stress, mitochondrial dysfunction, neuroinflammation, and abnormal protein accumulation adversely affect sleep architecture in these conditions. The interaction among these pathological processes is believed to modify sleep-regulating circuits, consequently worsening clinical symptoms. This review examines the cellular and molecular mechanisms that impair sleep regulation in experimental models of these four disorders, emphasizing how oxidative stress, neuroinflammation and synaptic dysfunction contribute to sleep fragmentation and alterations in rapid eye movement (REM) sleep and slow-wave sleep (SWS) phases. In Parkinson’s disease models (6-OHDA and MPTP), dopaminergic degeneration and damage to sleep-regulating nuclei result in daytime somnolence and disrupted sleep patterns. Epilepsy models (kainate, pentylenetetrazole, and kindling) provoke hyperexcitability and oxidative damage, compromising both REM and SWS. Huntington’s disease models (R6/2 and 3-NP) demonstrate reduced sleep duration, circadian irregularities, and oxidative damage in the hypothalamus and suprachiasmatic nucleus. In Alzheimer’s disease (AD) models (APP/PS1, 3xTg-AD, and Tg2576), early sleep problems include diminished SWS and REM sleep, increased awakenings, and circadian rhythm disruption. These changes correlate with β-amyloid and tau deposition, glial activation, chronic inflammation, and mitochondrial damage in the hypothalamus, hippocampus, and prefrontal cortex. Sleep disturbances across these neurodegenerative disease models share common underlying mechanisms like oxidative stress, neuroinflammation, and mitochondrial dysfunction. Understanding these pathways may reveal therapeutic targets to improve both motor symptoms and sleep quality in neurodegenerative disorders.
1. Introduction
Sleep is regulated by a complex network of brain regions, including the brainstem, hypothalamus, thalamus, and cerebral cortex, which interact through specific neural circuits to control the sleep–wake cycle [1,2]. GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus (VLPO) of the hypothalamus suppress wake-promoting areas such as the locus coeruleus and tuberomammillary nucleus, facilitating sleep onset and maintenance [3,4]. Multiple neurotransmitter systems, including gamma-aminobutyric acid (GABA), glutamate, acetylcholine, orexin, noradrenaline, norepinephrine, serotonine and histamine, modulate sleep architecture through precisely balanced excitatory and inhibitory synaptic pathways [5,6,7]. The degeneration of hypocretin-producing neurons in the lateral hypothalamus contributes to narcolepsy and the sleep disturbances commonly observed in neurodegenerative disorders such as Parkinson’s and Alzheimer’s disease [8,9]. In Parkinson’s disease, alterations in the mesocorticolimbic dopaminergic system disrupt circadian rhythms and sleep architecture, leading to insomnia, periodic limb movements, and excessive daytime somnolence [10,11]. Epilepsy involves the dysfunction of the thalamocortical networks that normally regulate sleep spindles, potentially promoting epileptiform discharges during non-REM sleep [12,13,14]. Huntington’s disease (HD) features circadian rhythm disturbances and the altered expression of clock genes such as Per1 and Bmal1, associated with neurodegeneration in the suprachiasmatic nucleus and other sleep-regulating regions [15,16,17]. In Alzheimer’s disease, β-amyloid and tau aggregation in the hypothalamus, brainstem, and prefrontal cortex correlates with sleep fragmentation and reduced SWS and REM sleep activity, thereby accelerating disease progression [18,19]. These findings highlight that sleep regulation involves specific neuronal and molecular mechanisms that become disrupted in major neurological disorders. Oxidative stress, neuroinflammation, mitochondrial dysfunction, and abnormal protein accumulation impair sleep-regulating circuits in neurodegenerative disease models. Sleep is a fundamental biological process essential to brain homeostasis, neuronal repair, and cognitive function, making it a critical subject of study in the context of neurological health. In neurodegenerative diseases, sleep disturbances are not merely secondary symptoms but active contributors to disease progression. Despite the high prevalence of sleep disorders in conditions such as Parkinson’s disease, epilepsy, Huntington’s disease, and Alzheimer’s disease, there is a lack of comprehensive studies examining how sleep-regulating circuits are affected in neurological experimental models. Present research mainly focuses on clinical symptoms or behavioral observations, with limited exploration of the cellular and molecular mechanisms underlying sleep disruption in animal models. This review addresses that critical gap by analyzing how oxidative stress, neuroinflammation, mitochondrial dysfunction, and synaptic impairments contribute to sleep fragmentation and alterations in REM and slow-wave sleep phases. Unraveling these interconnected mechanisms is crucial for developing targeted therapies that not only address motor and cognitive deficits and restore healthy sleep architecture. Improving sleep quality may represent a powerful strategy to mitigate neurological decline and enhance the overall quality of life of patients with neurodegenerative disorders, especially considering how these pathways disrupt sleep regulation in experimental models of Parkinson’s disease, epilepsy, Huntington’s disease, and Alzheimer’s disease.
We conducted a comprehensive literature search using Google Scholar and PubMed to identify relevant studies examining sleep’s role in experimental models of neurodegenerative diseases, specifically Parkinson’s disease, Alzheimer’s disease, epilepsy, and Huntington’s disease. The search employed multiple keyword combinations including SWS and REM, glymphatic system, protein clearance, β-amyloid, tau, α-synuclein, mutant huntingtin, oxidative stress, neuroinflammation, synaptic plasticity, signaling pathways, Nrf2, NF-κB, GABA, and glutamate. We included experimental studies (in vivo and in vitro) and review papers that utilized established models of the specified neurodegenerative disorders. Studies were selected based on their methodological quality, scientific rigor, and relevance to the physiological and molecular mechanisms through which sleep facilitates neuroprotection and disease modulation. We analyzed the selected literature to identify both the common and disease-specific mechanisms by which sleep influences protein aggregation, oxidative and inflammatory responses, neurotransmitter regulation, and the activation of survival and plasticity-related signaling pathways. We synthesized these findings to better understand sleep’s role in mitigating neurodegenerative processes.
2. Sleep and the Diseased Brain: Experimental Models in Neurodegenerative and Neurological Disorders
2.1. Models of Parkinson’s Disease and Sleep Disruptions
In Parkinson’s disease, experimental models such as 6-hydroxydopamine (6-OHDA) and 1-Methyl-4-phenyl-pyridine (MPP+) have proven essential for investigating dopaminergic neurodegeneration and associated sleep disturbances. Neurodegeneration directly affects the neuronal networks responsible for sleep–wake control, including the locus coeruleus, tuberomammillary nucleus, lateral hypothalamus, and suprachiasmatic nucleus. This leads to sleep fragmentation, reduced SWS, and the altered REM sleep patterns commonly observed in Parkinson’s patients [20,21,22,23,24]. Mitochondrial dysfunction and oxidative stress, central features of Parkinson’s disease that are replicated in these models, impair sleep by damaging critical neurons and altering the electrical excitability necessary for normal sleep architecture [25,26,27,28,29]. MPP+, the toxic metabolite derived from MPTP, blocks mitochondrial complex I, increasing reactive oxygen species (ROS) generation, which causes oxidative damage to neuronal proteins, lipids, and DNA in brain regions controlling sleep regulation [30,31,32,33,34,35]. Similarly, 6-OHDA induces severe oxidative stress through autooxidation in dopaminergic neurons, promoting apoptosis and glial activation, which intensify neuroinflammation and disrupt circadian rhythms [36,37,38,39,40]. Abnormal α-synuclein aggregation, a hallmark of Parkinson’s disease, correlates with sleep disturbances. These aggregates appear early in the brainstem nuclei involved in sleep regulation, as evidenced by the strong association between REM sleep behavior disorders and disease progression [41,42,43,44]. Oxidative stress and impaired cellular degradation systems promote protein aggregation [45,46,47,48], creating a vicious cycle that affects both neuropathology and sleep-related clinical symptoms. Disruptions in calcium homeostasis, excitotoxicity, and axonal transport, worsened by mitochondrial dysfunction and oxidative stress, damage orexinergic neurons in the lateral hypothalamus [7]. These neurons are crucial for maintaining stable wakefulness, and their impairment contributes to excessive daytime sleepiness and sleep fragmentation in both animal models and patients [22,49,50,51]. Neuroinflammation triggered by activated microglia and astrocytes leads to pro-inflammatory cytokine release (IL-1β, TNF-α, IL-6), which disrupts sleep-associated neurotransmitter regulation, worsening sleep fragmentation and reducing REM sleep [52,53,54,55,56]. Numerous drugs, including dopaminergic medicines like levodopa and dopamine agonists (like pramipexole and ropinirole), melatonin, and clonazepam, have been researched in this model to treat sleep–wake disorders, especially REM sleep behavior disorder [57,58]. These therapies try to change the circadian and REM-related circuits that are impacted in Parkinson’s disease or restore dopaminergic tone.
Genetic mutations in SNCA (α-synuclein), PARK2, PINK1, DJ-1, and LRRK2 affect protein aggregation and mitochondrial function, contributing to Lewy body formation and non-motor symptoms, including sleep disturbances [59,60,61,62]. Additionally, polymorphisms affecting oxidative stress and inflammatory responses may influence sleep disruption severity, motor symptoms and disease progression [63,64]. Dysfunction in signaling pathways including MAPK, NF-κB, and Nrf2, which are essential for oxidative stress responses and neuroinflammation, affects neuronal survival in sleep-regulating nuclei and influences sleep–wake cycle disturbances in Parkinson’s disease [65,66,67,68,69].
The 6-OHDA and MPTP models effectively recapitulate both the characteristic dopaminergic neurodegeneration of Parkinson’s disease and its non-motor symptoms, particularly sleep disturbances. These models demonstrate how oxidative stress, mitochondrial dysfunction, calcium dysregulation, neuroinflammation, and protein aggregation impact sleep-regulating neural circuits. They provide valuable experimental platforms for investigating pathogenic mechanisms and evaluating therapies targeting both motor and non-motor symptoms [70,71,72,73] (Figure 1).
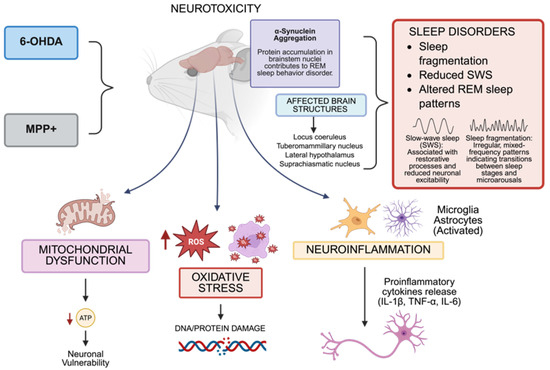
Figure 1.
In murine models, the administration of neurotoxins, including 6-hydroxydopamine (6-OHDA) and 1MPP+, leads to neurotoxicity characterized by mitochondrial injury, reactive oxygen species (ROS), oxidative stress, and neuroinflammation. These changes impact important brain centers that control sleep, including the locus coeruleus, tuberomammillary nucleus, lateral hypothalamus, and suprachiasmatic nucleus, causing poor sleep quality, a decrease in slow-wave sleep (SWS), and changes in rapid eye movement (REM) sleep. The accumulation of misfolded α-synuclein in brainstem nuclei and the production of pro-inflammatory cytokines (e.g., IL-1β, TNF-α, IL-6) are the causes of dysfunction in sleep–wake-regulating circuits. The dates show ↓ = Decreased; ↑ = Increased (Created with https://BioRender.com). By Emiliano Gonzalez and Alexis Ponce, 2025.
