

Clinical and Genetic Features of Autosomal Recessive Bestrophinopathy: A Case Series from a Vietnamese Cohort

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Individuals
2.2. Fundus Examination
2.3. Variant Screening of the BEST1 Gene
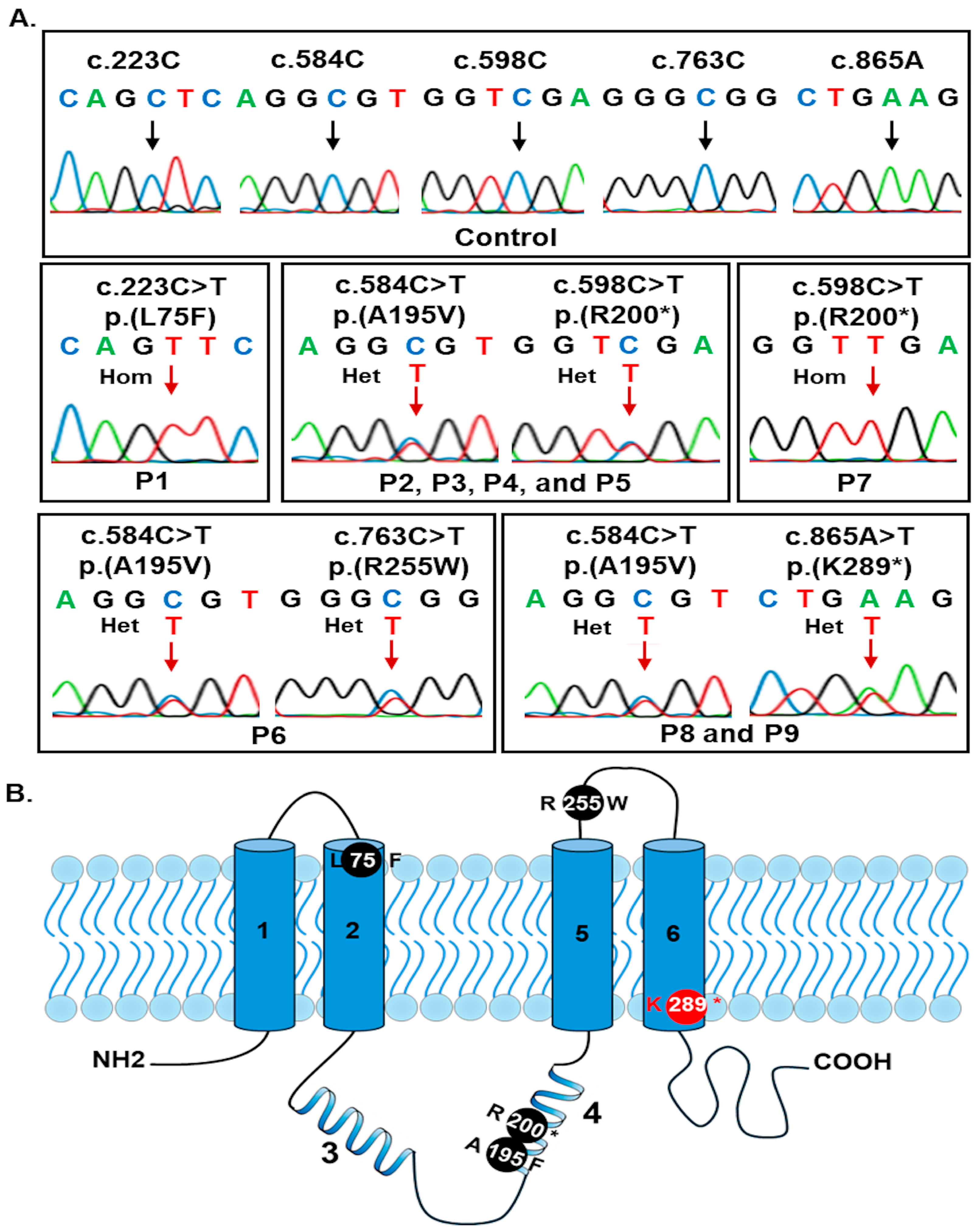
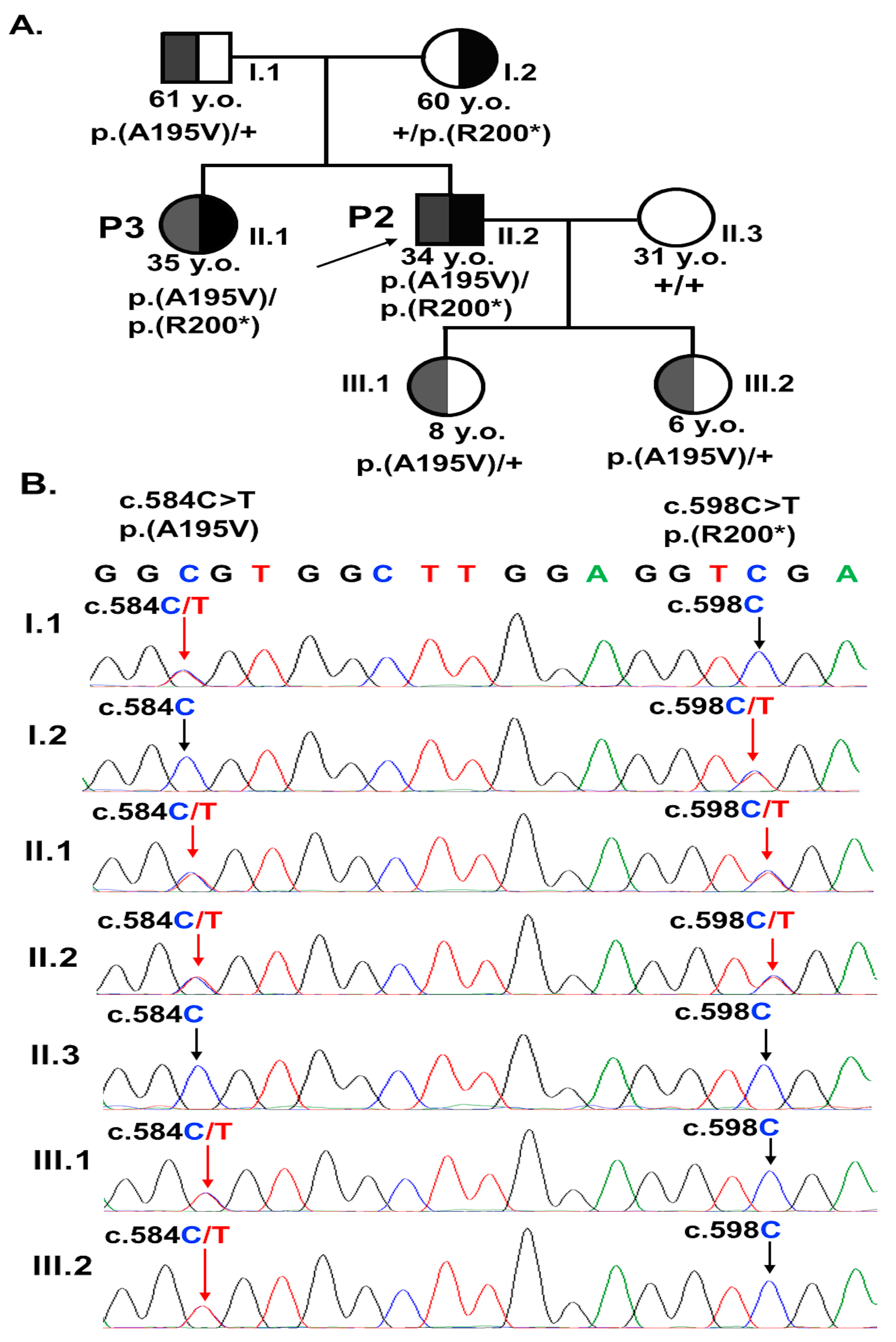
3. Results
3.1. Clinical Findings
3.2. Analyses of BEST1 Variants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ARB | Autosomal recessive bestrophinopathy |
| CaCC | Calcium-activated chloride channel |
| BVMD | Best vitelliform macular dystrophy |
| EOG | Electrooculography |
| FAF | Fundus autofluorescence |
| FFA | Fundus fluorescein angiography |
| OCT | Optical coherence tomography |
| PCR | Polymerase chain reaction |
| gnomAD | Genome Aggregation Database |
| CADD | Combined annotation dependent depletion |
| SIFT | Sorting intolerant from tolerant |
| PolyPhen_2 | Polymorphism phenotyping v2 |
| ACMG | American College of Medical Genetics and Genomics |
| BCVA | Best corrected visual acuity |
| OD | Oculus dexter (right eye) |
| OS | Oculus sinister (left eye) |
| RPE | Retinal pigment epithelium |
References
- Singh Grewal, S.; Smith, J.J.; Carr, A.-J.F. Bestrophinopathies: Perspectives on Clinical Disease, Bestrophin-1 Function and Developing Therapies. Ther. Adv. Ophthalmol. 2021, 13, 2515841421997191. [Google Scholar] [CrossRef] [PubMed]
- Doumanov, J.A.; Zeitz, C.; Dominguez Gimenez, P.; Audo, I.; Krishna, A.; Alfano, G.; Diaz, M.L.B.; Moskova-Doumanova, V.; Lancelot, M.-E.; Sahel, J.-A.; et al. Disease-Causing Mutations in BEST1 Gene Are Associated with Altered Sorting of Bestrophin-1 Protein. Int. J. Mol. Sci. 2013, 14, 15121–15140. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Guziewicz, K.E.; Lee, C.J.; Kalathur, R.C.; Pulido, J.S.; Marmorstein, L.Y.; Marmorstein, A.D. Bestrophin 1 and Retinal Disease. Prog. Retin. Eye Res. 2017, 58, 45–69. [Google Scholar] [CrossRef]
- Zhang, Y.; Kittredge, A.; Ward, N.; Ji, C.; Chen, S.; Yang, T. ATP Activates Bestrophin Ion Channels through Direct Interaction. Nat. Commun. 2018, 9, 3126. [Google Scholar] [CrossRef]
- Owji, A.P.; Wang, J.; Kittredge, A.; Clark, Z.; Zhang, Y.; Hendrickson, W.A.; Yang, T. Structures and Gating Mechanisms of Human Bestrophin Anion Channels. Nat. Commun. 2022, 13, 3836. [Google Scholar] [CrossRef]
- Yang, T.; Liu, Q.; Kloss, B.; Bruni, R.; Kalathur, R.; Kloppmann, E.; Rost, B.; Colecraft, H.M.; Hendrickson, W.A. Structure and Selectivity in Bestrophin Ion Channels. Science 2014, 346, 355–359. [Google Scholar] [CrossRef]
- Dickson, V.K.; Pedi, L.; Long, S.B. Structure and Insights into the Function of a Ca2+-Activated Cl− Channel. Nature 2014, 516, 213–218. [Google Scholar] [CrossRef]
- Boon, C.J.F.; Theelen, T.; Hoefsloot, E.H.; van Schooneveld, M.J.; Keunen, J.E.E.; Cremers, F.P.M.; Klevering, B.J.; Hoyng, C.B. Clinical and Molecular Genetic Analysis of Best Vitelliform Macular Dystrophy. Retina 2009, 29, 835–847. [Google Scholar] [CrossRef]
