
A Comprehensive Review of Somatic and Germline Biomarkers Associated with Childhood B-Cell Precursor Acute Lymphoblastic Leukemia: From Biological Significance to Precision Medicine Opportunities
 , ,
, ,
Abstract
1. Introduction
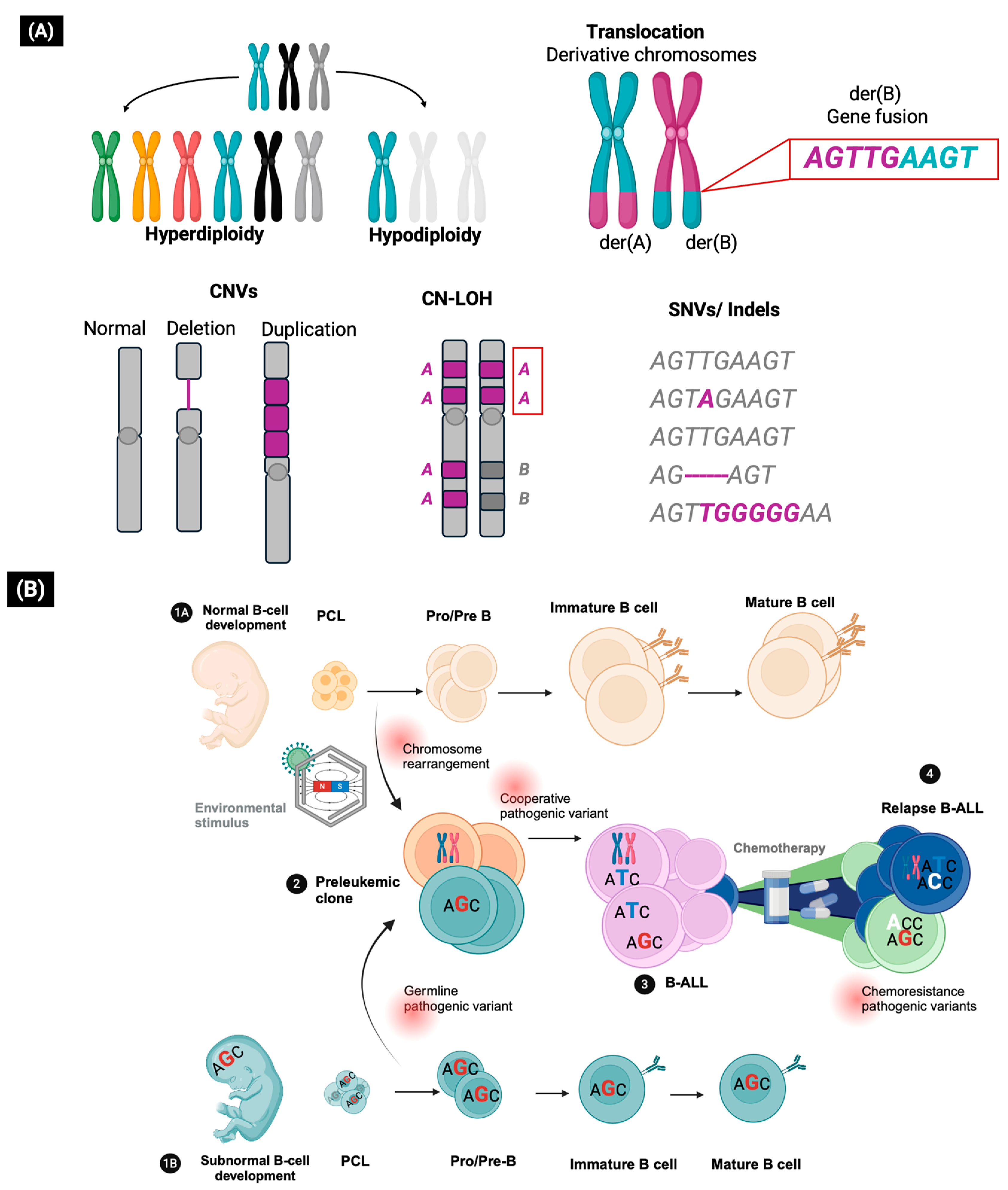
2. The Role of Genetic Variants in the Development of B-ALL
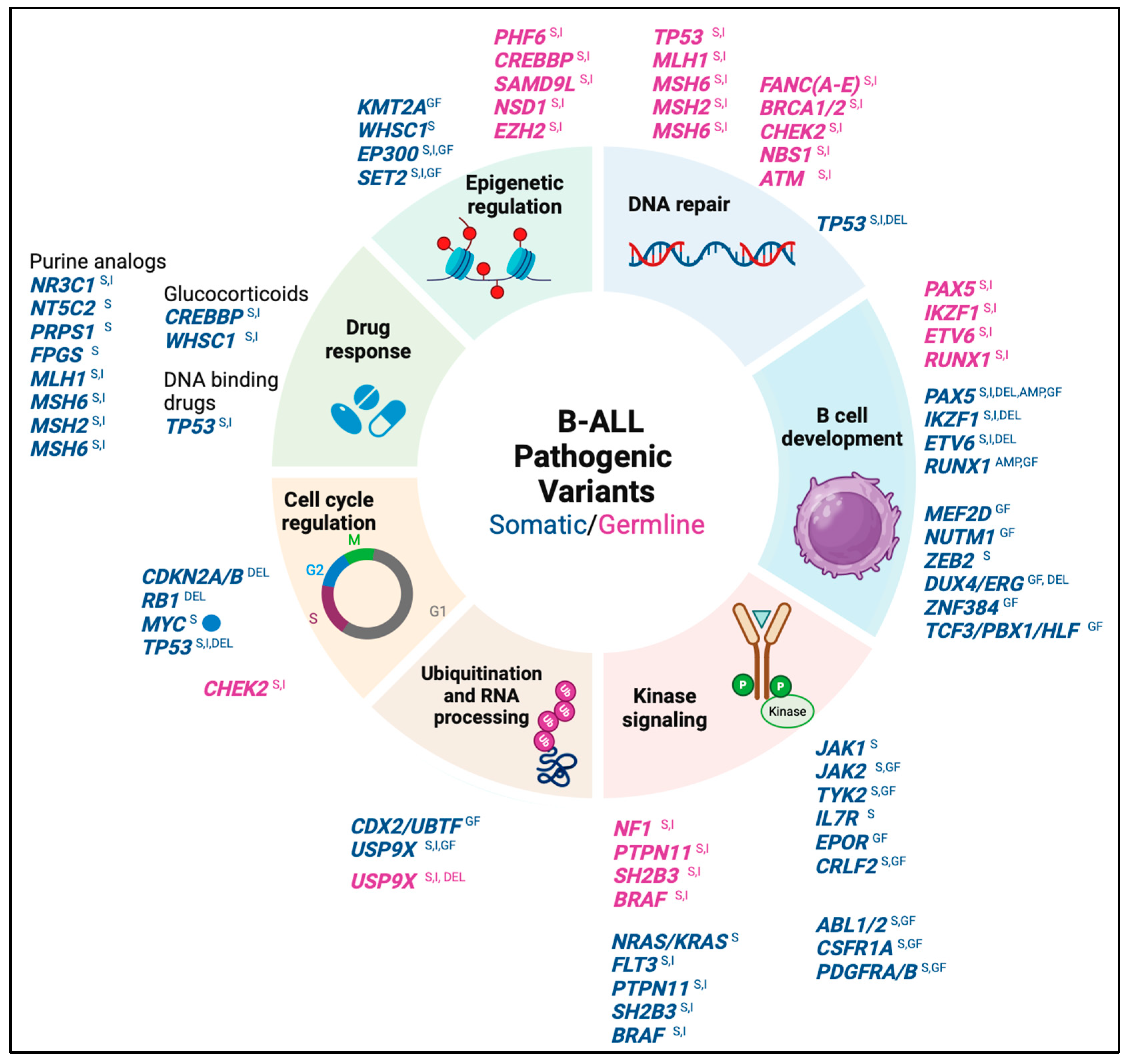
3. The Somatic Pathogenic Variants as Clinical Biomarkers for B-ALL
- Tier I (with strong clinical significance): These variants constitute the molecular taxonomy of B-ALL and produce a biochemical or immunological phenotype associated with specific gene expression profiles, thus, allowing the identification of molecular subtypes with prognostic value [2,22]. Some of these variants have therapeutic value because they allow the prediction of FDA-approved therapies in pediatric patients with B-ALL. Based on the level of evidence, tier 1A variants are described in the World Health Organization (WHO) 2022 classification of lymphoid tumors and/or in professional clinical management guidelines for B-ALL (e.g., COG guidelines), while level 1B variants are recognized by consensus of disease experts based on multiple studies from different groups [10,11].
