

Beyond Muscle Weakness: Unraveling Endocrine and Metabolic Dysfunctions in Duchenne Muscular Dystrophy, a Narrative Review

 ,
,  ,
,  , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
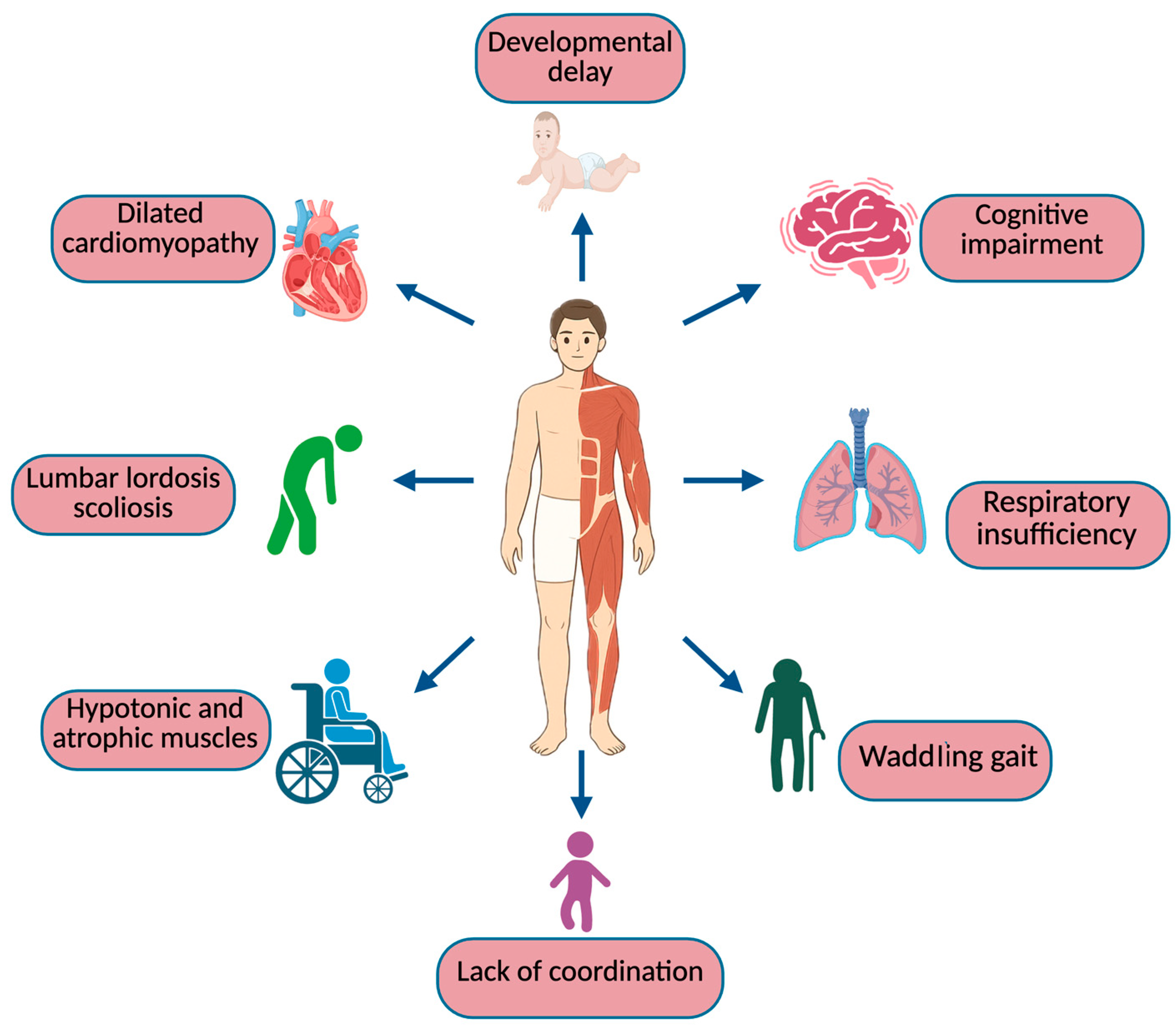
1. Introduction
2. Materials and Methods
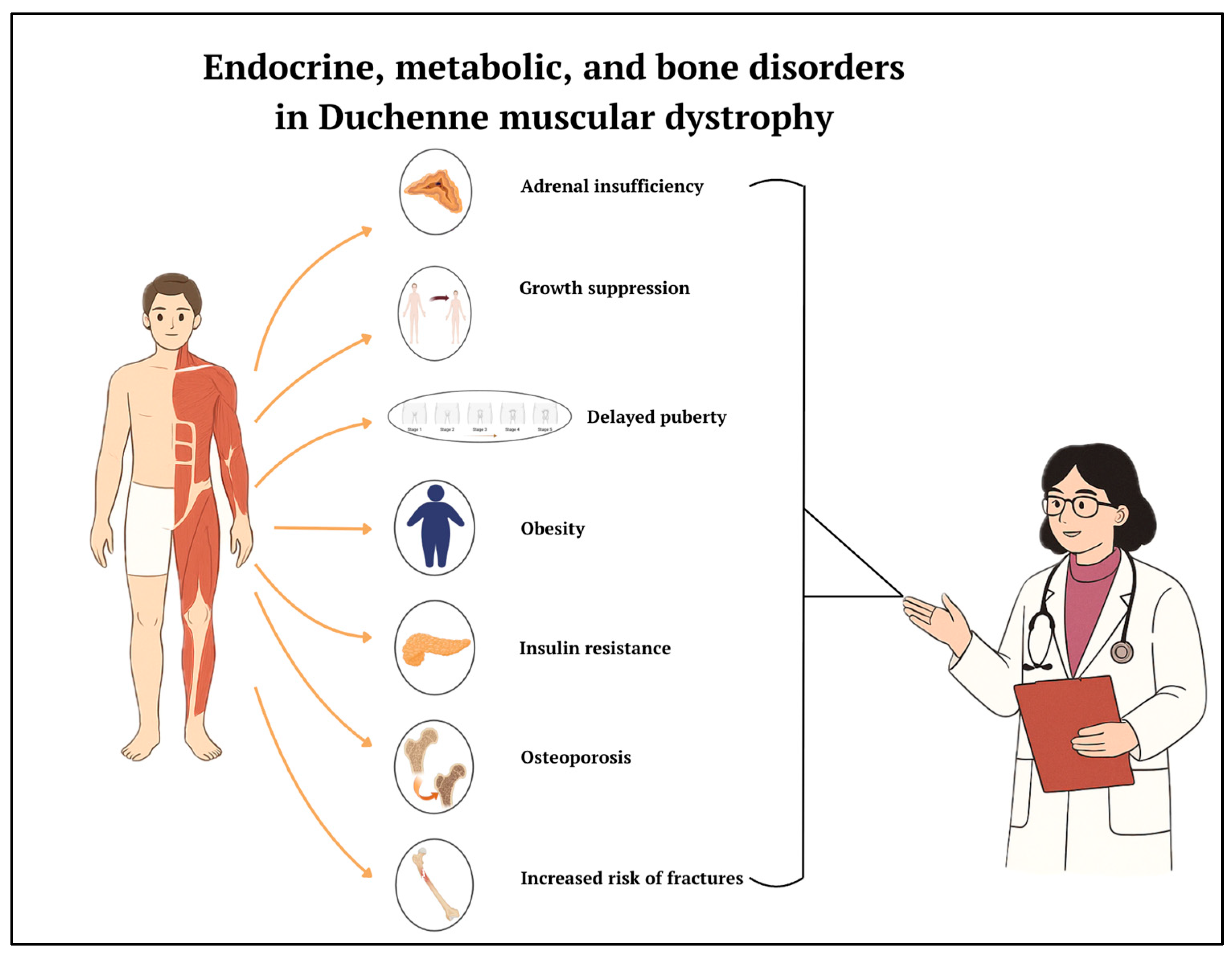
3. Results and Discussion
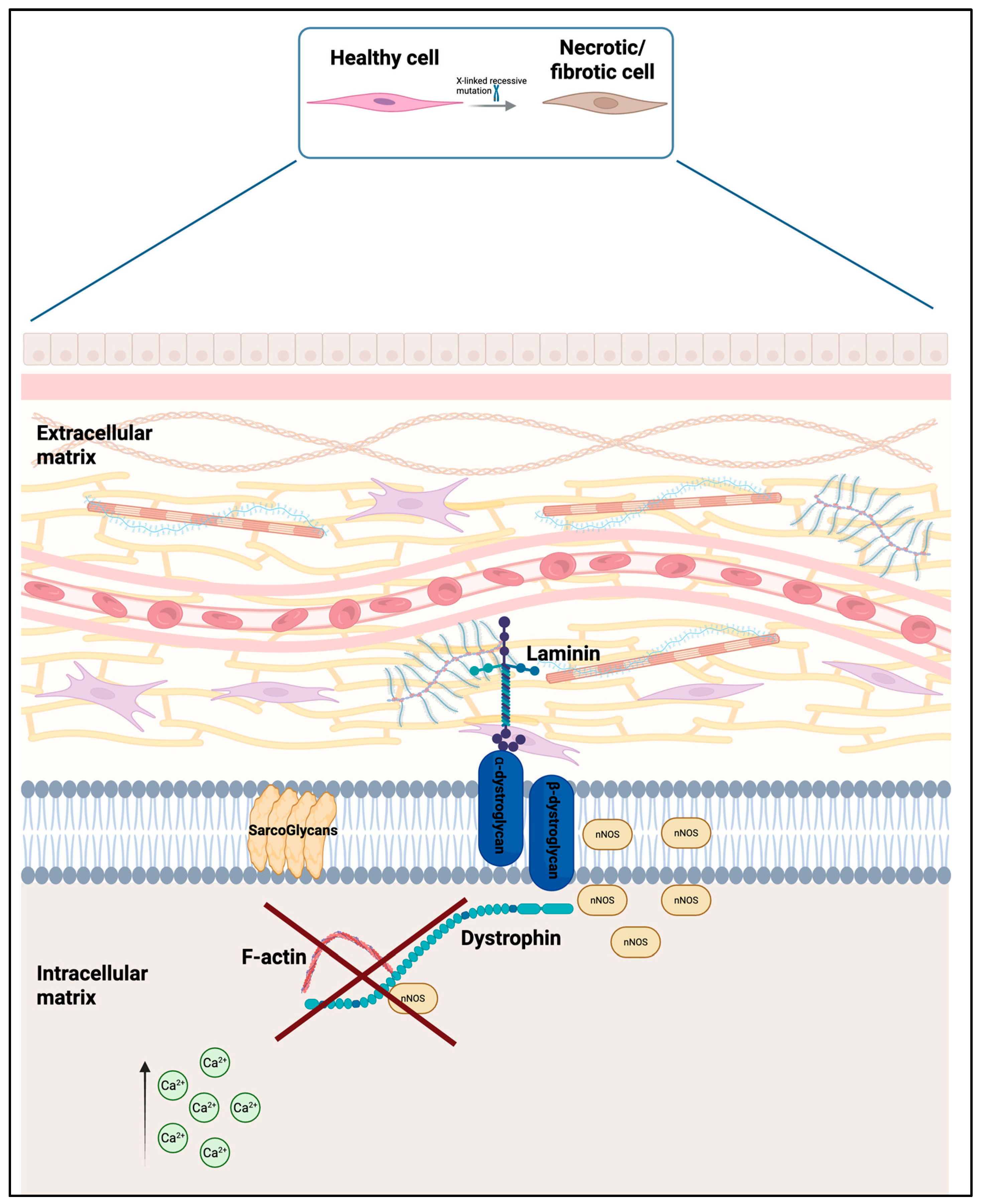
3.1. The Role of Glucocorticoids in Duchenne Muscular Dystrophy (DMD)
3.2. Adrenal Insufficiency Secondary to Chronic Glucocorticoid Treatment and Management
3.3. Effects of Glucorticoids on GH and Bone Growth
3.4. Causes of Short Stature in Duchenne Muscular Dystrophy (DMD) and Treatment
3.5. Pubertal Delay and Treatment
3.6. Obesity, Metabolic Derangements, and Management
3.7. Bone Disorders and Their Management in DMD
3.7.1. Skeletal Abnormalities and Fracture Risk in Duchenne Muscular Dystrophy (DMD)
3.7.2. New Drugs for Bone Health in DMD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Messina, S.; Trifirò, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; De Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; Al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, V.; Pavlakis, S. Duchenne Muscular Dystrophy. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Orso, M.; Migliore, A.; Polistena, B.; Russo, E.; Gatto, F.; Monterubbianesi, M.; d’Angela, D.; Spandonaro, F.; Pane, M. Duchenne muscular dystrophy in Italy: A systematic review of epidemiology, quality of life, treatment adherence, and economic impact. PLoS ONE 2023, 18, e0287774. [Google Scholar] [CrossRef]
- Akat, A.; Karaöz, E. Cell Therapy Strategies on Duchenne Muscular Dystrophy: A Systematic Review of Clinical Applications. Stem Cell Rev. Rep. 2024, 20, 138–158. [Google Scholar] [CrossRef]
- Caballero-Sánchez, N.; Alonso-Alonso, S.; Nagy, L. Regenerative inflammation: When immune cells help to re-build tissues. FEBS J. 2024, 291, 1597–1614. [Google Scholar] [CrossRef]
- Taglietti, V.; Kefi, K.; Rivera, L.; Bergiers, O.; Cardone, N.; Coulpier, F.; Gioftsidi, S.; Drayton-Libotte, B.; Hou, C.; Authier, F.-J.; et al. Thyroid-stimulating hormone receptor signaling restores skeletal muscle stem cell regeneration in rats with muscular dystrophy. Sci. Transl. Med. 2023, 15, eadd5275. [Google Scholar] [CrossRef]