- Owji, A.P.; Kittredge, A.; Zhang, Y.; Yang, T. Structure and Function of the Bestrophin Family of Calcium-Activated Chloride Channels. Channels 2021, 15, 604–623. [Google Scholar] [CrossRef]
- Ji, C.; Li, Y.; Kittredge, A.; Hopiavuori, A.; Ward, N.; Yao, P.; Fukuda, Y.; Zhang, Y.; Tsang, S.H.; Yang, T. Investigation and Restoration of BEST1 Activity in Patient-Derived RPEs with Dominant Mutations. Sci. Rep. 2019, 9, 19026. [Google Scholar] [CrossRef]
- Marquardt, A.; Stöhr, H.; Passmore, L.A.; Krämer, F.; Rivera, A.; Weber, B.H. Mutations in a Novel Gene, VMD2, Encoding a Protein of Unknown Properties Cause Juvenile-Onset Vitelliform Macular Dystrophy (Best’s Disease). Hum. Mol. Genet. 1998, 7, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Seddon, J.M.; Bernstein, P.S.; Hutchinson, A.; Atkinson, A.; Sharma, S.; Gerrard, B.; Li, W.; Metzker, M.L.; Wadelius, C.; et al. Evaluation of the Best Disease Gene in Patients with Age-Related Macular Degeneration and Other Maculopathies. Hum. Genet. 1999, 104, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.J.; Goldberg, M.F.; Orth, D.H.; Fishman, G.A.; Tessler, H.; Mizuno, K. Autosomal Dominant Vitreoretinochoroidopathy. Arch. Ophthalmol. 1982, 100, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.; Millar, I.D.; Leroy, B.P.; Urquhart, J.E.; Fearon, I.M.; De Baere, E.; Brown, P.D.; Robson, A.G.; Wright, G.A.; Kestelyn, P.; et al. Biallelic Mutation of BEST1 Causes a Distinct Retinopathy in Humans. Am. J. Hum. Genet. 2008, 82, 19–31. [Google Scholar] [CrossRef]
- Davidson, A.E.; Millar, I.D.; Urquhart, J.E.; Burgess-Mullan, R.; Shweikh, Y.; Parry, N.; O’Sullivan, J.; Maher, G.J.; McKibbin, M.; Downes, S.M.; et al. Missense Mutations in a Retinal Pigment Epithelium Protein, Bestrophin-1, Cause Retinitis Pigmentosa. Am. J. Hum. Genet. 2009, 85, 581–592. [Google Scholar] [CrossRef]
- Tawfik, C.A.; Roshdy, M.M.; Morris, N.M. Prevalence of Inherited Retinal Diseases in a Large Egyptian Cohort. BMC Ophthalmol. 2023, 23, 422. [Google Scholar] [CrossRef]
- Habibi, I.; Falfoul, Y.; Todorova, M.G.; Wyrsch, S.; Vaclavik, V.; Helfenstein, M.; Turki, A.; Matri, K.E.; Matri, L.E.; Schorderet, D.F. Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy (ARB). Genes 2019, 10, 953. [Google Scholar] [CrossRef]
- Hufendiek, K.; Hufendiek, K.; Jägle, H.; Stöhr, H.; Book, M.; Spital, G.; Rustambayova, G.; Framme, C.; Weber, B.H.F.; Renner, A.B.; et al. Clinical Heterogeneity in Autosomal Recessive Bestrophinopathy with Biallelic Mutations in the BEST1 Gene. Int. J. Mol. Sci. 2020, 21, 9353. [Google Scholar] [CrossRef]
- Bittmann, S.; Luchter, E.; Villalon, G.; Moschuring-Alieva, E.; Bittmann, L.; Weissenstein, A. First Pediatric Case of Autosomal Recessive Homozygotic Bestrophinopathy Due to Homozygous Mutation c.187G>C p. in Two Brothers. J. Clin. Med. Res. 2022, 14, 174–176. [Google Scholar] [CrossRef]