- Tier II variants (with potential clinical significance): These variants are recurrent in different hematological and non-hematological neoplasms, and some of them may integrate a profile of alterations that, in coexistence, have prognostic value. The presence of tier 2 variants may explain the heterogeneity in clinical response among patients with the same molecular subtype of B-ALL [10,11,16,22].
3.1. Biomarkers Associated with Poor Prognosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B-ALL Molecular Subtypes/Profiles | AMP Classification † | General Frequency | Detection Technique | Common Underlying Pathogenic Variants | Therapeutic Approaches | References |
|---|---|---|---|---|---|---|
| POOR PROGNOSIS | ||||||
| Hypodiploidy (Patterns of multiple chromosome losses) | Tier 1A Dx/Px | 1–2% | Karyotype, FISH, CMA, WGS RNA-seqGEP | TP53 germline variants | Intensive chemotherapy and HSCT | [2,25,26,27] |
| TCF3::HLF fusion | Tier 1A Dx/Px | <1% | Karyotype, FISH RT-PCR, WGS, RNA-seq GF/GEP | PAX5, BTG1, CDKN2A/B deletions and RAS signaling activating variants | Venetoclax § NCT05292664 Intensive chemotherapy | [27,28,29] |
| MYC rearrangement MYC juxtaposed with IGH, IGK, IGL | Tier 2 | <1% | Karyotype FISH WGS RNA- seqGF/GEP | CDKN2A/B, RB1 deletions RAS signaling activating variants | Mature B-cell leukemia/lymphoma chemotherapeutic regimen | [27,30] |
| BCR::ABL1 fusion | Tier 1A Dx/Px/Tx | 2–4% | Karyotype, FISH, RT-PCR WGS, RNA-seqGF/GEP | IKZF1 deletion | Intensive chemotherapy and Imatinib mesylate | [26,27] |
| Ph-like | SNVs, CNVs and GFs involving kinase genes | 10–15% | CMA WGS RNA-seq | IKZF1 deletions | Intensive chemotherapy and tyrosine kinase inhibitors | [22,27] |
| Ph-like positive to CRLF2 rearrangement or mutation (See supplementary Table S1) | Tier 1A Dx/Px/Tx | ~50% Ph-like | FISHrearrangement CMA P2RY8::CRLF2 WGSrearrangement, PM RNA-seq GF/PM/GEP. | IKZF1 deletions and RAS/JAK-STAT signaling activating variants | Intensive chemotherapy and Ruxolitinib (JAK inhibitor) §NCT02723994 §NCT04996160 §NCT03117751 | [3,27,28,31] |
| Ph-like positive to JAK-STAT class fusions (See supplementary Table S1) | Tier 1A Dx/Px/Tx | ~7–10% Ph-like | WGSrearrangements RNA- seqGF FISHrearrangements | Deletions in lymphoid differentiation genes. RAS signaling activating variants. | Intensive chemotherapy and Ruxolitinib §NCT02723994 §NCT04996160 §NCT03117751 | [3,22,28,31] |
| Ph-like positive to ABL class fusions (See supplementary Table S1) | Tier 1A Dx/Px/Tx | ~12% Ph-like | WGSrearrangements FISHrearrangements CMA NUP214::ABL1 RNA- seqGF | Deletions in lymphoid differentiation genes RAS signaling activating variants | Intensive chemotherapy and Dasatinib (Abl/Src inhibitor) §NCT02883049 §NCT03117751 §NCT03020030 | [3,22,28,31] |
| Ph-like positive to JAK-STAT and RAS activating point mutations | Tier 2 | ~6–11% Ph-like | WGSPM WESPM RNA- seqPM | Deletions in lymphoid differentiation genes | Intensive chemotherapy and Ruxolitinib § NCT02723994 § NCT04996160 § NCT03117751 | [3,22,28,31] |
| Ph-like positive to other kinase fusions (See supplementary Table S1) | Tier 1A Dx/Px/ Tx (NTRK3 fusion) | ~1% Ph-like | WGS, FISH RNA- seqGF | Deletions in lymphoid differentiation genes RAS signaling activating variants | Intensive chemotherapy and NTRK3 inhibitors §NCT03066661 Entrectinib § NCT03834961 Larotrectinib | [3,22,28,31] |
| iAMP21 (21q22.12 chromotripsis with multiple RUNX1 copies) | Tier 1A Dx/Px | ~2% | Karyotype, FISH CMA, WGS, RNA-seqGEP | SH2B3 loss of function variants | Intensive chemotherapy | [27,32] |
| KMT2A rearrangements | Tier 1A Dx/Px | 1–2% | Karyotype, FISH, RT-PCR, WGS, RNA-seqGF/GEP | Subclonal RAS signaling activating variants | Azacitidine and Histone Methyltransferase inhibitors § NCT03724084 | [27,33] |
| MEF2D rearrangement (See supplementary Table S2) | Tier 1B Dx/Px | ~4–5% | FISH WGS RNA- seq GF/GEP | Loss of function variants in PHF6 | HDACs Vorinostat Quisinostat (preclinical evidence) | [27,34,35] |
| CDX2/UBTF rearrangements | Tier 1B Dx/Px | <1% | WGS FISH RNA- seqGF/GEP | ETV6 monoallelic deletion PAX5::ZCCHC7 estructural variant | Intensive chemotherapy | [27,36,37,38] |
| IKZF1 c.475A>T p. N159Y IKZF1 Deletion IKZF1plus Profile | Tier 1B Dx/Px Tier 1B Px (IKZF1 deletions) Tier 2 Px (IKZF1 plus Concomitant deletions) | <1% ~15% ~6% | WGS, RNA- seqPM/GEP WGS CMA FISH | Unknown IKZF1 whole or intragenic deletion are underlying lesions in various B-ALL molecular subtypes Concomintant deletions in PAX5, CDKN2A, CDKN2B (homozygous) or PAR1 in absence of ERG deletion | Focal adhesion kinase inhibitors and rethinoids (preclinical evidence) | [27,39] |
| GOOD PROGNOSIS | ||||||
| Hyperdiploidy (Patterns of multiple chromosome gains) | Tier 1A Dx/Px | 20–30% | Karyotype FISH CMA WGS RNA-seq GEP | RAS signaling activating variants | Antimetabolite based chemotherapy | [9,22,40] |
| TCF3::PBX1 fusion | Tier 1A Dx/Px | 2–6% | Karyotype FISH RT-PCR WGS RNA-seq GF/GEP | PAX5, CTCF and SET2 amplifications. CDKN2A deletions | Intensive chemotherapy | [27,41] |
| ETV6::RUNX1 fusion | Tier 1A Dx/Px | 15–25% | Karyotype FISH RT-PCR WGS RNA-seq GF/GEP | PAX5 deletions and WHSC1 PM | Standard chemotherapy | [27,40] |
| DUX4/ERG rearrangements | Tier 1B Dx/Px | 4–6% | WGS FISH RNA-seq GEP | ERG and IKZF1 deletions | Intensive chemotherapy | [22,27,42,43,44] |
| NUTM1 rearrangement (See supplementary Table S2) | Tier 1B Dx/Px | <2% | WGS FISH RNA-seq GF/GEP | Deletions in lymphoid differentiation genes | Standard chemotherapy | [27,45] |
| INTERMEDIATE PROGNOSIS | ||||||