2.2. Epilepsy Models and Sleep Architecture
Animal models have been essential for understanding epilepsy pathophysiology, epileptogenesis, and therapeutic development. Kainate (KA) and pentylenetetrazol (PTZ) seizure models effectively replicate various epileptic seizures, enabling a detailed investigation of the underlying cellular and molecular mechanisms and associated sleep disorders. These models serve as vital tools for understanding the complex relationships among the key of components neuronal dysfunction, oxidative stress, and sleep architecture in human epilepsy, particularly in chronic and treatment-resistant cases.
The kainate model employs an AMPA/KA receptor agonist that induces severe seizures and neuronal damage, primarily in the hippocampus, mimicking human temporal lobe epilepsy [74,75,76]. Glutamatergic excitotoxicity causes intracellular calcium overload, activating enzymes like phospholipase A2 and nitric oxide synthase. This leads to excessive reactive oxygen and nitrogen species production, causing significant oxidative damage and cell death through necrosis and apoptosis [77,78,79,80].
This model demonstrates how metabolic alterations lead to functional sleep changes, including reduced SWS, diminished REM sleep, and increased sleep fragmentation. These changes result from neuronal damage in critical regions such as the hippocampus, thalamus, and cortex, accompanied by inflammatory and oxidative processes [81,82,83,84]. The KA model enables causal investigation of epilepsy, oxidative stress, and sleep disruption from both molecular and functional perspectives.
The PTZ model induces generalized seizures by antagonizing GABAA receptors, disrupting excitatory–inhibitory balance and triggering neuronal hyperexcitability [85,86]. This hyperactive state promotes ROS formation through oxidative enzyme activation and mitochondrial dysfunction, compromising cellular integrity [87,88]. The resulting oxidative stress response affects antioxidant protein production (superoxide dismutase and catalase) and activates the NF-κB-controlled inflammatory pathways involved in epileptogenesis [89,90,91]. The PTZ model shows sleep architecture changes characterized by reduced sleep efficiency, decreased REM sleep, and prolonged sleep latency. These modifications relate to alterations in key neurotransmitters (serotonin and dopamine) and increased oxidative stress [92,93,94]. By improving GABAergic tone and controlling circadian rhythms, drugs like benzodiazepines (such as diazepam and clonazepam) and melatonin have been investigated as ways to improve the quality of sleep in these models [95,96]. Although their effects on sleep can vary depending on the dosage and treatment length, antiepileptic medications such as levetiracetam and valproate have also demonstrated the ability to alter sleep architecture while reducing the frequency of seizures [97].
The kindling paradigm uses repetitive subconvulsive electrical stimulation in limbic areas like the hippocampus or amygdala, providing a crucial tool for investigating progressive epileptogenesis and associated sleep architecture changes [98,99,100,101]. This model demonstrates substantial decreases in SWS and REM sleep, with notable increases in sleep fragmentation [102,103,104], disrupting sleep quality and restorative capacity. These sleep disorders correlate with kindling-induced neuronal hyperexcitability, driven by neurotransmitter dysregulation including increased glutamate release and GABAergic system dysfunction [105,106]. At the molecular level, repetitive stimulation activates signaling pathways such as MAPK and JNK, promoting synaptic plasticity and hyperexcitability. Kindling also markedly increases ROS production, causing oxidative damage to lipids and DNA while compromising mitochondrial function, exacerbating epileptogenesis [107,108,109,110]. Oxidative stress disrupts sleep architecture by triggering inflammatory processes through microglial activation and pro-inflammatory cytokine production, affecting circadian rhythms [111,112]. These processes alter the neurotransmitters governing the sleep–wake cycle, leading to fragmentation and reduced SWS and REM sleep phases. Sleep deprivation impairs memory consolidation and neuronal repair, intensifying hyperexcitability and promoting chronic epilepsy progression [113].
Optogenetic models have revolutionized epilepsy research by enabling precise neuronal population control through light stimulation in genetically modified animals [114,115]. Prolonged excitatory neuron activation or inhibitory neuron suppression can trigger seizures, promote abnormal plasticity, and alter functional connectivity, directly affecting sleep architecture and quality [116]. The optogenetic stimulation of specific circuits, including the lateral hypothalamus and ventrolateral preoptic nucleus, substantially alters sleep duration and quality, reducing REM sleep and promoting prolonged wakefulness [117,118,119]. These sleep abnormalities create a detrimental cycle where sleep disturbance intensifies neuronal hyperexcitability and seizure frequency. These models reveal cellular-level calcium homeostasis disruption, crucial in neuronal excitotoxicity during seizures. This alteration increases ROS generation, causing oxidative damage that compromises neuronal function [114]. Oxidative stress exacerbates neuronal inflammation and synaptic dysfunction, also affecting circadian rhythms and sleep architecture [113].
Research on genetic epileptic channelopathy models has revealed significant sleep architecture and regulation changes. Animal models with ion channel gene mutations (SCN1A for sodium, KCNQ2 for potassium, or CACNA1H for calcium) show marked reductions in SWS, increased sleep fragmentation, and prolonged nocturnal wakefulness, suggesting impaired thalamocortical circuits and sleep-regulating structures, including the hypothalamus [120,121,122,123]. These changes demonstrate the strong relationship between abnormal neuronal excitability and sleep–wake cycles, indicating that channelopathic mutations not only cause seizures but also disrupt sleep homeostasis from early developmental stages [124]. These models help understand how chronic ionic dysregulation affects sleep physiology through both direct and indirect pathways, including oxidative stress, mitochondrial dysfunction, and neuroimmune activation processes [125,126,127,128]. These processes may worsen sleep problems by creating pro-inflammatory environments in the brain regions essential for circadian and sleep–wake control (Figure 2).
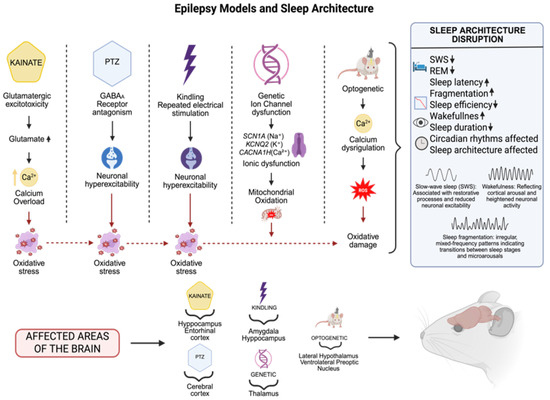
Figure 2.
Experimental models of epilepsy replicate distinct seizure mechanisms: in the case of kainate, a glutamatergic agonist, (increases glutamate and serum calcium levels),temporal lobe seizures are induced at a cellular level by excitotoxicity; pentylenetetrazole (PTZ), a gamma-aminobutyric acid A (GABAA) receptor antagonist: generalized seizures; kindling: progressive epileptogenesis; genetic models: inherited epilepsies such as ion channel mutations; and optogenetics: manipulation of sleep circuits. These models affect important brain areas involving the hippocampus, entorhinal and cerebral cortex, amygdala, thalamus, and hypothalamus, and are accompanied by changes in sleep features (decreased SWS REM, sleep efficiency, sleep duration, enhanced fragmentation, prolonged latency, altered circadian rhythm and sleep architecture) The dates show ↓ = Decreased; ↑ = Increased (Created with BioRender.com). By Emiliano Gonzalez and Alexis Ponce, 2025.
2.3. Huntington’s Disease Models and Sleep Disruptions
Sleep disturbances represent early and persistent manifestations of Huntington’s disease (HD), often appearing before motor symptoms and highlighting their potential as clinical biomarkers and therapeutic targets [15,129]. These abnormalities include reduced sleep quality, decreased REM and SWS, increased sleep fragmentation, prolonged sleep latency, and circadian rhythm disruption [130].
HD animal models, including transgenic mice such as YAC128 and R6/2, as well as knock-in animals, demonstrate substantial alterations in sleep architecture and circadian rhythms. These changes encompass sleep fragmentation, reduced SWS, decreased overall sleep duration, and disrupted circadian gene expression in critical brain regions including the suprachiasmatic nucleus, lateral hypothalamus, and striatum [15,131,132,133]. R6/2 mice expressing mutant huntingtin fragments exhibit progressive sleep–wake cycle disturbances, characterized by significant reductions in REM and SWS, increased sleep fragmentation, and prolonged sleep onset times [134]. These sleep alterations correlate with dysfunction in the hypothalamic and brainstem nuclei governing circadian rhythm and sleep regulation, where oxidative stress and neuroinflammation cause neuronal impairment [131,134,135,136,137]. Several pharmaceutical treatments have been tested to lessen sleep disturbances in these HD models. While sedative antidepressants like trazodone have been investigated for the treatment of insomnia [138], melatonin and its receptor agonists have demonstrated promise in enhancing sleep quality and re-establishing circadian rhythms [139]. Compounds that target adenosinergic transmission or anti-inflammatory drugs like minocycline are also being studied for their dual function of lowering neuroinflammation and regulating sleep [140]. Animal models, including transgenic sheep and monkeys, have been developed and show circadian dysfunction with more complex phenotypes, enhancing their translational relevance for evaluating sleep restoration therapies [141,142,143,144]. However, these models are costly and require extended periods for symptom manifestation.
Toxin-induced models using 3-nitropropionic acid (3-NP), an irreversible mitochondrial succinate dehydrogenase inhibitor, show substantial sleep architecture disruptions like R6/2 mice, including reduced REM sleep, increased awakenings, and sleep fragmentation [145,146]. These disturbances are linked to glial inflammation, mitochondrial dysfunction, and oxidative stress in sleep-regulating brain regions such as the hypothalamus and brainstem [147,148]. Quinolinic acid administration, a neurotoxic tryptophan pathway metabolite causing excitotoxicity, disrupts sleep cycles by altering neurotransmitters and promoting neuroglial inflammation, leading to sleep fragmentation and circadian rhythm disturbances [149,150,151,152].
HD results from abnormal CAG repeat expansion in the HTT gene, producing a mutant huntingtin protein variant [153,154,155]. This abnormal protein triggers multiple pathogenic processes including mitochondrial dysfunction, oxidative stress, DNA damage, calcium homeostasis disruption, synaptic dysfunction, and apoptotic pathway activation [156,157]. Oxidative stress serves as a central element in HD pathology, characterized by elevated ROS, a reduced cellular antioxidant capacity, and oxidative damage to lipids, proteins, and nucleic acids [158,159,160]. These metabolic alterations affect brain regions controlling sleep, exacerbating circadian rhythm and sleep homeostasis problems. At the molecular and cellular levels, these models demonstrate that oxidative stress in sleep-regulatory regions impairs neurochemical signaling relevant to sleep–wake regulation, affecting critical neurotransmitters including GABA, glutamate, adenosine, and monoamines [137,161,162,163]. Mitochondrial failure generates excessive ROS, disrupting neuronal and glial homeostasis and promoting neuronal death in circadian rhythm-regulating areas [164,165].
Chronic microglial and astrocyte activation leads to pro-inflammatory cytokine release (IL-1β and TNF-α), disrupting synaptic transmission and sleep homeostasis [166,167]. This pro-inflammatory and oxidative environment alters circadian clock gene expression and function (Clock, Bmal1, Per, and Cry), directly affecting sleep–wake cycle synchronization [131,137,168]. Disrupted purinergic transmission, particularly reduced adenosine levels, a sleep-promoting compound, further exacerbates the fragmentation and poor sleep quality observed in these models [137,169,170]. In cellular models using neuronal and stem cell cultures, mutant huntingtin expression induces mitochondrial dysfunction, oxidative stress, impaired axonal transport, and toxic protein aggregate accumulation [171,172,173]. Mitochondrial damage, oxidative stress, excitotoxicity, glial inflammation, and circadian gene dysfunction collectively form a pathogenic network underlying HD sleep disorders. These findings emphasize the importance of targeting oxidative stress and neuroinflammation as potential approaches to improve sleep disturbances in this condition [164,166,174] (Figure 3).