- Ricotti, V.; Ridout, D.A.; Scott, E.; Quinlivan, R.; Robb, S.A.; Manzur, A.Y.; Muntoni, F.; Manzur, A.; Muntoni, F.; Robb, S.; et al. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 2013, 84, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Porter, K.; Donovan, J.M.; Scavina, M.T.; Armstrong, N.; Denger, B.; Hasham, S.; Peay, H. A Mixed-Method Study Exploring Patient-Experienced and Caregiver-Reported Benefits and Side Effects of Corticosteroid Use in Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2023, 10, 593–613. [Google Scholar] [CrossRef]
- Landfeldt, E.; Alemán, A.; Abner, S.; Zhang, R.; Werner, C.; Tomazos, I.; Lochmüller, H.; Quinlivan, R.M.; Wahbi, K. Predictors of cardiac disease in duchenne muscular dystrophy: A systematic review and evidence grading. Orphanet J. Rare Dis. 2024, 19, 359. [Google Scholar] [CrossRef]
- Pascual-Morena, C.; Lucerón-Lucas-Torres, M.; Martínez-García, I.; Rodríguez-Gutiérrez, E.; Patiño-Cardona, S.; Sequí-Domínguez, I. Efficacy and Safety of Vamorolone in Duchenne Muscular Dystrophy: A Systematic Review. Pediatr. Drugs 2024, 26, 695–707. [Google Scholar] [CrossRef]
- Kinnett, K.; Noritz, G. The PJ Nicholoff Steroid Protocol for Duchenne and Becker Muscular Dystrophy and Adrenal Suppression. PLoS Curr. 2017, 9. [Google Scholar] [CrossRef]
- Weber, D.R.; Hadjiyannakis, S.; McMillan, H.J.; Noritz, G.; Ward, L.M. Obesity and Endocrine Management of the Patient With Duchenne Muscular Dystrophy. Pediatrics 2018, 142, S43–S52. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.M.; Collins, J.; Rose, S.R.; Woo, J.G.; Sucharew, H.; Sawnani, H.; Hor, K.N.; Cripe, L.H.; Wong, B.L. Growth hormone treatment in boys with Duchenne muscular dystrophy and glucocorticoid-induced growth failure. Neuromuscul. Disord. 2012, 22, 1046–1056. [Google Scholar] [CrossRef]