- Boon, C.J.F.; van den Born, L.I.; Visser, L.; Keunen, J.E.E.; Bergen, A.A.B.; Booij, J.C.; Riemslag, F.C.; Florijn, R.J.; van Schooneveld, M.J. Autosomal Recessive Bestrophinopathy: Differential Diagnosis and Treatment Options. Ophthalmology 2013, 120, 809–820. [Google Scholar] [CrossRef]
- Jaffal, L.; Joumaa, W.H.; Assi, A.; Helou, C.; Condroyer, C.; El Dor, M.; Cherfan, G.; Zeitz, C.; Audo, I.; Zibara, K.; et al. Novel Missense Mutations in BEST1 Are Associated with Bestrophinopathies in Lebanese Patients. Genes 2019, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Tekin, K.; Dulger, S.C.; Horozoglu Ceran, T.; Inanc, M.; Ozdal, P.C.; Teke, M.Y. Multimodal Imaging and Genetic Characteristics of Autosomal Recessive Bestrophinopathy. J. Fr. Ophtalmol. 2024, 47, 104097. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Han, J.; Kim, Y.J.; Kang, H.G.; Byeon, S.H.; Kim, S.S.; Lee, C.S. Clinical Features and Genetic Findings of Autosomal Recessive Bestrophinopathy. Genes 2022, 13, 1197. [Google Scholar] [CrossRef] [PubMed]
- Haque, O.I.; Chandrasekaran, A.; Nabi, F.; Ahmad, O.; Marques, J.P.; Ahmad, T. A Novel Compound Heterozygous BEST1 Gene Mutation in Two Siblings Causing Autosomal Recessive Bestrophinopathy. BMC Ophthalmol. 2022, 22, 493. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Poornachandra, B.; Verma, A.; Mehta, R.A.; Phalke, S.; Battu, R.; Ramprasad, V.L.; Peterson, A.S.; Ghosh, A.; Seshagiri, S. Next Generation Sequencing Identifies Novel Disease-Associated BEST1 Mutations in Bestrophinopathy Patients. Sci. Rep. 2018, 8, 10176. [Google Scholar] [CrossRef]
- Ye, P.; Xu, J.; Luo, Y.; Su, Z.; Yao, K. Familial Autosomal Recessive Bestrophinopathy: Identification of a Novel Variant in BEST1 Gene and the Specific Metabolomic Profile. BMC Med. Genet. 2020, 21, 16. [Google Scholar] [CrossRef]
- Tian, L.; Sun, T.; Xu, K.; Zhang, X.; Peng, X.; Li, Y. Screening of BEST1 Gene in a Chinese Cohort with Best Vitelliform Macular Dystrophy or Autosomal Recessive Bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3366–3375. [Google Scholar] [CrossRef]
- Casalino, G.; Khan, K.N.; Armengol, M.; Wright, G.; Pontikos, N.; Georgiou, M.; Webster, A.R.; Robson, A.G.; Grewal, P.S.; Michaelides, M. Autosomal Recessive Bestrophinopathy: Clinical Features, Natural History, and Genetic Findings in Preparation for Clinical Trials. Ophthalmology 2021, 128, 706–718. [Google Scholar] [CrossRef]
- Zhao, D.; Gu, V.Y.; Wang, Y.; Peng, J.; Lyu, J.; Fei, P.; Xu, Y.; Zhang, X.; Zhao, P. Clinical and Genetic Features in Autosomal Recessive Bestrophinopathy in Chinese Cohort. BMC Ophthalmol. 2024, 24, 308. [Google Scholar] [CrossRef]
- Gao, T.; Tian, C.; Hu, Q.; Liu, Z.; Zou, J.; Huang, L.; Zhao, M. Clinical and Mutation Analysis of Patients with Best Vitelliform Macular Dystrophy or Autosomal Recessive Bestrophinopathy in Chinese Population. Biomed. Res. Int. 2018, 2018, 4582816. [Google Scholar] [CrossRef]