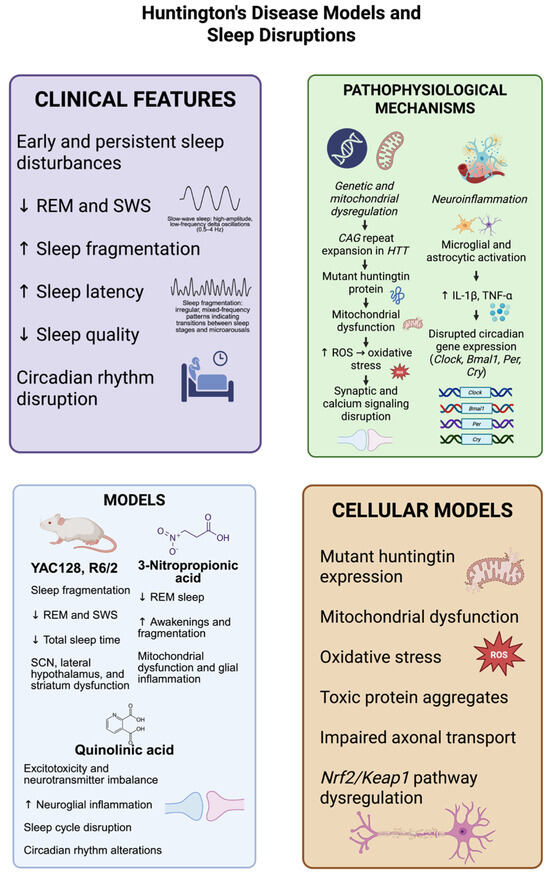
Figure 3.
Models of Huntington’s disease (HD) reliably reproduce the major sleep abnormalities seen in the disease, which consist of decreased rapid eye movement (REM) sleep, the loss of slow-wave sleep (SWS), increased sleep fragmentation, prolonged sleep onset latency, and disrupted circadian rhythms. Transgenic mouse models, including YAC128 and R6/2, and neurotoxin-based models, using 3-nitropropionic acid and quinolinic acid, display mitochondrial dysfunction, neuroinflammatory responses, and changes in the brain areas crucial for sleep regulation, such as the suprachiasmatic nucleus, hypothalamus, and striatum. Molecularly, the expression of mutant huntingtin protein driven by CAG repeat expansion results in oxidative stress via an increase in reactive oxygen species (ROS), disturbed synaptic function, and the defective expression of circadian genes (Clock, Bmal1, Per, Cry). Complementary cellular models further support these observations, showing the presence of cytotoxic protein aggregates, abnormalities in axonal transport, and the Nrf2/Keap1 antioxidant pathway disruption. These pathophysiological mechanisms together lead to sleep–wake disturbances, (decreased SWS, REM, sleep quality and total sleep time, increased awakenings, fragmentation and sleep latency), which can accelerate neurodegeneration. The dates show ↓ = Decreased; ↑ = Increased. (Created with BioRender.com). By Emiliano Gonzalez and Alexis Ponce, 2025.
2.4. Sleep as a Critical Factor in Alzheimer’s Disease
Evidence has highlighted the crucial role of sleep disruptions in Alzheimer’s disease etiology [175,176]. These disruptions appear as early clinical signs and significantly influence disease progression. Experimental models have been essential for understanding the cellular and molecular mechanisms linking sleep with key AD pathology, including amyloid-β accumulation, tau hyperphosphorylation, neuroinflammation, oxidative stress, and synaptic dysfunction [177,178,179].
Transgenic mouse models, including APP/PS1 and 3xTg-AD, have proven highly valuable for replicating the neuropathological features and sleep-related abnormalities observed in human patients [180,181]. In the 3xTg-AD model, chronic sleep deprivation for eight weeks caused significant memory deficits, increased tau phosphorylation, reduced synaptic protein levels (including PSD-95), and altered CREB expression, an essential transcription factor for synaptic plasticity [181,182]. Additionally, sleep deprivation triggers substantial astrogliosis, evidenced by elevated GFAP levels, suggesting that disrupted sleep intensifies glial activation and neuroinflammatory responses [183,184,185]. The 5xFAD model, characterized by rapid amyloid-β (Aβ) plaque accumulation, also demonstrates spontaneous sleep fragmentation. These mice show significant decreases in SWS and shorter, more fragmented sleep episodes, particularly in females, reflecting the sex-specific sleep disturbances observed in women with AD [186,187]. When 3xTg-AD mice undergo experimental sleep fragmentation, they display elevated hippocampal Aβ40 and Aβ42 levels, alongside intensified neuroinflammatory responses, indicating the increased vulnerability of the AD brain to sleep disturbances [188]. From a metabolic perspective, sleep fragmentation in AD models reduces mitochondrial activity. In 3xTg-AD mice, six weeks of sleep disruption downregulates mitochondrial biogenesis and electron transport chain complex expression, accompanied by decreased AMPK/SIRT1/PGC-1α pathway activity [189]. These changes result in a diminished antioxidant capacity and increased oxidative damage. However, the partial reversal of these alterations occurs with sleep recovery, highlighting the therapeutic potential of sleep normalization for restoring mitochondrial homeostasis [190]. Sleep disruption significantly affects tau pathology in experimental animals. PS19 mice expressing P301S mutant tau exhibit substantial sleep disorders, characterized by reduced SWS and REM sleep, a loss of REM atonia, and disrupted sleep spindles [191]. These changes closely resemble those seen in individuals with REM sleep behavior disorder, a prodromal feature of tauopathies. Pharmacological intervention using dual orexin receptor antagonists (e.g., DORA-12) restores normal sleep architecture in these animals, suggesting potential therapeutic approaches for early-stage intervention [18,192].
Melatonin supplementation, which has been demonstrated to increase SWS and lower oxidative stress, and GABAergic drugs like zolpidem, which promote sleep continuity, are further pharmaceutical strategies able to optimize sleep and lessen neurodegeneration in AD animals [193,194]. Orexin receptor antagonists and anti-inflammatory drugs are also being researched to address sleep issues and the neuroinflammatory pathways that contribute to the development of AD [195,196].
Animal studies demonstrate that even one night of sleep deprivation increases tau levels in the interstitial fluid and cerebrospinal fluid (CSF). This increase depends on neuronal activity and orexin signaling, as orexin inhibition reduces tau accumulation, supporting the concept that sleep deprivation promotes tau pathology spread [191]. Acute experimental models, including the intracerebroventricular (ICV) injection of Aβ oligomers or streptozotocin (STZ-ICV) administration, induce notable sleep disturbances. Aβ injection causes sleep fragmentation, microglial activation, and increased oxidative stress. The STZ-ICV model, which mimics sporadic AD and cerebral insulin resistance, shows cognitive impairments, circadian rhythm disturbances, and fragmented sleep, all associated with mitochondrial dysfunction and reactive oxygen species accumulation [197,198,199,200].
Non-mammalian models have provided valuable insights. In Drosophila melanogaster, neuronal Aβ expression alters circadian and sleep–wake cycles [201]. Transgenic AD models in Danio rerio (zebrafish) enable real-time sleep behavior observation in vivo, utilizing larval transparency and genetic manipulability [202]. These models offer unique perspectives on evolutionarily conserved sleep-regulating mechanisms and their relationship with neurodegenerative diseases. At the molecular level, experimental models show that sleep deprivation impairs the glymphatic clearance of metabolites, including Aβ, during SWS [203]. Disrupted sleep alters essential circadian clock gene expression (BMAL1, CLOCK, PER2), impairing the sleep–wake cycle and desynchronizing critical brain regions such as the suprachiasmatic nucleus [204]. Furthermore, chronic glial activation and inflammation from sleep deprivation create a neurotoxic environment that accelerates dementia progression [34,205]. These experimental models are crucial for both replicating AD neuropathology and validating the pathogenic and therapeutic significance of sleep disorders. They enable mechanistic studies of disease progression, the evaluation of pharmacological and genetic interventions, and the identification of early molecular biomarkers. These models demonstrate that maintaining sleep quality could serve as a powerful neuroprotective strategy against AD progression and provide an essential experimental framework for developing sleep-targeted therapeutics in neurodegenerative disorders (Figure 4).
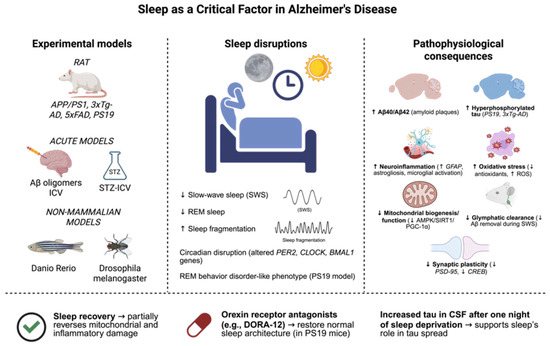
Figure 4.
Sleep disturbances are involved in the pathophysiology of Alzheimer’s disease (AD), as they are active participants in the development of the disease and a clinical manifestation of the disease. Several experimental models, including transgenic mouse lines (APP/PS1, 3xTg-AD, 5xFAD, PS19), acute models of Aβ oligomers, or the intracerebroventricular administration of streptozotocin (STZ-ICV) injected into the brain, and non-mammalian models including Danio rerio and Drosophila melanogaster, have been useful for the reproduction of sleep-related alterations that frequently appear in patients. These consist of decreased slow-wave sleep (SWS) and rapid eye movement (REM) sleep, increased sleep fragmentation, and circadian rhythm disturbances. Equally, some models (e.g., PS19) also demonstrate hallmarks of REM behavior disorder. Notably, sleep deprivation was demonstrated to aggravate several hallmarks of the pathological process, such as Aβ40/42 deposition, tau hyperphosphorylation, neuroinflammation, oxidative stress, mitochondrial dysfunction, glymphatic clearance deficit, as well as synaptic plasticity loss. On the other hand, interventions, e.g., involving sleep recovery or the administration of orexin receptor antagonists (e.g., DORA-12), have been shown to be able to reverse sleep architecture to physiological conditions and reduce cellular damage. These results suggest the possible protective action of sleep and point to its potential as a therapeutic target in Alzheimer’s disease. The dates show ↓ = Decreased; ↑ = Increased (Created with BioRender.com). By Emiliano Gonzalez and Alexis Ponce, 2025.