- Matsumoto, M.; Awano, H.; Lee, T.; Takeshima, Y.; Matsuo, M.; Iijima, K. Patients with Duchenne muscular dystrophy are significantly shorter than those with Becker muscular dystrophy, with the higher incidence of short stature in Dp71 mutated subgroup. Neuromuscul. Disord. 2017, 27, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Stimpson, G.; Raquq, S.; Chesshyre, M.; Fewtrell, M.; Ridout, D.; Sarkozy, A.; Manzur, A.; Ayyar Gupta, V.; De Amicis, R.; Muntoni, F.; et al. Growth pattern trajectories in boys with Duchenne muscular dystrophy. Orphanet J. Rare Dis. 2022, 17, 20. [Google Scholar] [CrossRef]
- Lavi, E.; Cohen, A.; Libdeh, A.A.; Tsabari, R.; Zangen, D.; Dor, T. Growth hormone therapy for children with Duchenne muscular dystrophy and glucocorticoid induced short stature. Growth Horm. IGF Res. 2023, 72–73, 101558. [Google Scholar] [CrossRef]
- Kao, K.-T.; Joseph, S.; Capaldi, N.; Brown, S.; Di Marco, M.; Dunne, J.; Horrocks, I.; Shepherd, S.; Ahmed, S.F.; Wong, S.C. Skeletal disproportion in glucocorticoid-treated boys with Duchenne muscular dystrophy. Eur. J. Pediatr. 2019, 178, 633–640. [Google Scholar] [CrossRef]
- Cirillo, F.; Lazzeroni, P.; Sartori, C.; Street, M. Inflammatory Diseases and Growth: Effects on the GH–IGF Axis and on Growth Plate. Int. J. Mol. Sci. 2017, 18, 1878. [Google Scholar] [CrossRef] [PubMed]
- Boccanegra, B.; Cappellari, O.; Mantuano, P.; Trisciuzzi, D.; Mele, A.; Tulimiero, L.; De Bellis, M.; Cirmi, S.; Sanarica, F.; Cerchiara, A.G.; et al. Growth hormone secretagogues modulate inflammation and fibrosis in mdx mouse model of Duchenne muscular dystrophy. Front. Immunol. 2023, 14, 1119888. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Radley, H.G.; Gebski, B.L.; Bogoyevitch, M.A.; Shavlakadze, T. Implications of cross-talk between tumour necrosis factor and insulin-like growth factor-1 signalling in skeletal muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 846–851. [Google Scholar] [CrossRef] [PubMed]
- McCarrison, S.; Denker, M.; Dunne, J.; Horrocks, I.; McNeilly, J.; Joseph, S.; Wong, S.C. Frequency of Delayed Puberty in Boys with Contemporary Management of Duchenne Muscular Dystrophy. J. Clin. Res. Pediatr. Endocrinol. 2024, 16, 458–465. [Google Scholar] [CrossRef]