- Shi, J.; Tian, L.; Sun, T.; Zhang, X.; Xu, K.; Xie, Y.; Peng, X.; Tang, X.; Jin, Z.-B.; Li, Y. Comprehensive Genetic Analysis Unraveled the Missing Heritability and a Founder Variant of BEST1 in a Chinese Cohort with Autosomal Recessive Bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2023, 64, 37. [Google Scholar] [CrossRef] [PubMed]
- Soto-Sierra, M.; Morillo-Sánchez, M.J.; Martín-Sánchez, M.; Ramos-Jiménez, M.; López-Domínguez, M.; Ponte-Zuñiga, B.; Antiñolo, G.; Rodríguez-de-la-Rúa, E. Novel BEST1 Mutations and Clinical Characteristics of Autosomal Recessive Bestrophinopathy in a Spanish Patient. Eur. J. Ophthalmol. 2022, 32, NP77–NP81. [Google Scholar] [CrossRef]
- Li, J.-X.; Meng, L.-R.; Hou, B.-K.; Hao, X.-L.; Wang, D.-J.; Qu, L.-H.; Li, Z.-H.; Zhang, L.; Jin, X. Detection of Novel BEST1 Variations in Autosomal Recessive Bestrophinopathy Using Third-Generation Sequencing. Curr. Med. Sci. 2024, 44, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; Alföldi, J.; Watts, N.A.; Vittal, C.; Gauthier, L.D.; et al. A Genomic Mutational Constraint Map Using Variation in 76,156 Human Genomes. Nature 2024, 625, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.; Zhang, H.; Wang, Q.; Villamarin, R.; Hefferon, T.; Ramanathan, A.; Kattman, B. The Evolution of dbSNP: 25 Years of Impact in Genomic Research. Nucleic Acids Res. 2025, 53, D925–D931. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Kaur, K.; Brown, G.; Chen, C.; Hart, J.; Hoffman, D.; Jang, W.; Liu, C.; Maddipatla, Z.; et al. ClinVar: Updates to Support Classifications of Both Germline and Somatic Variants. Nucleic Acids Res. 2025, 53, D1313–D1321. [Google Scholar] [CrossRef]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Schubach, M.; Maass, T.; Nazaretyan, L.; Röner, S.; Kircher, M. CADD v1.7: Using Protein Language Models, Regulatory CNNs and Other Nucleotide-Level Scores to Improve Genome-Wide Variant Predictions. Nucleic Acids Res. 2024, 52, D1143–D1154. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, V.M.; Röhrl, E.; Weber, B.H.F.; Strauss, O. Disease-Associated Missense Mutations in Bestrophin-1 Affect Cellular Trafficking and Anion Conductance. J. Cell Sci 2011, 124, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.L.M.; Hou, P.; Choy, K.-W.; Chiang, S.W.Y.; Tam, P.O.S.; Li, H.; Chan, W.-M.; Lam, D.S.C.; Pang, C.-P.; Lai, T.Y.Y. Novel and Homozygous BEST1 Mutations in Chinese Patients with Best Vitelliform Macular Dystrophy. Retina 2010, 30, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, S.; Preising, M.N.; Ciccolella, N.; Causio, F.; Lorenz, B.; Fischetto, R. Autosomal Recessive Bestrophinopathy: New Observations on the Retinal Phenotype—Clinical and Molecular Report of an Italian Family. Ophthalmologica 2011, 225, 228–235. [Google Scholar] [CrossRef]