3. Conclusions
The clearance of waste from the brain is significantly more efficient during sleep than during wake periods, as the convective clearance of neurotoxic compounds that accumulate during wakefulness is highly increased. This might boost brain restoration in many neurological disorders (Parkinson’s disease, epilepsy, Alzheimer’s disease, and Huntington’s syndrome). Their pathogenic mechanisms are all heavily influenced by sleep and include protein aggregation, mitochondrial dysfunction, oxidative stress, excitotoxicity, and neuroinflammation, despite their clinical heterogeneity. The glymphatic system is a primary pathway for CNS waste clearance during SWS, removing potentially harmful misfolded proteins (e.g., tau, α-synuclein, β-amyloid, and mutant huntingtin). The accumulation of harmful proteins plays a central role in causing neuronal damage in neurodegenerative diseases [189,203]. Additionally, sleep affects the expression of genes linked to energy consumption, neurogenesis, synaptic plasticity, and DNA repair, which all are crucial functions to maintain the integrity of neural networks and brain homeostasis [206,207]. Sleep has a profound influence on major cellular signaling pathways such as PI3K/Akt/mTOR, BDNF/TrkB, ERK/MAPK, and CREB, which are essential for memory consolidation, synaptic remodeling, and neuronal survival functions, which are impaired in these illnesses [208]. The progression and stabilization of neurodegenerative diseases are deeply influenced by the balance between NF-κB (a pro-inflammatory mediator) and Nrf2 (a key antioxidant regulator), especially during sleep. This balance is crucial: tipping it toward sustained inflammation and oxidative stress (as with chronic sleep loss) accelerates neurodegenerative processes [209], while restoring or maintaining it (via healthy sleep) offers neuroprotection and can help stabilize the progression of neurodegenerative diseases. Sleep helps lower seizure risk by enhancing GABAergic inhibition, which suppresses excessive neuronal firing and reduces glutamatergic excitation, lowering the chance that neurons will synchronize and “ignite” a seizure [12,210,211]. Many sleep-dependent processes are common denominators across brain diseases; improving sleep could have broad, multi-level therapeutic effects, reducing many damaging disease mechanisms at once. Focusing on sleep/wake regulation might be promising because sleep is a central “control hub” that influences many of the molecular and cellular pathways disrupted in neurological disorders. Sleep is a natural process in which the brain repairs, recalibrates, and clears debris. By targeting and improving specific aspects of sleep, some therapies might slow, stabilize, or even reverse the biological processes driving those diseases and represent a common therapeutic axis. Ongoing research into how sleep architecture is affected in each disorder might create unified, effective neurorestorative treatments that benefit many disorders and a shared opportunity for intervention across brain diseases (Table 1).

Table 1.
Summary of sleep disturbances observed in animal models of neurological diseases. Arrows indicate the direction of change in specific sleep parameters based on experimental findings. ↓ = Decreased; ↑ = Increased; ± = Altered; — = Not reported.
Author Contributions
C.R.: Original Draft Preparation, E.G.-S.: Figures and tables, Á.L.: Review and Editing, N.S.-G.: Review and Editing, A.P.-J.: Figures and tables, M.R.-O.: Original Draft Preparation, Conceptualization, Review and Editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. The APC was funded by the authors.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 3-NP | 3-nitropropionic acid |
| 3xTg-AD | Triple-transgenic mouse model of Alzheimer’s disease |
| 5xFAD | Transgenic mouse model used to study Alzheimer’s disease |
| 6-OHDA | 6-hydroxydopamine |
| AD | Alzheimer’s disease |
| Akt | Protein kinase B |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| AMPK | AMP-activated protein kinase |
| APP/PS1 | Amyloid precursor protein/Presenilin 1 transgenic mice |
| BDNF/TrkB | Brain-derived neurotrophic factor/Tropomyosin receptor kinase B |
| Bmal1 | Brain and muscle ARNT-like protein-1 |
| CAG | Cytosine–adenine–guanine repeat |
| CACNA1H | Calcium voltage-gated channel subunit alpha1 H |
| Clock | Circadian locomotor output cycles kaput gene |
| CREB | cAMP response element-binding protein |
| CSF | Cerebrospinal fluid |
| Cry | Cryptochrome circadian regulator gene |
| DJ-1 | Parkinsonism associated deglycase |
| DORA-12 | Dual orexin receptor antagonist-12 |
| ERK/MAPK | Extracellular signal-regulated kinase/Mitogen-activated protein kinase |
| GABA | Gamma-aminobutyric acid |
| GFAP | Glial fibrillary acidic protein |
| HD | Huntington’s disease |
| HTT | Huntingtin gene |
| IL-1β | Interleukin-1 beta |
| IL-6 | Interleukin-6 |
| JNK | c-Jun N-terminal kinase |
| KA | Kainic acid |
| KCNQ2 | Potassium voltage-gated channel subfamily Q member 2 |
| Kindling | Experimental epilepsy model through repeated stimulation |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAPK | Mitogen-activated protein kinase |
| mTOR | Mechanistic target of rapamycin |
| MPP+ | 1-methyl-4-phenylpyridinium ion (active metabolite of MPTP) |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| PARK2 | Parkin RBR E3 ubiquitin protein ligase |
| Per | Period circadian protein |
| PGC-1a | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | Phosphoinositide 3-kinase |
| PINK1 | PTEN-induced kinase 1 |
| PS19 | Transgenic mouse model that overexpresses mutant human tau |
| PSD-95 | Postsynaptic density protein 95 |
| PTZ | Pentylenetetrazole |
| R6/2 | Transgenic mouse line model of Huntington’s disease |
| REM | Rapid eye movement |
| ROS | Reactive oxygen species |
| SCN1A | Sodium voltage-gated channel alpha subunit 1 |
| SCN | Suprachiasmatic nucleus |
| SIRT1 | NAD-dependent deacetylase sirtuin-1 |
| SNCA | Synuclein alpha gene (α-synuclein) |
| STZ-ICV | Intracerebroventricular administration of streptozotocin |
| SWS | Slow-wave sleep |
| Tg2576 | Transgenic mouse overexpressing Swedish APP mutation (Alzheimer’s model) |
| TNF-α | Tumor necrosis factor alpha |
| VLPO | Ventrolateral preoptic nucleus |
| YAC128 | Yeast artificial chromosome mouse model of Huntington’s disease |
References
- Saper, C.B.; Scammell, T.E.; Lu, J. Hypothalamic regulation of sleep and circadian rhythms. Nature 2005, 437, 1257–1263. [Google Scholar] [CrossRef]
- Shi, J.X.; Wang, Z.Y.; Wang, S.W.; Shen, Q.; Tan, X. Exercise-mediated muscle-hypothalamus crosstalk: Improvement for cognitive dysfunction caused by disrupted circadian rhythm. Life Sci. 2025, 373, 123657. [Google Scholar] [CrossRef] [PubMed]
- Sherin, J.E.; Shiromani, P.J.; McCarley, R.W.; Saper, C.B. Activation of ventrolateral preoptic neurons during sleep. Science 1996, 271, 216–219. [Google Scholar] [CrossRef]
- Su, Y.J.; Yi, P.L.; Chang, F.C. Transcranial Direct Current Stimulation (tDCS) Ameliorates Stress-Induced Sleep Disruption via Activating Infralimbic-Ventrolateral Preoptic Projections. Brain Sci. 2024, 14, 105. [Google Scholar] [CrossRef]
- Brown, R.E.; Basheer, R.; McKenna, J.T.; Strecker, R.E.; McCarley, R.W. Control of sleep and wakefulness. Physiol. Rev. 2012, 92, 1087–1187. [Google Scholar] [CrossRef]
- Osorio-Forero, A.; Cardis, R.; Vantomme, G.; Guillaume-Gentil, A.; Katsioudi, G.; Devenoges, C.; Fernandez, L.M.J.; Lüthi, A. Noradrenergic circuit control of non-REM sleep substates. Curr. Biol. 2021, 31, 5009–5023.e7. [Google Scholar] [CrossRef]
- Sun, Y.; Tisdale, R.K.; Kilduff, T.S. Hypocretin/Orexin Receptor Pharmacology and Sleep Phases. Front. Neurol. Neurosci. 2021, 45, 22–37. [Google Scholar]
- Thannickal, T.C.; Moore, R.Y.; Nienhuis, R.; Ramanathan, L.; Gulyani, S.; Aldrich, M.; Cornford, M.; Siegel, J.M. Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000, 27, 469–474. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, M. Hypothalamic MCH neuron activity dynamics during cataplexy of narcolepsy. eNeuro 2020, 7, ENEURO.0017-20.2020. [Google Scholar] [CrossRef] [PubMed]
- Arnulf, I. Sleep and wake disturbances in Parkinson’s disease. J. Neural Transm. 2005, 112, 453–460. [Google Scholar]
- Ozawa, M.; Murakami, H.; Muraoka, Y.; Ibukuro, M.; Shiraishi, T.; Onda, A.; Matsuno, H.; Bono, K.; Umehara, T.; Omoto, S.; et al. Putamen dopaminergic dysfunction is associated with sleep disturbance in drug-naïve patients with Parkinson’s disease. Sleep Med. 2025, 129, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Nobili, L.; de Gennaro, L.; Proserpio, P.; Moroni, F.; Sarasso, S.; Pigorini, A. Local aspects of sleep: Observations from intracerebral recordings in humans. Prog. Brain Res. 2012, 199, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Assenza, G.; Lanzone, J.; Insola, A.; Amatori, G.; Ricci, L.; Tombini, M.; Di Lazzaro, V. Thalamo-cortical network dysfunction in temporal lobe epilepsy. Clin. Neurophysiol. 2020, 131, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Pati, S.; Agashe, S.; Kheder, A.; Riley, K.; Gavvala, J.; McGovern, R.; Suresh, S.; Chaitanya, G.; Thompson, S. Stereoelectroencephalography of the Deep Brain: Basal Ganglia and Thalami. J. Clin. Neurophysiol. 2024, 41, 423–429. [Google Scholar] [CrossRef]
- Morton, A.J.; Wood, N.I.; Hastings, M.H.; Hurelbrink, C.; Barker, R.A.; Maywood, E.S. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 2005, 25, 157–163. [Google Scholar] [CrossRef]
- Diago, E.B.; Martínez-Horta, S.; Lasaosa, S.S.; Alebesque, A.V.; Pérez-Pérez, J.; Kulisevsky, J.; Del Val, J.L. Circadian rhythm, cognition, and mood disorders in Huntington’s disease. J. Huntingt. Dis. 2018, 7, 193–198. [Google Scholar] [CrossRef]
- van Wamelen, D.J.; Aziz, N.A. Hypothalamic pathology in Huntington disease. Handb. Clin. Neurol. 2021, 182, 245–255. [Google Scholar]