- Lee, S.L.-K.; Lim, A.; Munns, C.; Simm, P.J.; Zacharin, M. Effect of Testosterone Treatment for Delayed Puberty in Duchenne Muscular Dystrophy. Horm. Res. Paediatr. 2020, 93, 108–118. [Google Scholar] [CrossRef]
- Wood, C.L.; Page, J.; Foggin, J.; Guglieri, M.; Straub, V.; Cheetham, T.D. The impact of testosterone therapy on quality of life in adolescents with Duchenne muscular dystrophy. Neuromuscul. Disord. 2021, 31, 1259–1265. [Google Scholar] [CrossRef]
- Sodero, G.; Cipolla, C.; Rigante, D.; Arzilli, F.; Mercuri, E.M. Pubertal induction therapy in pediatric patients with Duchenne muscular dystrophy. J. Pediatr. Endocrinol. Metab. 2025. [Google Scholar] [CrossRef]
- Manzur, A.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2004, 2004, CD003725. [Google Scholar] [CrossRef]
- Billich, N.; Adams, J.; Carroll, K.; Truby, H.; Evans, M.; Ryan, M.M.; Davidson, Z.E. The Relationship between Obesity and Clinical Outcomes in Young People with Duchenne Muscular Dystrophy. Nutrients 2022, 14, 3304. [Google Scholar] [CrossRef]
- Davidson, Z.E.; Ryan, M.M.; Kornberg, A.J.; Sinclair, K.; Cairns, A.; Walker, K.Z.; Truby, H. Observations of body mass index in Duchenne muscular dystrophy: A longitudinal study. Eur. J. Clin. Nutr. 2014, 68, 892–897. [Google Scholar] [CrossRef]
- Wernio, E.; Wasilewska, E.; Czaja-Stolc, S.; Śledzińska, K.; Wierzba, J.; Szlagatys-Sidorkiewicz, A.; Małgorzewicz, S. Nutritional Issues among Children with Duchenne Muscular Dystrophy—Incidence of Deficiency and Excess Body Mass. Nutrients 2024, 16, 2143. [Google Scholar] [CrossRef] [PubMed]
- Krishna, L.; Prashant, A.; Kumar, Y.H.; Paneyala, S.; Patil, S.J.; Ramachandra, S.C.; Vishwanath, P. Molecular and Biochemical Therapeutic Strategies for Duchenne Muscular Dystrophy. Neurol. Int. 2024, 16, 731–760. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Cai, J.; Tan, J. Adverse reactions of drugs specifically used for treatment of SARS-CoV-2 infection. Med. Clin. 2021, 156, 576. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cruz, M.; Sanchez, R.; Escobar, R.E.; Cruz-Guzmán Odel, R.; López-Alarcón, M.; García, M.B.; Coral-Vázquez, R.; Matute, G.; Wong, A.C.V. Evidence of Insulin Resistance and Other Metabolic Alterations in Boys with Duchenne or Becker Muscular Dystrophy. Int. J. Endocrinol. 2015, 2015, 867273. [Google Scholar] [CrossRef]