- Luo, J.; Lin, M.; Guo, X.; Xiao, X.; Li, J.; Hu, H.; Xiao, H.; Xu, X.; Zhong, Y.; Long, S.; et al. Novel BEST1 Mutations and Special Clinical Characteristics of Autosomal Recessive Bestrophinopathy in Chinese Patients. Acta Ophthalmol. 2019, 97, 247–259. [Google Scholar] [CrossRef]
- Yang, S.; Li, Z.; Cheng, W.; Ma, M.; Qi, R.; Rui, X.; Ren, Y.; Sheng, X.; Rong, W. BEST1 Novel Mutation Causes Bestrophinopathies in Six Families with Distinct Phenotypic Diversity. Mol. Genet. Genomic. Med. 2023, 11, e2095. [Google Scholar] [CrossRef]
- Pfister, T.A.; Zein, W.M.; Cukras, C.A.; Sen, H.N.; Maldonado, R.S.; Huryn, L.A.; Hufnagel, R.B. Phenotypic and Genetic Spectrum of Autosomal Recessive Bestrophinopathy and Best Vitelliform Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 22. [Google Scholar] [CrossRef]
- Boon, C.J.F.; Klevering, B.J.; Leroy, B.P.; Hoyng, C.B.; Keunen, J.E.E.; den Hollander, A.I. The Spectrum of Ocular Phenotypes Caused by Mutations in the BEST1 Gene. Prog. Retin. Eye Res. 2009, 28, 187–205. [Google Scholar] [CrossRef]
- Kinnick, T.R.; Mullins, R.F.; Dev, S.; Leys, M.; Mackey, D.A.; Kay, C.N.; Lam, B.L.; Fishman, G.A.; Traboulsi, E.; Iezzi, R.; et al. Autosomal Recessive Vitelliform Macular Dystrophy in a Large Cohort of Vitelliform Macular Dystrophy Patients. Retina 2011, 31, 581–595. [Google Scholar] [CrossRef]
- MacDonald, I.M.; Gudiseva, H.V.; Villanueva, A.; Greve, M.; Caruso, R.; Ayyagari, R. Phenotype and Genotype of Patients with Autosomal Recessive Bestrophinopathy. Ophthalmic Genet. 2012, 33, 123–129. [Google Scholar] [CrossRef]
- Miller, A.N.; Vaisey, G.; Long, S.B. Molecular Mechanisms of Gating in the Calcium-Activated Chloride Channel Bestrophin. Elife 2019, 8, e43231. [Google Scholar] [CrossRef]
- Navinés-Ferrer, A.; Ruiz-Nogales, S.; Navarro, R.; Pomares, E. Impaired Bestrophin Channel Activity in an iPSC-RPE Model of Best Vitelliform Macular Dystrophy (BVMD) from an Early Onset Patient Carrying the P77S Dominant Mutation. Int. J. Mol. Sci. 2022, 23, 7432. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Yang, G.; Wang, J.; Chen, Y. Screening for BEST1 Gene Mutations in Chinese Patients with Bestrophinopathy. Mol. Vis. 2014, 20, 1594–1604. [Google Scholar] [PubMed]
- Lambertus, S.; Bax, N.M.; Groenewoud, J.M.M.; Cremers, F.P.M.; van der Wilt, G.J.; Klevering, B.J.; Theelen, T.; Hoyng, C.B. Asymmetric Inter-Eye Progression in Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6824–6830. [Google Scholar] [CrossRef] [PubMed]
- Charng, J.; Cideciyan, A.V.; Jacobson, S.G.; Sumaroka, A.; Schwartz, S.B.; Swider, M.; Roman, A.J.; Sheplock, R.; Anand, M.; Peden, M.C.; et al. Variegated yet Non-Random Rod and Cone Photoreceptor Disease Patterns in RPGR-ORF15-Associated Retinal Degeneration. Hum. Mol. Genet. 2019, 28, 175. [Google Scholar] [CrossRef]