- Ju, Y.E.S.; Lucey, B.P.; Holtzman, D.M. Sleep and Alzheimer disease pathology—A bidirectional relationship. Nat. Rev. Neurol. 2014, 10, 115–119. [Google Scholar] [CrossRef]
- Slutsky, I. Linking activity dyshomeostasis and sleep disturbances in Alzheimer disease. Nat. Rev. Neurosci. 2024, 25, 272–284. [Google Scholar] [CrossRef]
- Lima, M.M.; Andersen, M.L.; Reksidler, A.B.; Vital, M.A.; Tufik, S. The role of the substantia nigra pars compacta in regulating sleep patterns in rats. PLoS ONE 2007, 2, e513. [Google Scholar] [CrossRef]
- Cavelli, M.; Prunell, G.; Costa, G.; Velásquez, N.; Gonzalez, J.; Castro-Zaballa, S.; Lima, M.M.S.; Torterolo, P. Electrocortical high frequency activity and respiratory entrainment in 6-hydroxydopamine model of Parkinson’s disease. Brain Res. 2019, 1724, 146439. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, D.C.; Lopes Aguiar, C.; Moraes, M.F.D.; Fisone, G. Sleep Disorders in Rodent Models of Parkinson’s Disease. Front. Pharmacol. 2019, 10, 1414. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Bohlen, J.K.; Moore, C.; Nipper, M.A.; Finn, D.A.; Jones, C.E.; Lim, M.M.; Meshul, C.K. Effects of sleep disruption on stress, nigrostriatal markers, and behavior in a chronic/progressive MPTP male mouse model of parkinsonism. J. Neurosci. Res. 2019, 97, 1706–1719. [Google Scholar] [CrossRef]
- Gros, P.; Videnovic, A. Overview of Sleep and Circadian Rhythm Disorders in Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofer, K.F. Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J. 2012, 31, 3038–3062. [Google Scholar] [CrossRef]
- Schapira, A.H.; Patel, S. Targeting mitochondria for neuroprotection in Parkinson disease. JAMA Neurol. 2014, 71, 537–538. [Google Scholar] [CrossRef]
- Toulorge, D.; Schapira, A.H.; Hajj, R. Molecular changes in the postmortem parkinsonian brain. J. Neurochem. 2016, 139, 27–58. [Google Scholar] [CrossRef]
- Mauri, S.; Favaro, M.; Bernardo, G.; Mazzotta, G.M.; Ziviani, E. Mitochondrial autophagy in the sleeping brain. Front. Cell Dev. Biol. 2022, 10, 956394. [Google Scholar] [CrossRef]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by MPP+, a metabolite of the neurotoxin MPTP. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef]
- Gerlach, M.; Riederer, P.; Przuntek, H.; Youdim, M.B. MPTP mechanisms of neurotoxicity and their implications for Parkinson’s disease. Eur. J. Pharmacol. 1991, 208, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Naini, A.B.; Jakowec, M.; Petzinger, G.; Miller, R.; Akram, M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): A technical review of its utility and safety. J. Neurochem. 2001, 76, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 2016, 354, 1004–1008. [Google Scholar] [CrossRef]
- Wang, X.; Wang, M.; Zhi, H.; Li, J.; Guo, D. REV-ERBα inhibitor rescues MPTP/MPP+-induced ferroptosis of dopaminergic neuron through regulating FASN/SCD1 signaling pathway. Heliyon 2024, 10, e40388. [Google Scholar] [CrossRef]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.; Benabid, A.L.; Sadoul, R.; Verna, J.M. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Koprich, J.B.; Reske-Nielsen, C.; Mithal, P.; Isacson, O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflamm. 2008, 5, 8. [Google Scholar] [CrossRef]
- Hernandez-Baltazar, D.; Mendoza-Garrido, M.E.; Martinez-Fong, D. Activation of GSK-3β and caspase-3 occurs in Nigral dopamine neurons during the development of apoptosis activated by a striatal injection of 6-hydroxydopamine. PLoS ONE 2013, 8, e70951. [Google Scholar] [CrossRef]
- Masini, D.; Lopes-Aguiar, C.; Bonito-Oliva, A.; Papadia, D.; Andersson, R.; Fisahn, A.; Fisone, G. The histamine H3 receptor antagonist thioperamide rescues circadian rhythm and memory function in experimental parkinsonism. Transl. Psychiatry 2017, 7, e1088. [Google Scholar] [CrossRef]
- Yang, S.; Wan, Y.; Wu, N.; Song, L.; Liu, Z.; Zhao, J.; Liu, Y.; Liu, Z.; Gan, J. L-3,4-Dihydroxyphenylalanine Recovers Circadian Rhythm Disturbances in the Rat Models of Parkinson’s Disease by Regulating the D1R-ERK1/2-mTOR Pathway. Front. Aging Neurosci. 2021, 13, 719885. [Google Scholar] [CrossRef]
- Iranzo, A.; Tolosa, E.; Gelpi, E.; Molinuevo, J.L.; Valldeoriola, F.; Serradell, M.; Sanchez-Valle, R.; Vilaseca, I.; Lomeña, F.; Vilas, D.; et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: An observational cohort study. Lancet Neurol. 2013, 12, 443–453. [Google Scholar] [CrossRef]
- Raheel, K.; See, Q.R.; Munday, V.; Fakhroo, B.; Ivanenko, O.; Salvatelli, M.L.; Mutti, C.; Goadsby, P.J.; Delogu, A.; Naismith, S.L.; et al. Orexin and Sleep Disturbances in Alpha-Synucleinopathies: A Systematic Review. Curr. Neurol. Neurosci. Rep. 2024, 24, 389–412. [Google Scholar] [CrossRef] [PubMed]
- Canonichesi, J.; Bellingacci, L.; Rivelli, F.; Tozzi, A. Enhancing sleep quality in synucleinopathies through physical exercise. Front. Cell. Neurosci. 2025, 19, 1515922. [Google Scholar] [CrossRef] [PubMed]
- Pounders, J.D.; McCarter, S.J. Sleep and Prodromal Synucleinopathies. Semin. Neurol. 2025, 45, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, S.; Oku, M.; Tsuda, M.; Hoseki, J.; Sakai, Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014, 4, 5896. [Google Scholar] [CrossRef]
- Pukaß, K.; Goldbaum, O.; Richter-Landsberg, C. Mitochondrial impairment and oxidative stress compromise autophagosomal degradation of α-synuclein in oligodendroglial cells. J. Neurochem. 2015, 135, 194–205. [Google Scholar] [CrossRef]
- Lewis, F.W.; Fairooz, S.; Elson, J.L.; Hubscher-Bruder, V.; Brandel, J.; Soundararajan, M.; Smith, D.; Dexter, D.T.; Tétard, D.; Pienaar, I.S. Novel 1-hydroxypyridin-2-one metal chelators prevent and rescue ubiquitin proteasomal-related neuronal injury in an in vitro model of Parkinson’s disease. Arch. Toxicol. 2020, 94, 813–831. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Labandeira, C.M.; Guerra, M.J.; Rodriguez-Perez, A.I. The role of the brain renin-angiotensin system in Parkinson’s disease. Transl. Neurodegener. 2024, 13, 22. [Google Scholar] [CrossRef]
- Maggi, G.; Vitale, C.; Cerciello, F.; Santangelo, G. Sleep and wakefulness disturbances in Parkinson’s disease: A meta-analysis on prevalence and clinical aspects of REM sleep behavior disorder, excessive daytime sleepiness and insomnia. Sleep Med. Rev. 2023, 68, 101759. [Google Scholar] [CrossRef]
- Summa, K.C.; Jiang, P.; González-Rodríguez, P.; Huang, X.; Lin, X.; Vitaterna, M.H.; Dan, Y.; Surmeier, D.J.; Turek, F.W. Disrupted sleep-wake regulation in the MCI-Park mouse model of Parkinson’s disease. NPJ Park. Dis. 2024, 10, 54. [Google Scholar] [CrossRef]
- Käufer, C.; Stanojlović, M.; Schidlitzki, A.; Bonsberger, J.; Storch, A.; Richter, F. Alterations in non-REM sleep and EEG spectra precede REM-sleep deficits in a model of synucleinopathy. J. Park. Dis. 2025, 15, 311–328. [Google Scholar] [CrossRef]
- Lima, M.M.; Reksidler, A.B.; Vital, M.A. The neurobiology of the substantia nigra pars compacta: From motor to sleep regulation. J. Neural Transm. Suppl. 2009, 73, 135–145. [Google Scholar]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.R.; Olmstead, R.; Carroll, J.E. Sleep Disturbance, Sleep Duration, and Inflammation: A Systematic review and meta-analysis of cohort studies and experimental sleep deprivation. Biol. Psychiatry 2016, 80, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Ciric, J.; Kapor, S.; Perovic, M.; Saponjic, J. Alterations of sleep and sleep oscillations in the hemiparkinsonian rat. Front. Neurosci. 2019, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Niazi, N.U.K.; Huang, C.; Yang, Z.; Zhang, Y.; Song, C. Comparison between sub-chronic and chronic sleep deprivation-induced behavioral and neuroimmunological abnormalities in mice: Focusing on glial cell phenotype polarization. Behav. Brain Res. 2024, 470, 115067. [Google Scholar] [CrossRef]
- Scanga, A.; Lafontaine, A.L.; Kaminska, M. An overview of the effects of levodopa and dopaminergic agonists on sleep disorders in Parkinson’s disease. J. Clin. Sleep Med. 2023, 19, 1133–1144. [Google Scholar] [CrossRef]
- Hadi, F.; Agah, E.; Tavanbakhsh, S.; Mirsepassi, Z.; Mousavi, S.V.; Talachi, N.; Tafakhori, A.; Aghamollaii, V. Safety and efficacy of melatonin, clonazepam, and trazodone in patients with Parkinson’s disease and sleep disorders: A randomized, double-blind trial. Neurol. Sci. 2022, 43, 6141–6148. [Google Scholar] [CrossRef]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef]
- Cherian, A.; Divya, K.P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 2020, 120, 1297–1305. [Google Scholar] [CrossRef]
- Thangaleela, S.; Sivamaruthi, B.S.; Kesika, P.; Mariappan, S.; Rashmi, S.; Choeisoongnern, T.; Sittiprapaporn, P.; Chaiyasut, C. Neurological insights into sleep disorders in Parkinson’s disease. Brain Sci. 2023, 13, 1202. [Google Scholar] [CrossRef] [PubMed]
- Blagov, A.; Postnov, A.; Sukhorukov, V.; Popov, M.; Uzokov, J.; Orekhov, A. Significance of mitochondrial dysfunction in the pathogenesis of Parkinson’s disease. Front. Biosci. 2024, 29, 36. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Park. Relat. Disord. 2012, 18 (Suppl. S1), S210–S212. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Duan, Z.; Tan, J.; Liu, J.; Wang, Q.; Wang, C.; Zhang, Z.; Sun, X.; Liu, R.; Cui, Y. PHGDH-mediated serine synthesis in astrocytes supports neuroinflammation by sustaining NADH level to promote histone acetylation. Cell Death Dis. 2025, 16, 397. [Google Scholar] [CrossRef]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Chen, C.Y.; Weng, Y.H.; Chien, K.Y.; Lin, K.J.; Yeh, T.H.; Cheng, Y.P.; Lu, C.S.; Wang, H.L. (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 2012, 19, 1623–1633. [Google Scholar] [CrossRef]
- Guo, L.; Li, Y.; Li, W.; Qiu, J.; Du, J.; Wang, L.; Zhang, T. Shikonin ameliorates oxidative stress and neuroinflammation via the Akt/ERK/JNK/NF-κB signalling pathways in a model of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 2022, 49, 1221–1231. [Google Scholar] [CrossRef]