| PAX5 altered PAX5 fusions (See supplementary Table S2) PAX5 intragenic amplification (iAMP) PAX5 c.239C>G p.Pro80Arg | Tier 1B Dx/Px | 3.3–9.3% for PAX5 fusions or iAMP 0.4–1.9% for P80R mutation | WGS CMA FISH RNA-seq GF/PM/GEP | CDKN2A/B deletions RAS/JAK-STAT signaling activating variants. | Intensive chemotherapy | [15] |
| ETV6::RUNX1 like (See supplementary Table S2) | Tier 1B Dx/Px | <5% | WGS FISH RNA-seqGEP/GF | ETV6, IKZF1 and ARPP21 deletions ETV6 germline variants | Intensive chemotherapy | [27,42] |
| ZNF384 rearrangement (See supplementary Table S2) | Tier 1B Dx/Px | 1–6% | WGS WES FISH RNA-seqGEP/GF | Loss of function variants in CREBBP, EP300, KDM6A, CHD4 and CHD8 epigenetic regulators RAS signaling activating variants | Intensive chemotherapy | [27,46,47,48] |
| ZEB2 mutated and IGH::CEBPE fusion | Tier 1B Dx/Px | <1% | WGS, WES, RNA-seq GEP/PM | RAS signaling activating variants | Undefined | [27] |
3.2. Biomarkers Associated with Good Prognosis
3.3. Biomarkers Associated with Intermediate Prognosis
3.4. Biomarkers Associated with Chemoresistance
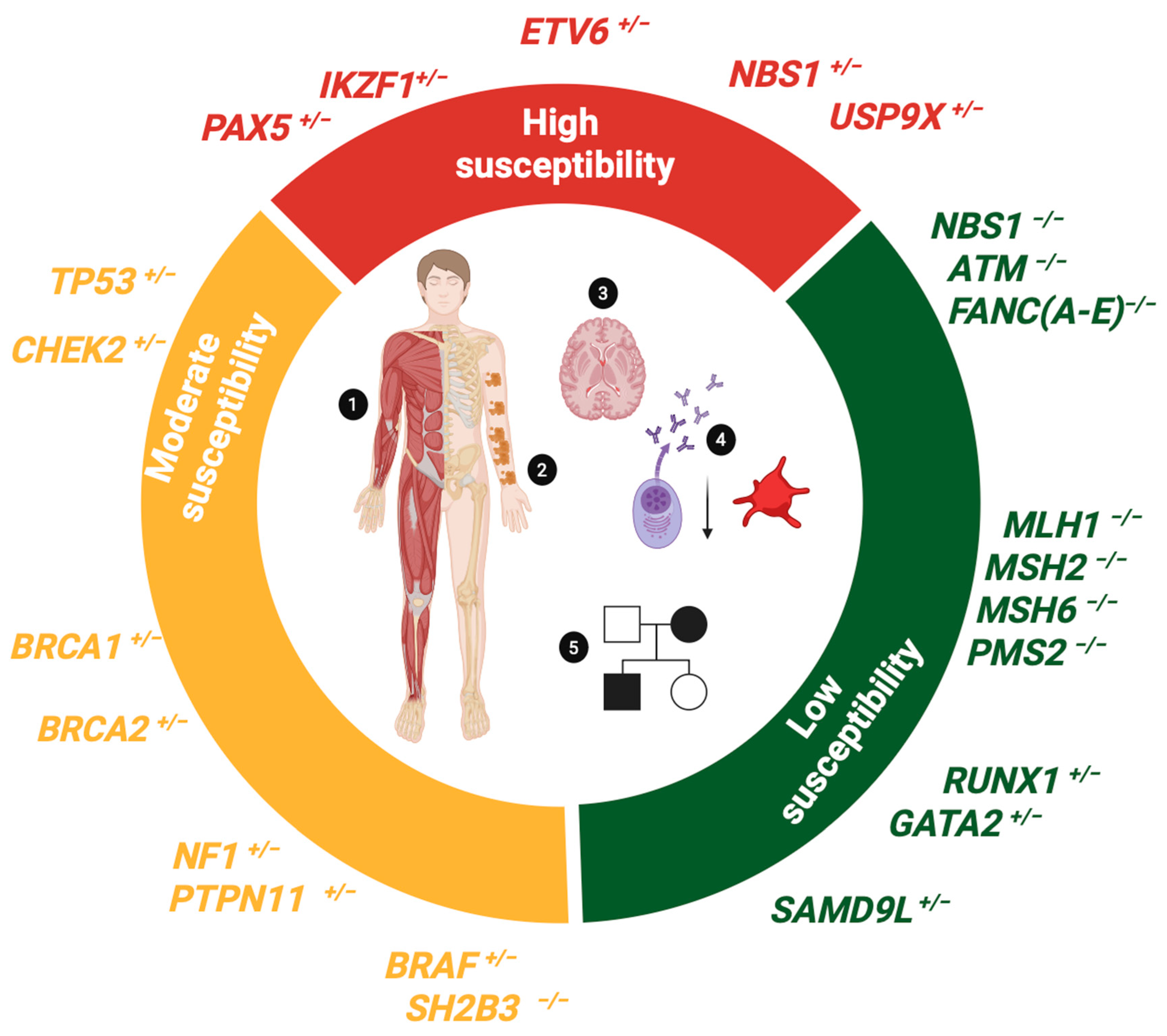
4. Germline Pathogenic Variants as Biomarkers of Susceptibility to B-ALL
4.1. Biomarkers Conferring High Susceptibility to B-ALL
| Genetic Syndrome (OMIM #ID) | Gene (Loci) | Inheritance | Neoplastic Phenotype | Main Non-Neoplastic Phenotypic Characteristics | References |
|---|---|---|---|---|---|
| HIGH SUSCEPTIBILITY SYNDROMES | |||||
| PAX5 Deficiency (615545) | PAX5 (9p13.2) | AD | B-ALL | Immunodeficiency with low levels of B cells and immunoglobulins | [16,74,75] |
| ETV6 Deficiency (616216) | ETV6 (12p13.2) | AD | Hyperdiploid or ETV6::RUNX1 like B-ALL | Thrombocytopenia | [56,76,77] |
| IKZF1 Deficiency (613065) | IKZF1 (7p12.2) | AD | B-ALL | Lymphopenia | [78] |
| USP9X Deficiency (300968) | USP9X (Xp11.4) | XLD | B-ALL restricted to female patients | Psychomotor delay and intellectual disability Congenital abnormalities | [79] |
| MODERATE SUSCEPTIBILITY SYNDROMES | |||||
| Li-Fraumeni syndrome (151623) | TP53 (17p13.1) | AD | Breast cancer, sarcoma, glioblastoma, adrenal gland cancer Hypodiploid B-ALL | NA | [80,81,82] |
| Neurofibromatosis type 1 (162200) | NF1 (17q11.2) | AD | Neurofibromas Gliomas, JMML B-ALL | CALMs Skeletal disorders Cognitive deficits | [83,84] |
| Noonan/Leopard syndrome (163950/15110) Cardiofaciocutaneous syndrome (115150) SH2B3 Deficiency (NA) | PTPN11 (12q24.13) (~50% cases) Other RAS genes (SOS, BRAF1, RAF1, MAPK1, KRAS, NRAS) BRAF (7q34) SH2B3 (12q24.12) | AD AD AR | JMML Neuroblastoma Rhabdomyosarcoma Hyperdiploid B-ALL Non-Hodgkin’s lymphoma Hepatoblastoma Rhabdomyosarcoma B-ALL MPS/JMML B-ALL | Craniofacial Anomalies, Congenital Heart Anomalies CALMs Short stature Thrombocytopenia Hepatosplenomegaly Autoimmunity Hepatosplenomegaly Autoimmunity | [85,86,87] |
| LOW SUSCEPTIBILITY SYNDROMES | |||||
| Nijmegen syndrome (615545) | NBN (NBS1) (8q21.3) | AR | T-cell lymphoproliferative disorders B-ALL * | Craniofacial dysmorphias Developmental delay Immunodeficiency | [88,89] |
| Constitutional mismatch repair deficiency syndrome. (CMMRD) (619069) | MLH1 (3p22.2) MSH2 (2p21-p16.3) MSH6 (2p16.3) PMS2 (7p22.1) | AR | Colorectal cancer Glioblastoma T-cell lymphoproliferative disorders B-ALL * | CALMs Immunodeficiency | [90,91] |
| Fanconi anemia (Various genes) | Genes FANC(A-E) (Múltiple loci) | AR, XLR | AML Head and neck cancer B-ALL * | Dysmorphological syndrome CALMs Musculoskeletal disorders Intellectual disability | [92,93] |
| Ataxia telangiectasia syndrome (208900) | ATM (11q22.3) | AR | T-cell lymphoproliferative disorders B-ALL * | Neurological disorders Telangiectatic disorders Immunodeficiency | [94,95] |
| RUNX1 Deficiency (601399) | RUNX1 (21q22.12) | AD | MDS and T-ALL B-ALL * | Thrombocytopenia | [96] |
| GATA2 Deficiency (614172) | GATA2 (3q21.3) | AD | MDS, AML, JMML, T-ALL B-ALL * | Immunodeficiency | [97] |
| Ataxia pancytopenia Syndrome (159550) | SAMD9L (7q21.2) | AD | MDS, AML, B-ALL * | Neurological disorders Cytopenia Immunodeficiency | [98,99] |
4.2. Biomarkers Conferring Moderate Susceptibility to B-ALL
4.3. Biomarkers Conferring Low Susceptibility to B-ALL
4.4. Clinical Management of Patients with B-ALL Associated with Germline Predisposition
- Consanguineous parents
- Presence of cytopenia in the patient and/or family members
- Dysmorphologic syndrome (musculoskeletal disorders)
- Pigmented lesions or other symptoms associated with RASopathies
- B-ALL of the hypodiploid or ETV6::RUNX1-like subtypes
- Family history of cancers included in the neoplastic spectrum of LFS
- Occurrence of a second primary neoplasm
- Siblings with childhood cancer or close relatives diagnosed with LFS spectrum cancers at age ≤ 45 years
5. Detection and Interpretation of Genetic Variants in B-ALL Patients
Genomic Techniques Applied to B-ALL Analysis
6. Clinical Significance and Etiology of Variants
7. Genetic Variants Applied to Clinical Practice
8. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACMG | American college of genetics and genomic medicine |
| AD | Autosomal dominant inheritance |
| ALL | Acute lymphoblastic leukemia |
| AMP | Association of molecular pathology |
| ASCO | American society of clinical oncology |
| BAF | B allele frequency |
| B-ALL | B-cell precursor acute lymphoblastic leukemia |
| CAR-T | Chimeric antigen receptor therapy |
| CAP | College of American pathologist |
| CGC | Cancer genomic consortium |
| CNV | Copy number variant |
| CN-LOH | Copy neutral loss of heterozygosity |
| CMA | Chromosomal microarray analysis |
| CMMRD | Constitutional mismatch repair deficiency syndrome |
| COG | Children oncology group |
| EFS | Event-free survival |
| FDA | Food and drug administration |
| FISH | Fluorescence in situ hybridization |
| HR | High-risk |
| HSCT | Hematopoietic stem cell transplant |