- Chakravarthy, H.; Georgyev, V.; Wagen, C.; Hosseini, A.; Matsubara, J. Blue Light-Induced Phototoxicity in Retinal Cells: Implications in Age-Related Macular Degeneration. Front. Aging Neurosci. 2024, 16, 1509434. [Google Scholar] [CrossRef]
- Antemie, R.-G.; Samoilă, O.C.; Clichici, S.V. Blue Light-Ocular and Systemic Damaging Effects: A Narrative Review. Int. J. Mol. Sci. 2023, 24, 5998. [Google Scholar] [CrossRef]
- Yu, K.; Qu, Z.; Cui, Y.; Hartzell, H.C. Chloride Channel Activity of Bestrophin Mutants Associated with Mild or Late-Onset Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4694–4705. [Google Scholar] [CrossRef]
- Xiao, Q.; Prussia, A.; Yu, K.; Cui, Y.; Hartzell, H.C. Regulation of Bestrophin Cl Channels by Calcium: Role of the C Terminus. J. Gen. Physiol. 2008, 132, 681–692. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Family History | Ophthalmic History | Treatment History |
|---|---|---|---|---|
| P1 | Male | None | Chronic central serous chorioretinopathy with CNV and open-angle glaucoma. | Trabeculectomy procedure for glaucoma and glaucoma eye drops. Received bevacizumab intravitreal injections without improvement. |
| P2 | Male | Sister (P3) | Maculopathy and hyperopic astigmatism. | None. |
| P3 | Female | Brother (P2) | Choroidal neovascularization, polypoidal choroidal vasculopathy, and hyperopia. | Received bevacizumab intravitreal injections. |
| P4 | Male | None | Maculopathy and open-angle glaucoma. | Glaucoma eye drops. |
| P5 | Male | None | Hyperopic, maculopathy, and exudative retinal detachment. | Artificial tears. |
| P6 | Female | None | Chronic central serous chorioretinopathy, closed-angle glaucoma, and hyperopia. | Laser peripheral iridotomy and glaucoma eye drops. |
| P7 | Male | None | Bullous central serous chorioretinopathy and hyperopia. | |
| P8 | Female | Brother (P9) | Hyperopic astigmatism and macular edema. | Artificial tears. |
| P9 | Male | Sister (P8) | Hyperopic astigmatism and macular edema. |
| Exon | Forward (3′–5′) | Reverse (3′–5′) | Length (bp) |
|---|---|---|---|
| 2 | GAGAGTTGAGGTCCAGAGCA | GCAGCCTCTCAGTCTGACTT | 484 |
| 3 | GTTTGGGGCTGTACAAGGAG | AGTCCGCACCTTTCCCTAC | 435 |
| 4 | TCTGGCGGATTTCTGGGAC | CACCCATCTTCCATTCCTGC | 550 |
| 5 | GCCCAGAACAGCACCTAGTA | ACCAGGACCTCACAGACTTG | 521 |
| 6 | AGCCAGGAATGGACCATAGG | ATTGCCTCTACTGGACTGGG | 393 |
| 7 | GCAAGTCAGAACAAGGCCTT | GCTTTCTACCCGTGAGACCT | 379 |
| 8 | TACACTCAGGGACAGCTGTG | ACAGTGGGGTCCTCTCTTTG | 399 |
| 9 | ATCTCCCCATTTCACAGGCA | CTGCACTAGGAGGGGCTTC | 329 |
| 10-1 | TCAGGAGAGAGGTGAGAGCT | CTGATACAGTGGGGCAGACT | 434 |
| 10-2 | CCAAACTACTGTGGCCCAAG | TTTTCGGGGATCTCTGGCAT | 421 |
| 10-3 | AAGACTGTGAGTTCTGGGGC | TGGCAGTGATGGAACCCTAG | 344 |
| 11 | TCAACCTTTGCCCTCCTACT | TAAGGTGTGGCTGTCTTGGA | 402 |
| Patient | Age (Years) | BCVA (OD/OS) | Fundus Findings (OU) | FFA (OU) | FAF (OU) | OCT Findings (OU) | BEST1 Variants | |
|---|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | |||||||