- Lim, H.S.; Lee, S.H.; Seo, H.; Park, G. Changes in RBM47 expression based on the timing of melatonin administration and its effects on Nrf2 activity in the hippocampus. Free Radic. Biol. Med. 2023, 208, 794–806. [Google Scholar] [CrossRef]
- Videnovic, A.; Breen, D.P.; Barker, R.A.; Zee, P.C. The central clock in patients with Parkinson disease-reply. JAMA Neurol. 2014, 71, 1456–1457. [Google Scholar] [CrossRef]
- Leng, Y.; Blackwell, T.; Cawthon, P.M.; Ancoli-Israel, S.; Stone, K.L.; Yaffe, K. Association of Circadian Abnormalities in Older Adults With an Increased Risk of Developing Parkinson Disease. JAMA Neurol. 2020, 77, 1270–1278. [Google Scholar] [CrossRef]
- Qiu, F.; Gu, P.; Liu, W.; Li, D. The spectrum characteristics of Parkinson’s disease (PD) patients with sleep disorders. Neurol. Sci. 2022, 43, 327–333. [Google Scholar] [CrossRef]
- Duan, W.X.; Xie, W.Y.; Ying, C.; Fen, W.; Cheng, X.Y.; Mao, C.J.; Liu, J.Y.; Liu, C.F. Butyrate improves abnormal sleep architecture in a Parkinson’s disease mouse model via BDNF/TrkB signaling. NPJ Park. Dis. 2025, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Limbic seizure and brain damage produced by kainic acid: Mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985, 14, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Lado, F.A. Chronic bilateral stimulation of the anterior thalamus of kainate-treated rats increases seizure frequency. Epilepsia 2006, 47, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.S.; Putra, M.; Meyer, C.; Parameswaran, S.; Thippeswamy, T. The effects of neuronal gyn knockdown in the hippocampus in the rat kainate Model of Temporal Lobe Epilepsy. Cells 2025, 14, 743. [Google Scholar] [CrossRef]
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free. Radic. Biol. Med. 2004, 37, 1951–1962. [Google Scholar] [CrossRef]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction in neurological disorders with epileptic phenotypes. J. Bioenerg. Biomembr. 2010, 42, 443–448. [Google Scholar] [CrossRef]
- Waldbaum, S.; Patel, M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010, 88, 23–45. [Google Scholar] [CrossRef]
- Su, Y.; Cao, N.; Zhang, D.; Wang, M. The effect of ferroptosis-related mitochondrial dysfunction in the development of temporal lobe epilepsy. Ageing Res. Rev. 2024, 96, 102248. [Google Scholar] [CrossRef]
- Mohd Sairazi, N.S.; Sirajudeen, K.N.; Asari, M.A.; Muzaimi, M.; Mummedy, S.; Sulaiman, S.A. Kainic Acid-Induced Excitotoxicity Experimental Model: Protective Merits of Natural Products and Plant Extracts. Evid.-Based Complement. Altern. Med. 2015, 2015, 972623. [Google Scholar] [CrossRef] [PubMed]
- Torolira, D.; Suchomelova, L.; Wasterlain, C.G.; Niquet, J. Widespread neuronal injury in a model of cholinergic status epilepticus in postnatal day 7 rat pups. Epilepsy Res. 2016, 120, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bertoglio, D.; Amhaoul, H.; Van Eetveldt, A.; Houbrechts, R.; Van De Vijver, S.; Ali, I.; Dedeurwaerdere, S. Kainic Acid-Induced Post-Status Epilepticus Models of Temporal Lobe Epilepsy with Diverging Seizure Phenotype and Neuropathology. Front. Neurol. 2017, 8, 588. [Google Scholar] [CrossRef] [PubMed]
- Peter-Derex, L.; Klimes, P.; Latreille, V.; Bouhadoun, S.; Dubeau, F.; Frauscher, B. Sleep disruption in epilepsy: Ictal and interictal epileptic activity matter. Ann. Neurol. 2020, 88, 907–920. [Google Scholar] [CrossRef]
- Meldrum, B.S. Identification and preclinical testing of novel antiepileptic compounds. Epilepsia 1997, 38, S7–S15. [Google Scholar] [CrossRef]
- Maugeri, A.; Citraro, R.; Leo, A.; Russo, C.; Navarra, M.; De Sarro, G. GABAA Receptors Are Involved in the Seizure Blockage Prompted by a Polyphenol-Rich Extract of White Grape Juice in Rodents. Pharmaceuticals 2025, 18, 186. [Google Scholar] [CrossRef]
- Kumar, M.; Kumar, P. Protective effect of spermine against pentylenetetrazole kindling epilepsy induced comorbidities in mice. Neurosci. Res. 2017, 120, 8–17. [Google Scholar] [CrossRef]
- Li, D.; Bai, X.; Jiang, Y.; Cheng, Y. Butyrate alleviates PTZ-induced mitochondrial dysfunction, oxidative stress and neuron apoptosis in mice via Keap1/Nrf2/HO-1 pathway. Brain Res. Bull. 2021, 168, 25–35. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef]
- Nazıroğlu, M.; Övey, İ.S. Involvement of apoptosis and calcium accumulation through TRPV1 channels in neurobiology of epilepsy. Neuroscience 2015, 293, 55–66. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Li, B.; Ayasoufi, K.; Simon, W.L.; Zhao, S.; Xie, M.; Thyen, G.; Hur, B.; Zheng, J.; Liang, Y.; et al. Microglial P2Y6 calcium signaling promotes phagocytosis and shapes neuroimmune responses in epileptogenesis. Neuron 2024, 112, 1959–1977. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.Y.; Zhu, M.J.; Song, Y.; Liu, X.M.; Tang, J.Y. Pentylenetetrazol-induced seizures are exacerbated by sleep deprivation through orexin receptor-mediated hippocampal cell proliferation. Neurol. Sci. 2014, 35, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.M.; Ali, D.A.; Kolieb, E.; Abdelaziz, E.Z. Ceftriaxone and selenium mitigate seizures and neuronal injury in pentylenetetrazole-kindled rats: Oxidative stress and inflammatory pathway. Int. Immunopharmacol. 2023, 120, 110304. [Google Scholar] [CrossRef] [PubMed]
- Akinduko, A.A.; Salawu, S.O.; Akinmoladun, A.C.; Akindahunsi, A.A.; Osemwegie, O.O. Assessment of the anxiolytic, antidepressant, and antioxidant potential of Parquetina nigrescens (Afzel.) Bullock in Wistar rats. J. Ethnopharmacol. 2024, 322, 117597. [Google Scholar] [CrossRef]
- Fountain, N.B.; Quigg, M.; Murchison, C.F.; Carrazana, E.; Rabinowicz, A.L. Analysis of seizure-cluster circadian periodicity from a long-term, open-label safety study of diazepam nasal spray. Epilepsia 2024, 65, 920–928. [Google Scholar] [CrossRef]
- Tomatsu, S.; Abbott, S.M.; Attarian, H. Clinical Chronobiology: Circadian Rhythms in Health and Disease. Semin. Neurol. 2025, 45, 317–332. [Google Scholar] [CrossRef]
- Carvalho, B.M.S.; Chaves, J.; da Silva, A.M. Effects of antiepileptic drugs on sleep architecture parameters in adults. Sleep Sci. 2022, 15, 224–244. [Google Scholar] [CrossRef]
- Goddard, G.V.; McIntyre, D.C.; Leech, C.K. A permanent change in brain function resulting from daily electrical stimulation. Exp. Neurol. 1969, 25, 295–330. [Google Scholar] [CrossRef]
- Raol, Y.H.; Meti, B.L. Sleep-wakefulness alterations in amygdala-kindled rats. Epilepsia 1998, 39, 1133–1137. [Google Scholar] [CrossRef]
- Mohamed, J.; Scott, B.W.; David, O.; McIntyre Burnham, W. Development of propagated discharge and behavioral arrest in hippocampal and amygdala-kindled animals. Epilepsy Res. 2018, 148, 78–89. [Google Scholar] [CrossRef]
- Ryu, B.; Nagappan, S.; Santos-Valencia, F.; Lee, P.; Rodriguez, E.; Lackie, M.; Takatoh, J.; Franks, K.M. Chronic loss of inhibition in piriform cortex following brief, daily optogenetic stimulation. Cell Rep. 2021, 35, 109001. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Naquet, R. Kindling effect and sleep organization in cats. Electroencephalogr. Clin. Neurophysiol. 1975, 39, 449–454. [Google Scholar] [CrossRef]
- Rubio, C.; Paz, C. Indomethacin reverts sleep disorders produced by ozone exposure in rats. Toxicology 2003, 191, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.L.; Tsai, C.H.; Lin, J.G.; Lee, C.C.; Chang, F.C. Kindling stimuli delivered at different times in the sleep-wake cycle. Sleep 2004, 27, 203–212. [Google Scholar] [CrossRef]
- Colangeli, R.; Morena, M.; Werner, A.; Thompson, R.J.; van der Stelt, M.; Pittman, Q.J.; Hill, M.N.; Teskey, G.C. 2-AG-Mediated Control of GABAergic Signaling Is Impaired in a Model of Epilepsy. J. Neurosci. 2023, 43, 571–583. [Google Scholar] [CrossRef]
- Sashindranath, M.; McLean, K.J.; Trounce, I.A.; Cotton, R.G.; Cook, M.J. Early hippocampal oxidative stress is a direct consequence of seizures in the rapid electrical amygdala kindling model. Epilepsy Res. 2010, 90, 285–294. [Google Scholar] [CrossRef]
- Rowley, S.; Patel, M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free. Radic. Biol. Med. 2013, 62, 121–131. [Google Scholar] [CrossRef]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. BioMed Res. Int. 2015, 2015, 745613. [Google Scholar] [CrossRef]
- Mohammed, H.S.; Ahmed, D.H.; Khadrawy, Y.A.; Madian, N.G. Neuroprotection in pentylenetetrazol kindling rat model: A synergistic approach with eugenol and photobiomodulation. Brain Res. 2025, 1858, 149645. [Google Scholar] [CrossRef]
- Uchino, E.; Sonoda, S.; Kinukawa, N.; Sakamoto, T. Alteration pattern of tear cytokines during the course of a day: Diurnal rhythm analyzed by multicytokine assay. Cytokine 2006, 33, 36–40. [Google Scholar] [CrossRef]
- Cutolo, M. Circadian rhythms and rheumatoid arthritis. Jt. Bone Spine 2019, 86, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Niu, R.; Guo, X.; Wang, J.; Yang, X. The hidden rhythms of epilepsy: Exploring biological clocks and epileptic seizure dynamics. Acta Epileptol. 2025, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Krook-Magnuson, E.; Armstrong, C.; Oijala, M.; Soltesz, I. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat. Commun. 2013, 4, 1376. [Google Scholar] [CrossRef] [PubMed]
- Christenson Wick, Z.; Krook-Magnuson, E. Specificity, versatility, and continual development: The power of optogenetics for epilepsy research. Front. Cell. Neurosci. 2018, 12, 151. [Google Scholar] [CrossRef]
- Wykes, R.C.; Kullmann, D.M.; Pavlov, I.; Magloire, V. Optogenetic approaches to treat epilepsy. J. Neurosci. Methods 2016, 260, 215–220. [Google Scholar] [CrossRef]
- Adamantidis, A.R.; Zhang, F.; Aravanis, A.M.; Deisseroth, K.; de Lecea, L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 2007, 450, 420–424. [Google Scholar] [CrossRef]