| LFS | Li Fraumeni syndrome |
| MRD | Minimal residual disease |
| NCI | National cancer institute |
| NFS | Neurofibromatosis syndrome |
| NLS | Noonan/Leopard syndrome |
| NGS | Next-generation sequencing |
| NJS | Nijmegen syndrome |
| RT-PCR | Reverse transcript polymerase chain reaction |
| SNV | Single nucleotide variant |
| SR | Standard risk |
| TKI | Tyrosine kinase inhibitor |
| VAF | Variant allele fraction |
| VUS | Variant of uncertain significance |
| WES | Whole exome sequencing |
| WBC | White blood cell |
| WHO | World health organization |
| WGS | Whole genome sequencing |
References
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet Lond. Engl. 2020, 395, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Hunger, S.P. Genomic characterization of paediatric acute lymphoblastic leukaemia: An opportunity for precision medicine therapeutics. Br. J. Haematol. 2017, 176, 867–882. [Google Scholar] [CrossRef]
- Tran, T.H.; Hunger, S.P. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin. Cancer Biol. 2022, 84, 144–152. [Google Scholar] [CrossRef]
- Heikamp, E.B.; Pui, C.-H. Next-Generation Evaluation and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Pediatr. 2018, 203, 14–24.e2. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. Genomic characterization of childhood acute lymphoblastic leukemia. Semin. Hematol. 2013, 50, 314–324. [Google Scholar] [CrossRef]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet Lond. Engl. 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Stuchly, J.; Winkowska, L.; Musilova, A.; Fiser, K.; Slamova, M.; Starkova, J.; Vaskova, M.; Hrusak, O.; Sramkova, L.; et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 2019, 104, 1396–1406. [Google Scholar] [CrossRef]
- Roberts, K.G.; Mullighan, C.G. Genomics in acute lymphoblastic leukaemia: Insights and treatment implications. Nat. Rev. Clin. Oncol. 2015, 12, 344–357. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. JMD 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Mikhail, F.M.; Biegel, J.A.; Cooley, L.D.; Dubuc, A.M.; Hirsch, B.; Horner, V.L.; Newman, S.; Shao, L.; Wolff, D.J.; Raca, G. Technical laboratory standards for interpretation and reporting of acquired copy-number abnormalities and copy-neutral loss of heterozygosity in neoplastic disorders: A joint consensus recommendation from the American College of Medical Genetics and Genomics (ACMG) and the Cancer Genomics Consortium (CGC). Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 1903–1916. [Google Scholar] [CrossRef]
- Cobaleda, C.; Vicente-Dueñas, C.; Nichols, K.E.; Sanchez-Garcia, I. Childhood B cell leukemia: Intercepting the paths to progression. BioEssays News Rev. Mol. Cell. Dev. Biol. 2024, 46, e2400033. [Google Scholar] [CrossRef]
- de Smith, A.J.; Spector, L.G. In Utero Origins of Acute Leukemia in Children. Biomedicines 2024, 12, 236. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Li, Y.; Qiu, X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: Mechanism and regulation. Immunology 2020, 160, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Pui, C.-H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, H.; Yang, W.; Yadav, R.; Morrison, A.C.; Qian, M.; Devidas, M.; Liu, Y.; Perez-Andreu, V.; Zhao, X.; et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat. Commun. 2015, 6, 7553. [Google Scholar] [CrossRef]
- Kratz, C.P.; Stanulla, M.; Cavé, H. Genetic predisposition to acute lymphoblastic leukemia: Overview on behalf of the I-BFM ALL Host Genetic Variation Working Group. Eur. J. Med. Genet. 2016, 59, 111–115. [Google Scholar] [CrossRef]
- Cobaleda, C.; Godley, L.A.; Nichols, K.E.; Wlodarski, M.W.; Sanchez-Garcia, I. Insights into the Molecular Mechanisms of Genetic Predisposition to Hematopoietic Malignancies: The Importance of Gene-Environment Interactions. Cancer Discov. 2024, 14, 396–405. [Google Scholar] [CrossRef]
- Gocho, Y.; Yang, J.J. Genetic defects in hematopoietic transcription factors and predisposition to acute lymphoblastic leukemia. Blood 2019, 134, 793–797. [Google Scholar] [CrossRef]
- Strom, S.P. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Harrison, C.J. Advances in B-cell Precursor Acute Lymphoblastic Leukemia Genomics. HemaSphere 2018, 2, e53. [Google Scholar] [CrossRef]
- Schwab, C.J.; Murdy, D.; Butler, E.; Enshaei, A.; Winterman, E.; Cranston, R.E.; Ryan, S.; Barretta, E.; Hawking, Z.; Murray, J.; et al. Genetic characterisation of childhood B-other-acute lymphoblastic leukaemia in UK patients by fluorescence in situ hybridisation and Multiplex Ligation-dependent Probe Amplification. Br. J. Haematol. 2022, 196, 753–763. [Google Scholar] [CrossRef]
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef]
- Shago, M. Recurrent Cytogenetic Abnormalities in Acute Lymphoblastic Leukemia. Methods Mol. Biol. Clifton NJ 2017, 1541, 257–278. [Google Scholar] [CrossRef]
- Duffield, A.S.; Mullighan, C.G.; Borowitz, M.J. International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch. Int. J. Pathol. 2023, 482, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef]
- Bomken, S.; Enshaei, A.; Schwalbe, E.C.; Mikulasova, A.; Dai, Y.; Zaka, M.; Fung, K.T.M.; Bashton, M.; Lim, H.; Jones, L.; et al. Molecular characterization and clinical outcome of B-cell precursor acute lymphoblastic leukemia with IG-MYC rearrangement. Haematologica 2023, 108, 717–731. [Google Scholar] [CrossRef]
- Yokota, T.; Kanakura, Y. Genetic abnormalities associated with acute lymphoblastic leukemia. Cancer Sci. 2016, 107, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Baughn, L.B.; Meredith, M.M.; Oseth, L.; Smolarek, T.A.; Hirsch, B. SH2B3 aberrations enriched in iAMP21 B lymphoblastic leukemia. Cancer Genet. 2018, 226–227, 30–35. [Google Scholar] [CrossRef]