| P1 | 79.6 | 20/40; 20/125 | Multiple yellow subretinal deposits in the macula and around the optic disc. Serous retinal detachment. RPE alteration. | Diffuse hyperfluorescence at the macula. Descending tract pattern of hyperautofluorescence. | Hyperautofluorescence corresponding to the yellow deposits. Mixed areas of hyper and hypoautofluorescence due to RPE alterations. | Retinal thickening and fluid. Disruption of photoreceptor elongation. Disruption in the epiretinal membrane. | c.223C>T p.(L75F) | c.223C>T p.(L75F) |
| P2 | 33.8 | 20/40; 20/50 | Yellow subretinal deposits in the macular and retinal area above the optic disc. | Diffuse hyperfluorescence at the macula. | Hyperautofluorescence corresponding to the yellow deposits. Hypoautofluorescence due to RPE alteration. | Subretinal fluid. Subretinal deposition of lipofuscin. | c.584C>T p.A195V | c.598C>T p.(R200*) |
| P3 | 34.8 | 20/100; 20/125 | Yellow subretinal deposits in the macular and retinal area above the optic disc. | Hyperfluores-cence in the macula at the late phase. | Hyperautofluorescence corresponding to the yellow deposits. Hypoautofluorescence due to RPE alteration. | Subretinal fluid. Subretinal deposition of lipofuscin. | c.584C>T p.A195V | c.598C>T p.(R200*) |
| P4 | 32.5 | 20/25; 20/40 | Diffuse RPE changes. Diffuse subretinal deposits at the macula and around the optic discs. | Hyperfluorescence due to RPE thinning and subretinal deposits. | Diffuse hyperautofluorescence at the macula and around the optic discs. | Subretinal fluid and diffuse deposits. Retinoschisis. RPE thinning. | c.584C>T p.A195V | c.598C>T p.(R200*) |
| P5 | 45.4 | 20/50; 20/50 | Diffuse subretinal deposits with some foci of deposits near the vascular arcade and above the optic disc. | Diffuse hyperfluorescence at the macula. | Hyperautofluorescence at the macular. Hyporautofluorescence in the centre in a ring pattern. | Subretinal fluid and deposits. Outer retinal thinning. Photoreceptor disruption and elongation. | c.584C>T p.A195V | c.598C>T p.(R200*) |
| P6 | 48.1 | 20/25; 20/32 | Yellow subretinal deposits at the macula. RPE alteration. | Diffuse hyperautofluorescence in the macular region. | Hyperautofluorescence at the macular. Hyporautofluorescence in the centre in a ring pattern. | Subretinal fluid and deposits. Outer retinal thinning. Photoreceptor disruption and elongation. | c.584C>T p.A195V | c.763C>T p.(R255W) |
| P7 | 42.2 | 20/100; 20/125 | Diffuse subretinal deposits. Foci of deposits around the optic discs. Peripheral and inferior retinal alteration. | Widespread hyperautofluorescence at the macular region. | Diffuse hyperautofluorescence. Descending streaks of hyperautofluorescence. | Subretinal fluid. Outer retinal thinning. | c.598C>T p.(R200*) | c.598C>T p.(R200*) |