- Ward, L.M.; Weber, D.R. Growth, pubertal development, and skeletal health in boys with Duchenne Muscular Dystrophy. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 39. [Google Scholar] [CrossRef]
- Bell, J.M.; Shields, M.D.; Watters, J.; Hamilton, A.; Beringer, T.; Elliott, M.; Quinlivan, R.; Tirupathi, S.; Blackwood, B. Interventions to prevent and treat corticosteroid-induced osteoporosis and prevent osteoporotic fractures in Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2017, 2017, CD010899. [Google Scholar] [CrossRef]
- Loscalzo, E.; See, J.; Bharill, S.; Yousefzadeh, N.; Gough, E.; Wu, M.; Crane, J.L. Growth hormone and testosterone delay vertebral fractures in boys with muscular dystrophy on chronic glucocorticoids. Osteoporos. Int. 2024, 35, 327–338. [Google Scholar] [CrossRef]
- Tung, J.Y.-L.; Lam, T.-P.; Chan, S.H.-S. Bone microarchitectural alterations in boys with Duchenne muscular dystrophy on long-term glucocorticoid treatment. J. Bone Miner. Metab. 2021, 39, 606–611. [Google Scholar] [CrossRef]
- Hurley-Novatny, A.; Chang, D.; Murakami, K.; Wang, L.; Li, H. Poor bone health in Duchenne muscular dystrophy: A multifactorial problem beyond corticosteroids and loss of ambulation. Front. Endocrinol. 2024, 15, 1398050. [Google Scholar] [CrossRef]
- Atilano-Miguel, S.; Barbosa-Cortés, L.; Ortiz-Muñiz, R. Duchenne muscular dystrophy: RANK/RANKL/OPG (receptor activator of nuclear factor-kB/RANK ligand/osteoprotegerin) system and glucocorticoids. Bol. Med. Hosp. Infant. Mex. 2022, 79, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.M.; Hadjiyannakis, S.; McMillan, H.J.; Noritz, G.; Weber, D.R. Bone Health and Osteoporosis Management of the Patient with Duchenne Muscular Dystrophy. Pediatrics 2018, 142, S34–S42. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moretti, A.; Liguori, S.; Paoletta, M.; Gimigliano, F.; Iolascon, G. Effectiveness of Neridronate in the Management of Bone Loss in Patients with Duchenne Muscular Dystrophy: Results from a Pilot Study. Adv. Ther. 2022, 39, 3308–3315. [Google Scholar] [CrossRef]