- Forgione, M.O.; McClure, B.J.; Eadie, L.N.; Yeung, D.T.; White, D.L. KMT2A rearranged acute lymphoblastic leukaemia: Unravelling the genomic complexity and heterogeneity of this high-risk disease. Cancer Lett. 2020, 469, 410–418. [Google Scholar] [CrossRef]
- Ohki, K.; Kiyokawa, N.; Saito, Y.; Hirabayashi, S.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yoshimi, A.; Ogata-Kawata, H.; et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 2019, 104, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Okuno, Y.; Kawashima, N.; Muramatsu, H.; Okuno, T.; Wang, X.; Kataoka, S.; Sekiya, Y.; Hamada, M.; Murakami, N.; et al. MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 3451–3459. [Google Scholar] [CrossRef] [PubMed]
- Passet, M.; Kim, R.; Gachet, S.; Sigaux, F.; Chaumeil, J.; Galland, A.; Sexton, T.; Quentin, S.; Hernandez, L.; Larcher, L.; et al. Concurrent CDX2 cis-deregulation and UBTF::ATXN7L3 fusion define a novel high-risk subtype of B-cell ALL. Blood 2022, 139, 3505–3518. [Google Scholar] [CrossRef]
- Kimura, S.; Montefiori, L.; Iacobucci, I.; Zhao, Y.; Gao, Q.; Paietta, E.M.; Haferlach, C.; Laird, A.D.; Mead, P.E.; Gu, Z.; et al. Enhancer retargeting of CDX2 and UBTF::ATXN7L3 define a subtype of high-risk B-progenitor acute lymphoblastic leukemia. Blood 2022, 139, 3519–3531. [Google Scholar] [CrossRef]
- Leeman-Neill, R.J.; Song, D.; Bizarro, J.; Wacheul, L.; Rothschild, G.; Singh, S.; Yang, Y.; Sarode, A.Y.; Gollapalli, K.; Wu, L.; et al. Noncoding mutations cause super-enhancer retargeting resulting in protein synthesis dysregulation during B cell lymphoma progression. Nat. Genet. 2023, 55, 2160–2174. [Google Scholar] [CrossRef]
- Churchman, M.L.; Evans, K.; Richmond, J.; Robbins, A.; Jones, L.; Shapiro, I.M.; Pachter, J.A.; Weaver, D.T.; Houghton, P.J.; Smith, M.A.; et al. Synergism of FAK and tyrosine kinase inhibition in Ph+ B-ALL. JCI Insight 2016, 1, e86082. [Google Scholar] [CrossRef]
- Roberts, K.G.; Mullighan, C.G. The Biology of B-Progenitor Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2020, 10, a034835. [Google Scholar] [CrossRef]
- Zhou, B.; Chu, X.; Tian, H.; Liu, T.; Liu, H.; Gao, W.; Chen, S.; Hu, S.; Wu, D.; Xu, Y. The clinical outcomes and genomic landscapes of acute lymphoblastic leukemia patients with E2A-PBX1: A 10-year retrospective study. Am. J. Hematol. 2021, 96, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Li, Z.; Lee, S.H.R.; Chin, W.H.N.; Lu, Y.; Jiang, N.; Lim, E.H.; Coustan-Smith, E.; Chiew, K.H.; Oh, B.L.Z.; Koh, G.S.; et al. Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv. 2021, 5, 5226–5238. [Google Scholar] [CrossRef]
- Rehn, J.A.; O’Connor, M.J.; White, D.L.; Yeung, D.T. DUX Hunting-Clinical Features and Diagnostic Challenges Associated with DUX4-Rearranged Leukaemia. Cancers 2020, 12, 2815. [Google Scholar] [CrossRef]
- Hormann, F.M.; Hoogkamer, A.Q.; Beverloo, H.B.; Boeree, A.; Dingjan, I.; Wattel, M.M.; Stam, R.W.; Escherich, G.; Pieters, R.; den Boer, M.L.; et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica 2019, 104, e455–e459. [Google Scholar] [CrossRef] [PubMed]
- Lilljebjörn, H.; Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1395–1401. [Google Scholar] [CrossRef]
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef]
- Zaliova, M.; Potuckova, E.; Lukes, J.; Winkowska, L.; Starkova, J.; Janotova, I.; Sramkova, L.; Stary, J.; Zuna, J.; Stanulla, M.; et al. Frequency and prognostic impact of ZEB2 H1038 and Q1072 mutations in childhood B-other acute lymphoblastic leukemia. Haematologica 2021, 106, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Weigert, O.; Lane, A.A.; Bird, L.; Kopp, N.; Chapuy, B.; Van Bodegom, D.; Toms, A.V.; Marubayashi, S.; Christie, A.L.; McKeown, M.; et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J. Exp. Med. 2012, 209, 259–273. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: Toward detecting high-risk ALL and implementing precision medicine. Blood 2015, 125, 3977–3987. [Google Scholar] [CrossRef]
- Gao, Q.; Ryan, S.L.; Iacobucci, I.; Ghate, P.S.; Cranston, R.E.; Schwab, C.; Elsayed, A.H.; Shi, L.; Pounds, S.; Lei, S.; et al. The genomic landscape of acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21. Blood 2023, 142, 711–723. [Google Scholar] [CrossRef]
- Moorman, A.V. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica 2016, 101, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Paolino, J.; Tsai, H.K.; Harris, M.H.; Pikman, Y. IKZF1 Alterations and Therapeutic Targeting in B-Cell Acute Lymphoblastic Leukemia. Biomedicines 2024, 12, 89. [Google Scholar] [CrossRef]
- Stanulla, M.; Cavé, H.; Moorman, A.V. IKZF1 deletions in pediatric acute lymphoblastic leukemia: Still a poor prognostic marker? Blood 2020, 135, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; Te Kronnie, G.; Bourquin, J.-P.; Bornhauser, B.; et al. IKZF1plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef]
- Nishii, R.; Baskin-Doerfler, R.; Yang, W.; Oak, N.; Zhao, X.; Yang, W.; Hoshitsuki, K.; Bloom, M.; Verbist, K.; Burns, M.; et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood 2021, 137, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Sanada, M.; Tsuzuki, S.; Hayakawa, F. Oncogenic lesions and molecular subtypes in adults with B-cell acute lymphoblastic leukemia. Cancer Sci. 2023, 114, 8–15. [Google Scholar] [CrossRef]
- Erichsen, H.C.; Chanock, S.J. SNPs in cancer research and treatment. Br. J. Cancer 2004, 90, 747–751. [Google Scholar] [CrossRef]