| P8 | 16.5 | 20/25; 20/32 | Diffuse subretinal deposits. Foci of deposits around the optic discs and retinal vascular arcade. | Diffuse hyperluorescence. Gravitational hyperfluorescence streak. | Hyperautofluorescence at the macula and around the optic discs. Foci of hyperautofluorescence. | Subretinal fluid and deposits. Retinoschisis. Photoreceptor elongation and disruption. | c.598C>T p.(R200*) | c.865A>T p.(K289*) |
| P9 | 14.1 | 20/20; 20/20 | Diffuse subretinal deposits. Foci of deposits around the optic discs and retinal vascular arcade. | Diffuse hyperluorescence. Gravitational hyperfluorescence streak. | Hyperautofluorescence at macula and around the optic discs. Foci of hyperautofluorescence. | Subretinal fluid and deposits. Retinoschisis. Photoreceptor elongation and disruption. | c.598C>T p.(R200*) | c.865A>T p.(K289*) |
| Exon | c.DNA Change | Amino Acid Change | MutationTaster2021 | CADD v1.7 | PolyPhen_2 | SIFT | GnomAD v4.1.0 | dbSNP157 | ClinVar | Literature | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | c.223C>T | p.(L75F) | D | D | B | T | 2/1614224 | rs1335203485 | 3767340 LP | [43] | LP (PM1, PM2, PM5, PP3 and PP5) |
| 5 | c.584C>T | p.(A195V) | D | D | D | D | 254/1614092 | rs200277476 | 99725 P | [18,23] | P (PS3, PM1, PM2, PP1, and PP3) |
| 5 | c.598C>T | p.(R200*) | D | D | 10/1613960 | rs121918286 | 2741 P | [14,44] | P (PVS1, PM2, PP1, PP3, and PP5) | ||
| 7 | c.763C>T | p.(R255W) | D | D | D | D | 44/1614060 | rs372989281 | 143127 P | [27,45] | LP (PM1, PM2, PP3, and PP5) |
| 7 | c.865A>T | p.(K289*) | D | D | 0 | 0 | 0 | 0 | P (PVS1, PM2, PP1, and PP3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.T.; Tran, V.K.; Nguyen, N.L.; Huy, N.V.; Tran, T.H.; Phuong, L.T.; Nguyen, P.L.; Nguyen, T.T.; Trang, T.T.Q.; Huong, D.T.; et al. Clinical and Genetic Features of Autosomal Recessive Bestrophinopathy: A Case Series from a Vietnamese Cohort. Biomedicines 2025, 13, 1625. https://doi.org/10.3390/biomedicines13071625
Nguyen TTT, Tran VK, Nguyen NL, Huy NV, Tran TH, Phuong LT, Nguyen PL, Nguyen TT, Trang TTQ, Huong DT, et al. Clinical and Genetic Features of Autosomal Recessive Bestrophinopathy: A Case Series from a Vietnamese Cohort. Biomedicines. 2025; 13(7):1625. https://doi.org/10.3390/biomedicines13071625
Chicago/Turabian StyleNguyen, Trang Thi Thu, Van Khanh Tran, Ngoc Lan Nguyen, Nguyen Van Huy, Thinh Huy Tran, Le Thi Phuong, Phan Long Nguyen, Thuy Thu Nguyen, Tran Thi Quynh Trang, Do Thanh Huong, and et al. 2025. "Clinical and Genetic Features of Autosomal Recessive Bestrophinopathy: A Case Series from a Vietnamese Cohort" Biomedicines 13, no. 7: 1625. https://doi.org/10.3390/biomedicines13071625
APA StyleNguyen, T. T. T., Tran, V. K., Nguyen, N. L., Huy, N. V., Tran, T. H., Phuong, L. T., Nguyen, P. L., Nguyen, T. T., Trang, T. T. Q., Huong, D. T., Huong, N. T. T., Pham, T. V., & Mai, Q. T. (2025). Clinical and Genetic Features of Autosomal Recessive Bestrophinopathy: A Case Series from a Vietnamese Cohort. Biomedicines, 13(7), 1625. https://doi.org/10.3390/biomedicines13071625