- Landfeldt, E.; Phung, K.; Zaman, F.; Åström, E.; Abner, S.; Lochmüller, H.; Sejersen, T.; Ward, L.M. Bisphosphonates in Glucocorticoid-Treated Patients with Duchenne Muscular Dystrophy: A Systematic Review and Grading of the Evidence. Neurology 2024, 102, e207948. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, G.; Biasucci, G.; Bertolini, L.; Patianna, V.; Petraroli, M.; Pilloni, S.; Esposito, S.; Street, M.E. Osteoporosis and Bone Fragility in Children: Diagnostic and Treatment Strategies. J. Clin. Med. 2024, 13, 4951. [Google Scholar] [CrossRef]
- Jayash, S.N.; Hamoudi, D.; Stephen, L.A.; Argaw, A.; Huesa, C.; Joseph, S.; Wong, S.C.; Frenette, J.; Farquharson, C. Anti-RANKL Therapy Prevents Glucocorticoid-Induced Bone Loss and Promotes Muscle Function in a Mouse Model of Duchenne Muscular Dystrophy. Calcif. Tissue Int. 2023, 113, 449–468. [Google Scholar] [CrossRef] [PubMed]
- Nasomyont, N.; Keefe, C.; Tian, C.; Hornung, L.; Khoury, J.; Tilden, J.C.; Hochwalt, P.; Jackson, E.; Rybalsky, I.; Wong, B.L.; et al. Safety and efficacy of teriparatide treatment for severe osteoporosis in patients with Duchenne muscular dystrophy. Osteoporos. Int. 2020, 31, 2449–2459. [Google Scholar] [CrossRef]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; Roux-Biejat, P.; Napoli, A.; Vantaggiato, C.; Cervia, D.; et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne Muscular Dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef]
- Saad, F.A.; Saad, J.F.; Siciliano, G.; Merlini, L.; Angelini, C. Duchenne Muscular Dystrophy Gene Therapy. Curr. Gene Ther. 2024, 24, 17–28. [Google Scholar] [CrossRef]
- Happi Mbakam, C.; Tremblay, J.P. Gene therapy for Duchenne muscular dystrophy: An update on the latest clinical developments. Expert Rev. Neurother. 2023, 23, 905–920. [Google Scholar] [CrossRef]