- Blanco-Centurion, C.; Liu, M.; Konadhode, R.P.; Zhang, X.; Pelluru, D.; van den Pol, A.N.; Shiromani, P.J. Optogenetic activation of melanin-concentrating hormone neurons increases non-rapid eye movement and rapid eye movement sleep during the night in rats. Eur. J. Neurosci. 2016, 44, 2846–2857. [Google Scholar] [CrossRef]
- Mondino, A.; Jadidian, A.; Toth, B.A.; Hambrecht-Wiedbusch, V.S.; Floran-Garduno, L.; Li, D.; York, A.K.; Torterolo, P.; Pal, D.; Burgess, C.R.; et al. Regulation of REM and NREM sleep by preoptic glutamatergic neurons. Sleep 2025, in press. [Google Scholar] [CrossRef]
- DuBois, D.W.; Murchison, D.A.; Mahnke, A.H.; Bang, E.; Winzer-Serhan, U.; Griffith, W.H.; Souza, K.A. Maintenance of optogenetic channel rhodopsin (ChR2) function in aging mice: Implications for pharmacological studies of inhibitory synaptic transmission, quantal content, and calcium homeostasis. Neuropharmacology 2023, 238, 109651. [Google Scholar] [CrossRef]
- Ahuja, A.S.; Rozen, T.D.; Atwal, P.S. A sleep modulated Channelopathy: A novel CACNA1A pathogenic variant identified in episodic Ataxia type 2 and a potential link to sleep alleviated migraine. BMC Neurol. 2019, 19, 246. [Google Scholar] [CrossRef]
- Paulhus, K.; Ammerman, L.; Glasscock, E. Clinical spectrum of KCNA1 mutations: New insights into episodic ataxia and epilepsy comorbidity. Int. J. Mol. Sci. 2020, 21, 2802. [Google Scholar] [CrossRef] [PubMed]
- Czekus, C.; Steullet, P.; Orero López, A.; Bozic, I.; Rusterholz, T.; Bandarabadi, M.; Do, K.Q.; Gutierrez Herrera, C. Alterations in TRN-anterodorsal thalamocortical circuits affect sleep architecture and homeostatic processes in oxidative stress vulnerable Gclm-/- mice. Mol. Psychiatry 2022, 27, 4394–4406. [Google Scholar] [CrossRef] [PubMed]
- Rusina, E.; Simonti, M.; Duprat, F.; Cestèle, S.; Mantegazza, M. Voltage-gated sodium channels in genetic epilepsy: Up and down of excitability. J. Neurochem. 2024, 168, 3872–3890. [Google Scholar] [CrossRef]
- Steinlein, O.K.; Bertrand, D. Nicotinic receptor channelopathies and epilepsy. Eur. J. Physiol. 2010, 460, 495–503. [Google Scholar] [CrossRef]
- Noebels, J.L. Exploring new gene discoveries in idiopathic generalized epilepsy. Epilepsia 2003, 44, 16–21. [Google Scholar] [CrossRef]
- Kullmann, D.M. Neurological channelopathies. Annu. Rev. Neurosci. 2010, 33, 151–172. [Google Scholar] [CrossRef]
- Hedrich, U.B.; Liautard, C.; Kirschenbaum, D.; Pofahl, M.; Lavigne, J.; Liu, Y.; Theiss, S.; Slotta, J.; Escayg, A.; Dihné, M.; et al. Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Na(V)1.1 mutation. J. Neurosci. 2014, 34, 14874–14889. [Google Scholar] [CrossRef]
- Riaz, M.; Abbasi, M.H.; Sheikh, N.; Saleem, T.; Virk, A.O. GABRA1 and GABRA6 gene mutations in idiopathic generalized epilepsy patients. Seizure 2021, 93, 88–94. [Google Scholar] [CrossRef]
- Kantor, S.; Szabo, L.; Varga, J.; Cuesta, M.; Morton, A.J. Progressive sleep and electroencephalogram changes in mice carrying the Huntington’s disease mutation. Brain 2013, 136, 2147–2158. [Google Scholar] [CrossRef]
- Fitzgerald, E.S.; Manousakis, J.E.; Glikmann-Johnston, Y.; Rankin, M.; Anderson, C.; Stout, J.C.; Jackson, M.L. Sleep fragmentation despite intact rest-activity patterns in premanifest Huntington’s disease: An actigraphy study. Sleep Med. 2024, 124, 16–29. [Google Scholar] [CrossRef]
- Kudo, T.; Schroeder, A.; Loh, D.H.; Kuljis, D.; Jordan, M.C.; Roos, K.P.; Colwell, C.S. Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Exp. Neurol. 2011, 228, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.; Gabery, S.; Soylu-Kucharz, R.; Cheong, R.Y.; Henningsen, J.B.; Englund, E.; McLean, C.; Kirik, D.; Halliday, G.; Petersén, Å. SIRT1 is increased in affected brain regions and hypothalamic metabolic pathways are altered in Huntington disease. Neuropathol. Appl. Neurobiol. 2019, 45, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Ouk, K.; Aungier, J.; Ware, M.; Morton, A.J. Abnormal photic entrainment to phase-delaying stimuli in the R6/2 mouse model of Huntington’s disease, despite retinal responsiveness to light. eNeuro 2019, 6, ENEURO.0088-19.2019. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.P.; Black, S.W.; Schwartz, M.D.; Wilk, A.J.; Chen, T.M.; Lincoln, W.U.; Liu, H.W.; Kilduff, T.S.; Morairty, S.R. Longitudinal analysis of the electroencephalogram and sleep phenotype in the R6/2 mouse model of Huntington’s disease. Brain 2013, 136, 2159–2172. [Google Scholar] [CrossRef]
- Morton, A.J. Circadian and sleep disorder in Huntington’s disease. Exp. Neurol. 2013, 243, 34–44. [Google Scholar] [CrossRef]
- Saade-Lemus, S.; Videnovic, A. Sleep Disorders and Circadian Disruption in Huntington’s Disease. J. Huntingt. Dis. 2023, 12, 121–131. [Google Scholar] [CrossRef]
- Morton, A.J. Sleep and circadian rhythm dysfunction in animal models of Huntington’s disease. J. Huntingt. Dis. 2023, 12, 133–148. [Google Scholar] [CrossRef]
- Mills, J. The Use of Trazodone for Insomnia and Other Sleep Disturbances. Issues Ment. Health Nurs. 2024, 45, 658–662. [Google Scholar] [CrossRef]
- Zisapel, N. New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation. Br. J. Pharmacol. 2018, 175, 3190–3199. [Google Scholar] [CrossRef]
- Romero-Miguel, D.; Lamanna-Rama, N.; Casquero-Veiga, M.; Gómez-Rangel, V.; Desco, M.; Soto-Montenegro, M.L. Minocycline in neurodegenerative and psychiatric diseases: An update. Eur. J. Neurol. 2021, 28, 1056–1081. [Google Scholar] [CrossRef]
- Yang, S.H.; Cheng, P.H.; Banta, H.; Piotrowska-Nitsche, K.; Yang, J.J.; Cheng, E.C.; Snyder, B.; Larkin, K.; Liu, J.; Orkin, J.; et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 2008, 453, 921–924. [Google Scholar] [CrossRef]
- Jacobsen, J.C.; Bawden, C.S.; Rudiger, S.R.; McLaughlan, C.J.; Reid, S.J.; Waldvogel, H.J.; MacDonald, M.E.; Gusella, J.F.; Walker, S.K.; Kelly, J.M.; et al. An ovine transgenic Huntington’s disease model. Hum. Mol. Genet. 2010, 19, 1873–1882. [Google Scholar] [CrossRef]
- Han, B.; Liang, W.; Li, X.J.; Li, S.; Yan, S.; Tu, Z. Large animal models for Huntington’s disease research. Zool. Res. 2024, 45, 275–283. [Google Scholar] [CrossRef]
- Handley, R.R.; Reid, S.J.; Burch, Z.; Jacobsen, J.C.; Gillis, T.; Correia, K.; Rudiger, S.R.; McLaughlin, C.J.; Bawden, C.S.; MacDonald, M.E.; et al. Somatic CAG Repeat Stability in a Transgenic Sheep Model of Huntington’s Disease. J. Huntingt. Dis. 2024, 13, 33–40. [Google Scholar] [CrossRef]
- Wüllner, U.; Young, A.B.; Penney, J.B.; Beal, M.F. 3-Nitropropionic acid toxicity in the striatum. J. Neurochem. 1994, 63, 1772–1781. [Google Scholar] [CrossRef]
- Kudo, T.; Loh, D.H.; Tahara, Y.; Truong, D.; Hernández-Echeagaray, E.; Colwell, C.S. Circadian dysfunction in response to in vivo treatment with the mitochondrial toxin 3-nitropropionic acid. ASN Neuro 2014, 6, e00133. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E. Mitochondria and Huntington’s disease pathogenesis: Insight from genetic and chemical models. Ann. N. Y. Acad. Sci. 2008, 1147, 358–382. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Chen, C.H.; Chen, Y.C.; Wu, M.T.; Lin, T.Y.; Hua, K.F.; Ju, T.C. Protective Effects of Antcin H Isolated from Antrodia cinnamomea Against Neuroinflammation in Huntington’s Disease via NLRP3 Inflammasome Inhibition. J. Neuroimmune Pharmacol. 2024, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Block, F.; Kunkel, M.; Schwarz, M. Quinolinic acid lesion of the striatum induces impairment in spatial learning and motor performance in rats. Neurosci. Lett. 1993, 149, 126–128. [Google Scholar] [CrossRef]
- Stoy, N.; Mackay, G.M.; Forrest, C.M.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J. Neurochem. 2005, 93, 611–623. [Google Scholar] [CrossRef]
- Tassan Mazzocco, M.; Murtaj, V.; Martins, D.; Schellino, R.; Coliva, A.; Toninelli, E.; Vercelli, A.; Turkheimer, F.; Belloli, S.; Moresco, R.M. Exploring the neuroprotective effects of montelukast on brain inflammation and metabolism in a rat model of quinolinic acid-induced striatal neurotoxicity. J. Neuroinflamm. 2023, 20, 34. [Google Scholar] [CrossRef] [PubMed]
- Rentschler, K.M.; Milosavljevic, S.; Baratta, A.M.; Wright, C.J.; Piroli, M.V.; Tentor, Z.; Valafar, H.; O’Reilly, C.; Pocivavsek, A. Reducing brain kynurenic acid synthesis precludes kynurenine-induced sleep disturbances. J. Sleep Res. 2024, 33, e14038. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Lin, C.H.; Tallaksen-Greene, S.; Chien, W.M.; Cearley, J.A.; Jackson, W.S.; Crouse, A.B.; Ren, S.; Li, X.J.; Albin, R.L.; Detloff, P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 137–144. [Google Scholar] [CrossRef]
- Portillo-Ledesma, S.; Hang, M.; Schlick, T. Regulation of Genome Architecture in Huntington’s Disease. Biochemistry 2025, 64, 2100–2115. [Google Scholar] [CrossRef]
- Browne, S.E.; Beal, M.F. Oxidative damage and mitochondrial dysfunction in neurodegenerative diseases. Biochem. Soc. Trans. 1994, 22, 1002–1006. [Google Scholar] [CrossRef]
- Beal, M.F. Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef]
- Polyzos, A.A.; McMurray, C.T. The chicken or the egg: Mitochondrial dysfunction as a cause or consequence of toxicity in Huntington’s disease. Mech. Ageing Dev. 2017, 161, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Chan, N.G.; Góngora-Alfaro, J.L.; Álvarez-Cervera, F.J.; Solís-Rodríguez, F.A.; Heredia-López, F.J.; Arankowsky-Sandoval, G. Quinolinic acid lesions of the pedunculopontine nucleus impair sleep architecture, but not locomotion, exploration, emotionality or working memory in the rat. Behav. Brain Res. 2011, 225, 482–490. [Google Scholar] [CrossRef]
- Kantor, S.; Varga, J.; Morton, A.J. A single dose of hypnotic corrects sleep and EEG abnormalities in symptomatic Huntington’s disease mice. Neuropharmacology 2016, 105, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.K.; Akkermans, J.; Lawson, M.; Syta, P.; Staelens, S.; Adhikari, M.H.; Morton, A.J.; Nitzsche, B.; Boltze, J.; Christou, C.; et al. Imaging glucose metabolism and dopaminergic dysfunction in sheep (Ovis aries) brain using positron emission tomography imaging reveals abnormalities in OVT73 Huntington’s disease sheep. ACS Chem. Neurosci. 2024, 15, 4082–4091. [Google Scholar] [CrossRef] [PubMed]