- Relling, M.V.; Ramsey, L.B. Pharmacogenomics of acute lymphoid leukemia: New insights into treatment toxicity and efficacy. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 126–130. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Pierro, J.; Saliba, J.; Narang, S.; Sethia, G.; Saint Fleur-Lominy, S.; Chowdhury, A.; Qualls, A.; Fay, H.; Kilberg, H.L.; Moriyama, T.; et al. The NSD2 p.E1099K Mutation Is Enriched at Relapse and Confers Drug Resistance in a Cell Context-Dependent Manner in Pediatric Acute Lymphoblastic Leukemia. Mol. Cancer Res. MCR 2020, 18, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Loh, M.L.; Ma, X.; Rusch, M.; Wu, G.; Harvey, R.C.; Wheeler, D.A.; Hampton, O.A.; Carroll, W.L.; Chen, I.-M.; et al. Comparison Of Mutational Profiles Of Diagnosis and Relapsed Pediatric B-Acute Lymphoblastic Leukemia: A Report From The COG ALL Target Project. Blood 2013, 122, 824. [Google Scholar] [CrossRef]
- Malinowska-Ozdowy, K.; Frech, C.; Schönegger, A.; Eckert, C.; Cazzaniga, G.; Stanulla, M.; Zur Stadt, U.; Mecklenbräuker, A.; Schuster, M.; Kneidinger, D.; et al. KRAS and CREBBP mutations: A relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia 2015, 29, 1656–1667. [Google Scholar] [CrossRef]
- Tamai, M.; Kasai, S.; Akahane, K.; Thu, T.N.; Kagami, K.; Komatsu, C.; Abe, M.; Watanabe, A.; Goi, K.; Miyake, K.; et al. Glucocorticoid receptor gene mutations confer glucocorticoid resistance in B-cell precursor acute lymphoblastic leukemia. J. Steroid Biochem. Mol. Biol. 2022, 218, 106068. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.A.; Wang, J.; Hogan, L.E.; Yang, J.J.; Dandekar, S.; Patel, J.P.; Tang, Z.; Zumbo, P.; Li, S.; Zavadil, J.; et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 290–294. [Google Scholar] [CrossRef]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation With Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults With First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 833–842. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Jabbour, E.; Advani, A. Recent Advances in Managing Acute Lymphoblastic Leukemia. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2020, 40, 330–342. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L. Chimeric Antigen Receptor T Cell Therapy for Pediatric B-ALL: Narrowing the Gap Between Early and Long-Term Outcomes. Front. Immunol. 2020, 11, 1985. [Google Scholar] [CrossRef]
- Mendes-de-Almeida, D.P.; Andrade, F.G.; Sampaio Carvalho, M.d.P.S.; Córdoba, J.C.; Souza, M.D.S.; Neto, P.C.; Spector, L.G.; Pombo-de-Oliveira, M.S. Identifying childhood leukemia with an excess of hematological malignancies in first-degree relatives in Brazil. Front. Oncol. 2023, 13, 1207695. [Google Scholar] [CrossRef]
- Douglas, S.P.M.; Lahtinen, A.K.; Koski, J.R.; Leimi, L.; Keränen, M.A.I.; Koskenvuo, M.; Heckman, C.A.; Jahnukainen, K.; Pitkänen, E.; Wartiovaara-Kautto, U.; et al. Enrichment of cancer-predisposing germline variants in adult and pediatric patients with acute lymphoblastic leukemia. Sci. Rep. 2022, 12, 10670. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-Y.; Lee, H.; Lee, S.-T.; Choi, J.R.; Jung, C.W.; Koo, H.H.; Kim, S.-H. Recurrent somatic mutations and low germline predisposition mutations in Korean ALL patients. Sci. Rep. 2021, 11, 8893. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Jamrog, L.A.; Fregona, V.; Hamelle, C.; Fenwarth, L.; Lejeune, S.; Helevaut, N.; Geffroy, S.; Caillault, A.; Marceau-Renaut, A.; et al. Germline PAX5 mutation predisposes to familial B-cell precursor acute lymphoblastic leukemia. Blood 2021, 137, 1424–1428. [Google Scholar] [CrossRef]
- Hyde, R.K.; Liu, P.P. Germline PAX5 mutations and B cell leukemia. Nat. Genet. 2013, 45, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, J.; Porter, C.C. ETV6-related thrombocytopenia and leukemia predisposition. Blood 2019, 134, 663–667. [Google Scholar] [CrossRef]
- Feurstein, S.; Godley, L.A. Germline ETV6 mutations and predisposition to hematological malignancies. Int. J. Hematol. 2017, 106, 189–195. [Google Scholar] [CrossRef]
- Churchman, M.L.; Qian, M.; Te Kronnie, G.; Zhang, R.; Yang, W.; Zhang, H.; Lana, T.; Tedrick, P.; Baskin, R.; Verbist, K.; et al. Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 2018, 33, 937–948.e8. [Google Scholar] [CrossRef]
- Sisoudiya, S.D.; Mishra, P.; Li, H.; Schraw, J.M.; Scheurer, M.E.; Salvi, S.; Doddapaneni, H.; Muzny, D.; Mitchell, D.; Taylor, O.; et al. Identification of USP9X as a leukemia susceptibility gene. Blood Adv. 2023, 7, 4563–4575. [Google Scholar] [CrossRef]
- Schmiegelow, K. Treatment-related toxicities in children with acute lymphoblastic leukaemia predisposition syndromes. Eur. J. Med. Genet. 2016, 59, 654–660. [Google Scholar] [CrossRef]
- Itov, A.; Ilyasova, K.; Soldatkina, O.; Kazakova, A.; Kozeev, V.; Semchenkova, A.; Osipova, E.; Boichenko, E.; Volchkov, E.; Popov, A.; et al. TP53 variants underlying pediatric low-hypodiploidy B-cell acute lymphoblastic leukemia demonstrate diverse origins and may persist as a hematopoietic clone in remission. EJHaem 2024, 5, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Cao, X.; Devidas, M.; Yang, W.; Cheng, C.; Dai, Y.; Carroll, A.; Heerema, N.A.; Zhang, H.; Moriyama, T.; et al. TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 591–599. [Google Scholar] [CrossRef]
- Heatley, S.L.; Page, E.C.; Eadie, L.N.; McClure, B.J.; Rehn, J.; Yeung, D.T.; Osborn, M.; Revesz, T.; Kirby, M.; White, D.L. Case Report: Precision Medicine Target Revealed by In Vitro Modeling of Relapsed, Refractory Acute Lymphoblastic Leukemia From a Child With Neurofibromatosis. Front. Oncol. 2022, 12, 851572. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primer 2017, 3, 17004. [Google Scholar] [CrossRef]