- Patterson, G.; Conner, H.; Groneman, M.; Blavo, C.; Parmar, M.S. Duchenne muscular dystrophy: Current treatment and emerging exon skipping and gene therapy approach. Eur. J. Pharmacol. 2023, 947, 175675. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Bragg, L.; Rossi, M.; Proietti, D.; Perani, L.; Bacigaluppi, M.; Tonlorenzi, R.; Sibanda, T.; Caffarini, M.; Talapatra, A.; et al. Cell-mediated exon skipping normalizes dystrophin expression and muscle function in a new mouse model of Duchenne Muscular Dystrophy. EMBO Mol. Med. 2024, 16, 927–944. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, E.S.; Mendell, J.R. Evolving Therapeutic Options for the Treatment of Duchenne Muscular Dystrophy. Neurotherapeutics 2023, 20, 1669–1681. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannalire, G.; Biasucci, G.; Sambati, V.; Toschetti, T.; Bellani, A.M.; Shulhai, A.-M.; Casadei, F.; Di Bari, E.R.; Ferraboschi, F.; Parenti, C.; et al. Beyond Muscle Weakness: Unraveling Endocrine and Metabolic Dysfunctions in Duchenne Muscular Dystrophy, a Narrative Review. Biomedicines 2025, 13, 1613. https://doi.org/10.3390/biomedicines13071613
Cannalire G, Biasucci G, Sambati V, Toschetti T, Bellani AM, Shulhai A-M, Casadei F, Di Bari ER, Ferraboschi F, Parenti C, et al. Beyond Muscle Weakness: Unraveling Endocrine and Metabolic Dysfunctions in Duchenne Muscular Dystrophy, a Narrative Review. Biomedicines. 2025; 13(7):1613. https://doi.org/10.3390/biomedicines13071613
Chicago/Turabian StyleCannalire, Giuseppe, Giacomo Biasucci, Vanessa Sambati, Tommaso Toschetti, Arianna Maria Bellani, Anna-Mariia Shulhai, Federica Casadei, Erika Rita Di Bari, Francesca Ferraboschi, Cecilia Parenti, and et al. 2025. "Beyond Muscle Weakness: Unraveling Endocrine and Metabolic Dysfunctions in Duchenne Muscular Dystrophy, a Narrative Review" Biomedicines 13, no. 7: 1613. https://doi.org/10.3390/biomedicines13071613
APA StyleCannalire, G., Biasucci, G., Sambati, V., Toschetti, T., Bellani, A. M., Shulhai, A.-M., Casadei, F., Di Bari, E. R., Ferraboschi, F., Parenti, C., Pera, M. C., Esposito, S., & Street, M. E. (2025). Beyond Muscle Weakness: Unraveling Endocrine and Metabolic Dysfunctions in Duchenne Muscular Dystrophy, a Narrative Review. Biomedicines, 13(7), 1613. https://doi.org/10.3390/biomedicines13071613