- Fonken, L.K.; Frank, M.G.; Kitt, M.M.; Barrientos, R.M.; Watkins, L.R.; Maier, S.F. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav. Immun. 2015, 45, 171–179. [Google Scholar] [CrossRef]
- Griffin, P.; Dimitry, J.M.; Sheehan, P.W.; Lananna, B.V.; Guo, C.; Robinette, M.L.; Hayes, M.E.; Cedeño, M.R.; Nadarajah, C.J.; Ezerskiy, L.A.; et al. Circadian clock protein Reverbα regulates neuroinflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 5102–5107. [Google Scholar] [CrossRef]
- Picard, K.; Corsi, G.; Decoeur, F.; Di Castro, M.A.; Bordeleau, M.; Persillet, M.; Layé, S.; Limatola, C.; Tremblay, M.È.; Nadjar, A. Microglial homeostasis disruption modulates non-rapid eye movement sleep duration and neuronal activity in adult female mice. Brain Behav. Immun. 2023, 107, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Kalsbeek, A.; Yi, C.X. Microglia, circadian rhythm and lifestyle factors. Neuropharmacology 2024, 257, 110029. [Google Scholar] [CrossRef]
- Gršković, P.; Korać, P. Circadian Gene Variants in Diseases. Genes 2023, 14, 1703. [Google Scholar] [CrossRef]
- Huang, Z.L.; Urade, Y.; Hayaishi, O. The role of adenosine in the regulation of sleep. Curr. Top. Med. Chem. 2011, 11, 1047–1057. [Google Scholar] [CrossRef]
- Petersen, N.; McCann, K.E.; Stavarache, M.A.; Kim, L.Y.; Weinshenker, D.; Winder, D.G. Adenosine A2A Receptors Link Astrocytic α1-Adrenergic Signaling to Wake-Promoting Dopamine Neurons. Biol. Psychiatry 2025, 97, 915–928. [Google Scholar] [CrossRef]
- Nithianantharajah, J.; Hannan, A.J. Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience-dependent mediators of cognitive and psychiatric symptoms in Huntington’s disease. Neuroscience 2013, 251, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Vezzoli, E.; Caron, I.; Talpo, F.; Besusso, D.; Conforti, P.; Battaglia, E.; Sogne, E.; Falqui, A.; Petricca, L.; Verani, M.; et al. Inhibiting pathologically active ADAM10 rescues synaptic and cognitive decline in Huntington’s disease. J. Clin. Investig. 2019, 129, 2390–2403. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Kim, G.W. Decreased SREBP2 of the striatal cell relates to disrupted protein degradation in Huntington’s disease. Brain Res. 2025, 1846, 149250. [Google Scholar] [CrossRef]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92. [Google Scholar] [CrossRef]
- Vitiello, M.V.; Prinz, P.N.; Williams, D.E.; Frommlet, M.S.; Ries, R.K. Sleep disturbances in patients with mild-stage Alzheimer’s disease. J. Gerontol. 1990, 45, M131–M138. [Google Scholar] [CrossRef]
- Lucey, B.P.; Bateman, R.J. Amyloid-β diurnal pattern: Possible role of sleep in Alzheimer’s disease pathogenesis. Neurobiol. Aging 2014, 35, S29–S34. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Awasthi, S.; Hindle, A.; Sawant, N.A.; George, M.; Vijayan, M.; Kshirsagar, S.; Morton, H.; Bunquin, L.E.; Palade, P.T.; Lawrence, J.J.; et al. RALBP1 in oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Cells 2021, 10, 3113. [Google Scholar] [CrossRef]
- Bilkei-Gorzo, A. Genetic mouse models of brain ageing and Alzheimer’s disease. Pharmacol. Ther. 2014, 142, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.; Bezprozvanny, I.; Wu, L.; Ryskamp, D.A.; Bar-Ner, N.; Natan, N.; Brandeis, R.; Elkon, H.; Nahum, V.; Gershonov, E.; et al. AF710B, a Novel M1/σ1 Agonist with Therapeutic Efficacy in Animal Models of Alzheimer’s Disease. Neuro-Degener. Dis. 2016, 16, 95–110. [Google Scholar] [CrossRef]
- Li, Y.Y.; Yu, K.Y.; Cui, Y.J.; Wang, Z.J.; Cai, H.Y.; Cao, J.M.; Wu, M.N. Orexin-A aggravates cognitive deficits in 3xTg-AD mice by exacerbating synaptic plasticity impairment and affecting amyloid β metabolism. Neurobiol. Aging 2023, 124, 71–84. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Wadhwa, M.; Kumari, P.; Chauhan, G.; Roy, K.; Alam, S.; Kishore, K.; Ray, K.; Panjwani, U. Sleep deprivation induces spatial memory impairment by altered hippocampus neuroinflammatory responses and glial cells activation in rats. J. Neuroimmunol. 2017, 312, 38–48. [Google Scholar] [CrossRef]
- Ji, S.; Dong, Y.; Wang, Z.; Zhu, R.; Jiang, Y.; Li, S.; Ma, X. Edaravone attenuates sleep deprivation-induced memory deficits by inhibiting oxidative stress and neuroinflammation in murine models. Biomedicines 2025, 13, 1047. [Google Scholar] [CrossRef]
- Satterfield, B.C.; Wisor, J.P.; Field, S.A.; Schmidt, M.A.; Van Dongen, H.P. TNFα G308A polymorphism is associated with resilience to sleep deprivation-induced psychomotor vigilance performance impairment in healthy young adults. Brain Behav. Immun. 2015, 47, 66–74. [Google Scholar] [CrossRef]
- Kang, J.; Park, M.; Kim, T. Vitamin D Reduces GABA-Positive Astrocytes in the 5xFAD Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2024, 97, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.J.; Guerriero, L.E.; Kohler, K.; Beechem, L.E.; Gillis, B.D.; Salisbury, F.; Wessel, C.; Wang, J.; Sunderam, S.; Bachstetter, A.D.; et al. Chronic fragmentation of the daily sleep-wake rhythm increases amyloid-beta levels and neuroinflammation in the 3xTg-AD mouse model of Alzheimer’s disease. Neuroscience 2022, 481, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef]
- Li, H.; Feng, F.; Xie, S.; Ma, Y.; Wang, Y.; Zhang, F.; Wu, H.; Huang, S. Identification of HIBCH and MGME1 as Mitochondrial Dynamics-Related Biomarkers in Alzheimer’s Disease Via Integrated Bioinformatics Analysis. IET Syst. Biol. 2025, 19, e70018. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef]
- Moriya, S.; Takahashi, H.; Masukawa, D.; Yamada, M.; Ishigooka, J.; Nishimura, K. Dual orexin receptor antagonist (DORA-12) treatment affects the overall levels of Net/maoA mRNA expression in the hippocampus. J. Pharmacol. Sci. 2021, 145, 198–201. [Google Scholar] [CrossRef]
- Ribeiro, R.F.N.; Santos, M.R.; Aquino, M.; De Almeida, L.P.; Cavadas, C.; Silva, M.M.C. The Therapeutic Potential of Melatonin and Its Novel Synthetic Analogs in Circadian Rhythm Sleep Disorders, Inflammation-Associated Pathologies, and Neurodegenerative Diseases. Med. Res. Rev. 2025, 45, 1515–1539. [Google Scholar] [CrossRef]
- Louzada, L.L.; Machado, F.V.; Quintas, J.L.; Ribeiro, G.A.; Silva, M.V.; Mendonça-Silva, D.L.; Gonçalves, B.S.B.; Nóbrega, O.T.; Camargos, E.F. The efficacy and safety of zolpidem and zopiclone to treat insomnia in Alzheimer’s disease: A randomized, triple-blind, placebo-controlled trial. Neuropsychopharmacology 2022, 47, 570–579. [Google Scholar] [CrossRef]
- Duncan, M.J.; Farlow, H.; Tirumalaraju, C.; Yun, D.; Wang, C.; Howard, J.A.; Sanden, M.N.; O’Hara, B.F.; McQuerry, K.J.; Bachstetter, A.D. Effects of the dual orexin receptor antagonist DORA-22 on sleep in 5XFAD mice. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 70–80. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimer’s Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. Insulin receptor signaling in rat hippocampus: A study in STZ (ICV) induced memory deficit model. Eur. Neuropsychopharmacol. 2011, 21, 261–273. [Google Scholar] [CrossRef]
- Musiek, E.S.; Xiong, D.D.; Holtzman, D.M. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp. Mol. Med. 2015, 47, e148. [Google Scholar] [CrossRef] [PubMed]
- Cavallucci, V.; D’Amelio, M.; Cecconi, F. Aβ toxicity in Alzheimer’s disease. Mol. Neurobiol. 2012, 45, 366–378. [Google Scholar] [CrossRef]
- Rigat, L.; Ouk, K.; Kramer, A.; Priller, J. Dysfunction of circadian and sleep rhythms in the early stages of Alzheimer’s disease. Acta Physiol. 2023, 238, e13970. [Google Scholar] [CrossRef]
- Kaldun, J.C.; Lone, S.R.; Humbert Camps, A.M.; Fritsch, C.; Widmer, Y.F.; Stein, J.V.; Tomchik, S.M.; Sprecher, S.G. Dopamine, sleep, and neuronal excitability modulate amyloid-β-mediated forgetting in Drosophila. PLoS Biol. 2021, 19, e3001412. [Google Scholar] [CrossRef]
- Choudhary, P.; Bansal, S.; Meena, P.; Verma, R.; Komal; Verma, R. Exploring the utility of zebrafish models for understanding neuropsychiatric disorders and advancement of drug discovery. J. Integr. Sci. Technol. 2025, 13, 1051. [Google Scholar] [CrossRef]
- Ghanizada, H.; Nedergaard, M. The glymphatic system. Handb. Clin. Neurol. 2025, 209, 161–170. [Google Scholar] [PubMed]
- Stevanovic, K.; Yunus, A.; Joly-Amado, A.; Gordon, M.; Morgan, D.; Gulick, D.; Gamsby, J. Disruption of normal circadian clock function in a mouse model of tauopathy. Exp. Neurol. 2017, 294, 58–67. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid-β peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, M.; de Vivo, L.; Chini, M.; Gilli, F.; Tononi, G.; Cirelli, C. Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. J. Neurosci. 2017, 37, 5263–5273. [Google Scholar] [CrossRef]
- Tononi, G.; Boly, M.; Cirelli, C. Consciousness and sleep. Neuron 2024, 112, 1568–1594. [Google Scholar] [CrossRef]
- Mao, J.Q.; Cheng, L.; Zhang, Y.D.; Xie, G.J.; Wang, P. Chinese formula Guben-Jiannao Ye alleviates the dysfunction of circadian and sleep rhythms in APP/PS1 mice implicated in activation of the PI3K/AKT/mTOR signaling pathway. J. Ethnopharmacol. 2024, 335, 118696. [Google Scholar] [CrossRef]
- Rábago-Monzón, Á.R.; Osuna-Ramos, J.F.; Armienta-Rojas, D.A.; Camberos-Barraza, J.; Camacho-Zamora, A.; Magaña-Gómez, J.A.; De la Herrán-Arita, A.K. Stress-Induced Sleep Dysregulation: The Roles of Astrocytes and Microglia in Neurodegenerative and Psychiatric Disorders. Biomedicines 2025, 13, 1121. [Google Scholar] [CrossRef]
- Bazil, C.W. Sleep and Epilepsy. Semin. Neurol. 2017, 37, 407–412. [Google Scholar] [CrossRef]
- González-Rueda, A.; Pedrosa, V.; Feord, R.C.; Clopath, C.; Paulsen, O. Activity-dependent downscaling of subthreshold synaptic inputs during slow-wave-sleep-like activity in vivo. Neuron 2018, 97, 1244–1252.e5. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).