- Carcavilla, A.; Suárez-Ortega, L.; Rodríguez Sánchez, A.; Gonzalez-Casado, I.; Ramón-Krauel, M.; Labarta, J.I.; Quinteiro Gonzalez, S.; Riaño Galán, I.; Ezquieta Zubicaray, B.; López-Siguero, J.P. [Noonan syndrome: Genetic and clinical update and treatment options]. An. Pediatr. 2020, 93, 61.e1–61.e14. [Google Scholar] [CrossRef] [PubMed]
- Bess, J.; Brown, T.; Bhala, S.; Faizer, A.; Ahmadzada, M.; Livinski, A.A.; Pemov, A.; Kim, J.; Rosenberg, P.S.; Ney, G.M.; et al. A Literature Review and Pooled Case Analysis of Cardiofaciocutaneous Syndrome to Estimate Cancer Risk. MedRxiv Prepr. Serv. Health Sci. 2024, 27, 101423. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Erlacher, M. SH2B3 alterations in a novel genetic condition, juvenile myelomonocytic leukemia, and myeloproliferative neoplasia. Haematologica 2024, 109, 2391–2394. [Google Scholar] [CrossRef]
- Boyarchuk, O.; Kostyuchenko, L.; Akopyan, H.; Bondarenko, A.; Volokha, A.; Hilfanova, A.; Savchak, I.; Nazarenko, L.; Yarema, N.; Urbas, O.; et al. Nijmegen breakage syndrome: 25-year experience of diagnosis and treatment in Ukraine. Front. Immunol. 2024, 15, 1428724. [Google Scholar] [CrossRef]
- Escherich, C.S.; Chen, W.; Li, Y.; Yang, W.; Nishii, R.; Li, Z.; Raetz, E.A.; Devidas, M.; Wu, G.; Nichols, K.E.; et al. Germ line genetic NBN variation and predisposition to B-cell acute lymphoblastic leukemia in children. Blood 2024, 143, 2270–2283. [Google Scholar] [CrossRef]
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.A.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.-M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium “care for CMMRD” (C4CMMRD). J. Med. Genet. 2014, 51, 355–365. [Google Scholar] [CrossRef]
- Oshrine, B.; Grana, N.; Moore, C.; Nguyen, J.; Crenshaw, M.; Edwards, M.; Sudhaman, S.; Forster, V.J.; Tabori, U. B-cell acute lymphoblastic leukemia with high mutation burden presenting in a child with constitutional mismatch repair deficiency. Blood Adv. 2019, 3, 1795–1798. [Google Scholar] [CrossRef]
- Fiesco-Roa, M.Ó.; García-de Teresa, B.; Leal-Anaya, P.; van ’t Hek, R.; Wegman-Ostrosky, T.; Frías, S.; Rodríguez, A. Fanconi anemia and dyskeratosis congenita/telomere biology disorders: Two inherited bone marrow failure syndromes with genomic instability. Front. Oncol. 2022, 12, 949435. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, N.; Wali, R.; Fadoo, Z.; Saleem, A.F. Acute lymphoblastic leukemia in a child with Fanconi’s anaemia. J. Coll. Physicians Surg.-Pak. JCPSP 2012, 22, 458–460. [Google Scholar]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Elitzur, S.; Shiloh, R.; Loeffen, J.; Pastorczak, A.; Takagi, M.; Bomken, S.; Baruchel, A.; Ducassou, S.; Mahlaoui, N.; Strullu, M.; et al. ATM Germline Pathogenic Variants Affect Treatment Outcomes in Children with Acute Lymphoblastic Leukemia/Lymphoma and Ataxia Telangiectasia. Blood 2023, 142 (Suppl. 1), 520. [Google Scholar] [CrossRef]
- Schlegelberger, B.; Heller, P.G. RUNX1 deficiency (familial platelet disorder with predisposition to myeloid leukemia, FPDMM). Semin. Hematol. 2017, 54, 75–80. [Google Scholar] [CrossRef]
- Calvo, K.R.; Hickstein, D.D. The spectrum of GATA2 deficiency syndrome. Blood 2023, 141, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Rundberg Nilsson, A.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef]
- Cheah, J.J.C.; Brown, A.L.; Schreiber, A.W.; Feng, J.; Babic, M.; Moore, S.; Young, C.-C.; Fine, M.; Phillips, K.; Guandalini, M.; et al. A novel germline SAMD9L mutation in a family with ataxia-pancytopenia syndrome and pediatric acute lymphoblastic leukemia. Haematologica 2019, 104, e318–e321. [Google Scholar] [CrossRef]
- Tomasik, B.; Pastorczak, A.; Fendler, W.; Bartłomiejczyk, M.; Braun, M.; Mycko, M.; Madzio, J.; Polakowska, E.; Ulińska, E.; Matysiak, M.; et al. Heterozygous carriers of germline c.657_661del5 founder mutation in NBN gene are at risk of central nervous system relapse of B-cell precursor acute lymphoblastic leukemia. Haematologica 2018, 103, e200–e203. [Google Scholar] [CrossRef]
- de Smith, A.J. NBN: Protein instability, ALL susceptibility. Blood 2024, 143, 2221–2222. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Kirschner-Schwabe, R.; Groeneveld-Krentz, S.; Escherich, G.; Möricke, A.; von Stackelberg, A.; Stanulla, M.; Bailey, S.; Richter, L.; Steinemann, D.; et al. Clinical and genetic characteristics of children with acute lymphoblastic leukemia and Li-Fraumeni syndrome. Leukemia 2021, 35, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.-M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.G.; Ewald, I.P.; Sapienza, M.; Pinheiro, M.; Peixoto, A.; de Nóbrega, A.F.; Carraro, D.M.; Teixeira, M.R.; Ashton-Prolla, P.; Achatz, M.I.W.; et al. Li-Fraumeni-like syndrome associated with a large BRCA1 intragenic deletion. BMC Cancer 2012, 12, 237. [Google Scholar] [CrossRef]
- Schütte, P.; Möricke, A.; Zimmermann, M.; Bleckmann, K.; Reismüller, B.; Attarbaschi, A.; Mann, G.; Bodmer, N.; Niggli, F.; Schrappe, M.; et al. Preexisting conditions in pediatric ALL patients: Spectrum, frequency and clinical impact. Eur. J. Med. Genet. 2016, 59, 143–151. [Google Scholar] [CrossRef]
- Bloom, M.; Maciaszek, J.L.; Clark, M.E.; Pui, C.-H.; Nichols, K.E. Recent advances in genetic predisposition to pediatric acute lymphoblastic leukemia. Expert Rev. Hematol. 2020, 13, 55–70. [Google Scholar] [CrossRef]
- McReynolds, L.J.; Savage, S.A. Pediatric leukemia susceptibility disorders: Manifestations and management. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 242–250. [Google Scholar] [CrossRef]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. Guideline Development Group, American College of Medical Genetics and Genomics Professional Practice and Guidelines Committee and National Society of Genetic Counselors Practice Guidelines Committee A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 70–87. [Google Scholar] [CrossRef]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Next-Generation Sequencing in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2019, 20, 2929. [Google Scholar] [CrossRef]
- Ni Chin, W.H.; Li, Z.; Jiang, N.; Lim, E.H.; Suang Lim, J.Y.; Lu, Y.; Chiew, K.H.; Yin Kham, S.K.; Zhi Oh, B.L.; Tan, A.M.; et al. Practical Considerations for Using RNA Sequencing in Management of B-Lymphoblastic Leukemia: Malaysia-Singapore Acute Lymphoblastic Leukemia 2020 Implementation Strategy. J. Mol. Diagn. JMD 2021, 23, 1359–1372. [Google Scholar] [CrossRef]
- Bařinka, J.; Hu, Z.; Wang, L.; Wheeler, D.A.; Rahbarinia, D.; McLeod, C.; Gu, Z.; Mullighan, C.G. RNAseqCNV: Analysis of large-scale copy number variations from RNA-seq data. Leukemia 2022, 36, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.V.; Such, E.; Sargas, C.; Simarro, J.; Miralles, A.; Pérez, G.; de Juan, I.; Palanca, S.; Avetisyan, G.; Santiago, M.; et al. Design and Validation of a Custom Next-Generation Sequencing Panel in Pediatric Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2023, 24, 4440. [Google Scholar] [CrossRef] [PubMed]
- Montaño, A.; Hernández-Sánchez, J.; Forero-Castro, M.; Matorra-Miguel, M.; Lumbreras, E.; Miguel, C.; Santos, S.; Ramírez-Maldonado, V.; Fuster, J.L.; de Las Heras, N.; et al. Comprehensive Custom NGS Panel Validation for the Improvement of the Stratification of B-Acute Lymphoblastic Leukemia Patients. J. Pers. Med. 2020, 10, 137. [Google Scholar] [CrossRef]
- Surrey, L.F.; MacFarland, S.P.; Chang, F.; Cao, K.; Rathi, K.S.; Akgumus, G.T.; Gallo, D.; Lin, F.; Gleason, A.; Raman, P.; et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019, 11, 32. [Google Scholar] [CrossRef]
- Schieck, M.; Lentes, J.; Thomay, K.; Hofmann, W.; Behrens, Y.L.; Hagedorn, M.; Ebersold, J.; Davenport, C.F.; Fazio, G.; Möricke, A.; et al. Implementation of RNA sequencing and array CGH in the diagnostic workflow of the AIEOP-BFM ALL 2017 trial on acute lymphoblastic leukemia. Ann. Hematol. 2020, 99, 809–818. [Google Scholar] [CrossRef]
- Burke, W.; Parens, E.; Chung, W.K.; Berger, S.M.; Appelbaum, P.S. The Challenge of Genetic Variants of Uncertain Clinical Significance: A Narrative Review. Ann. Intern. Med. 2022, 175, 994–1000. [Google Scholar] [CrossRef]
- Calò, V.; Bruno, L.; La Paglia, L.; Perez, M.; Margarese, N.; Di Gaudio, F.; Russo, A. The Clinical Significance of Unknown Sequence Variants in BRCA Genes. Cancers 2010, 2, 1644–1660. [Google Scholar] [CrossRef]
- Kraft, I.L.; Godley, L.A. Identifying potential germline variants from sequencing hematopoietic malignancies. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 219–227. [Google Scholar] [CrossRef]
- Padron, E.; Ball, M.C.; Teer, J.K.; Painter, J.S.; Yoder, S.J.; Zhang, C.; Zhang, L.; Moscinski, L.C.; Rollison, D.E.; Gore, S.D.; et al. Germ line tissues for optimal detection of somatic variants in myelodysplastic syndromes. Blood 2018, 131, 2402–2405. [Google Scholar] [CrossRef] [PubMed]
- Ipe, A.; Angiolillo, A.; Jacobsohn, D.; Cheng, J.; Bornhorst, M.; Turner, J.; Vatsayan, A. Case report: Tisagenlecleucel for treatment of relapsed B- acute lymphoblastic leukemia in a patient with CHEK2 mutation. Front. Pediatr. 2023, 11, 1067131. [Google Scholar] [CrossRef]
- Salzer, W.L.; Burke, M.J.; Devidas, M.; Dai, Y.; Hardy, K.K.; Kairalla, J.A.; Gore, L.; Hilden, J.M.; Larsen, E.; Rabin, K.R.; et al. Impact of Intrathecal Triple Therapy Versus Intrathecal Methotrexate on Disease-Free Survival for High-Risk B-Lymphoblastic Leukemia: Children’s Oncology Group Study AALL1131. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Nebral, K.; Gertzen, C.G.W.; Ganmore, I.; Haas, O.A.; Bhatia, S.; Fischer, U.; Kuhlen, M.; Gohlke, H.; Izraeli, S.; et al. JAK2 p.G571S in B-cell precursor acute lymphoblastic leukemia: A synergizing germline susceptibility. Leukemia 2019, 33, 2331–2335. [Google Scholar] [CrossRef] [PubMed]
- McLeod, C.; Gout, A.M.; Zhou, X.; Thrasher, A.; Rahbarinia, D.; Brady, S.W.; Macias, M.; Birch, K.; Finkelstein, D.; Sunny, J.; et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov. 2021, 11, 1082–1099. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez Anaya, D.; Rodriguez Ruiz, J.I.; Navarrete-Meneses, M.d.P.; Pérez-Vera, P. A Comprehensive Review of Somatic and Germline Biomarkers Associated with Childhood B-Cell Precursor Acute Lymphoblastic Leukemia: From Biological Significance to Precision Medicine Opportunities. Biomedicines 2025, 13, 1626. https://doi.org/10.3390/biomedicines13071626
Martínez Anaya D, Rodriguez Ruiz JI, Navarrete-Meneses MdP, Pérez-Vera P. A Comprehensive Review of Somatic and Germline Biomarkers Associated with Childhood B-Cell Precursor Acute Lymphoblastic Leukemia: From Biological Significance to Precision Medicine Opportunities. Biomedicines. 2025; 13(7):1626. https://doi.org/10.3390/biomedicines13071626
Chicago/Turabian StyleMartínez Anaya, Daniel, Johana Itzel Rodriguez Ruiz, María del Pilar Navarrete-Meneses, and Patricia Pérez-Vera. 2025. "A Comprehensive Review of Somatic and Germline Biomarkers Associated with Childhood B-Cell Precursor Acute Lymphoblastic Leukemia: From Biological Significance to Precision Medicine Opportunities" Biomedicines 13, no. 7: 1626. https://doi.org/10.3390/biomedicines13071626
APA StyleMartínez Anaya, D., Rodriguez Ruiz, J. I., Navarrete-Meneses, M. d. P., & Pérez-Vera, P. (2025). A Comprehensive Review of Somatic and Germline Biomarkers Associated with Childhood B-Cell Precursor Acute Lymphoblastic Leukemia: From Biological Significance to Precision Medicine Opportunities. Biomedicines, 13(7), 1626. https://doi.org/10.3390/biomedicines13071626