
GLP-1 and Its Role in Glycogen Production: A Narrative Review

 ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Clinical Disorders and Physiological Pathways Impacted by GLP-1
2.1. T2DM and Obesity
2.1.1. Introduction to T2DM and Obesity
2.1.2. Current Pharmacological Treatments for Obesity and T2DM and Their Mechanism of Action
2.1.3. Current Pharmacological GLP-1RAs with a Comparison of Their Efficacy Pharmacokinetics/Dynamics and Other Relevant Comparisons
2.2. Blood Glucose Homeostasis and Hepatic Glycogen Metabolism
2.3. Energy Balance and Thermogenesis
3. GLP-1 and Its Role in Glycogen Metabolism and Energy Balance
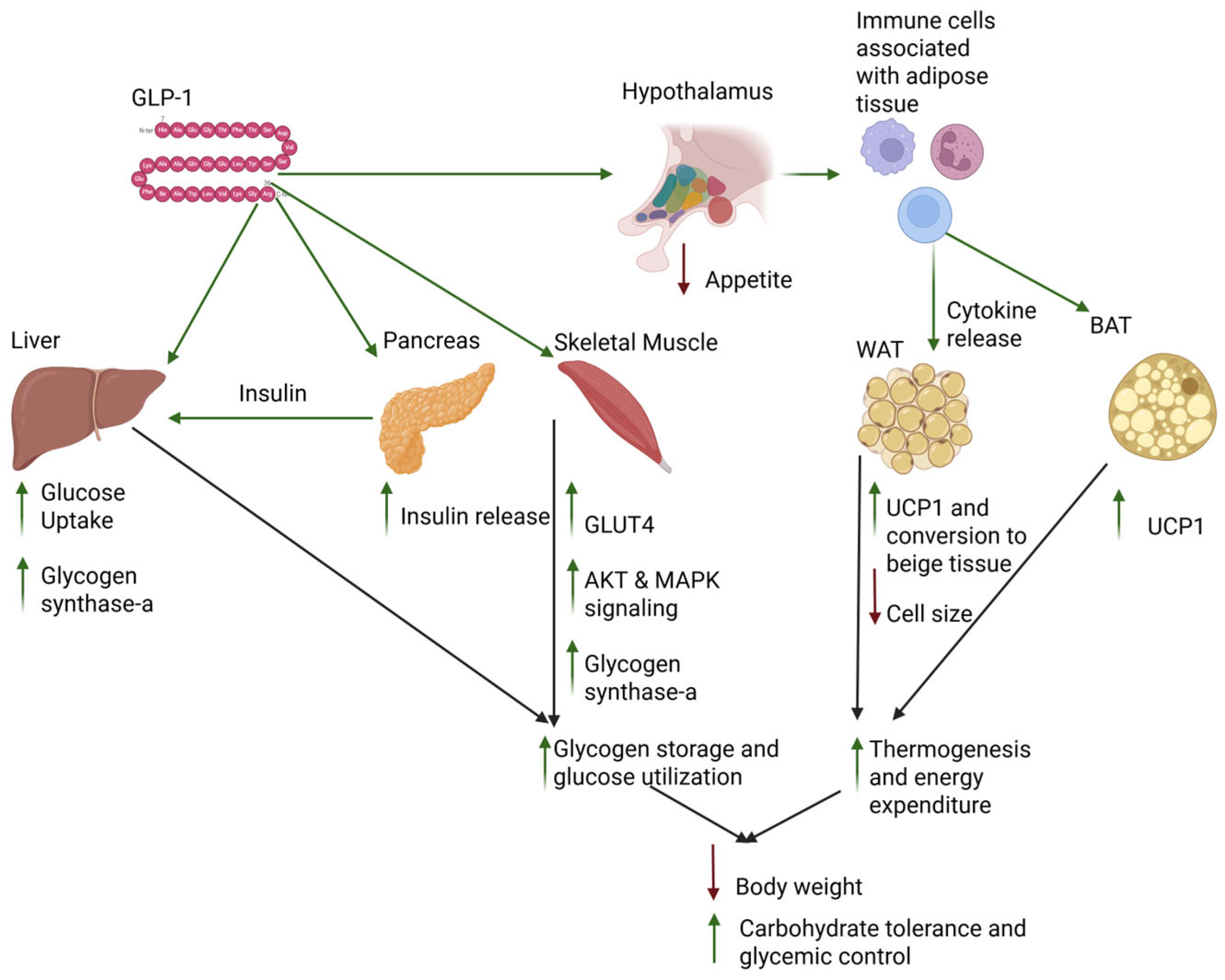
3.1. Introduction to GLP-1 Physiology
3.2. Tissue-Specific Effects of GLP-1 Activity
3.2.1. GLP-1 Effects on the Pancreas
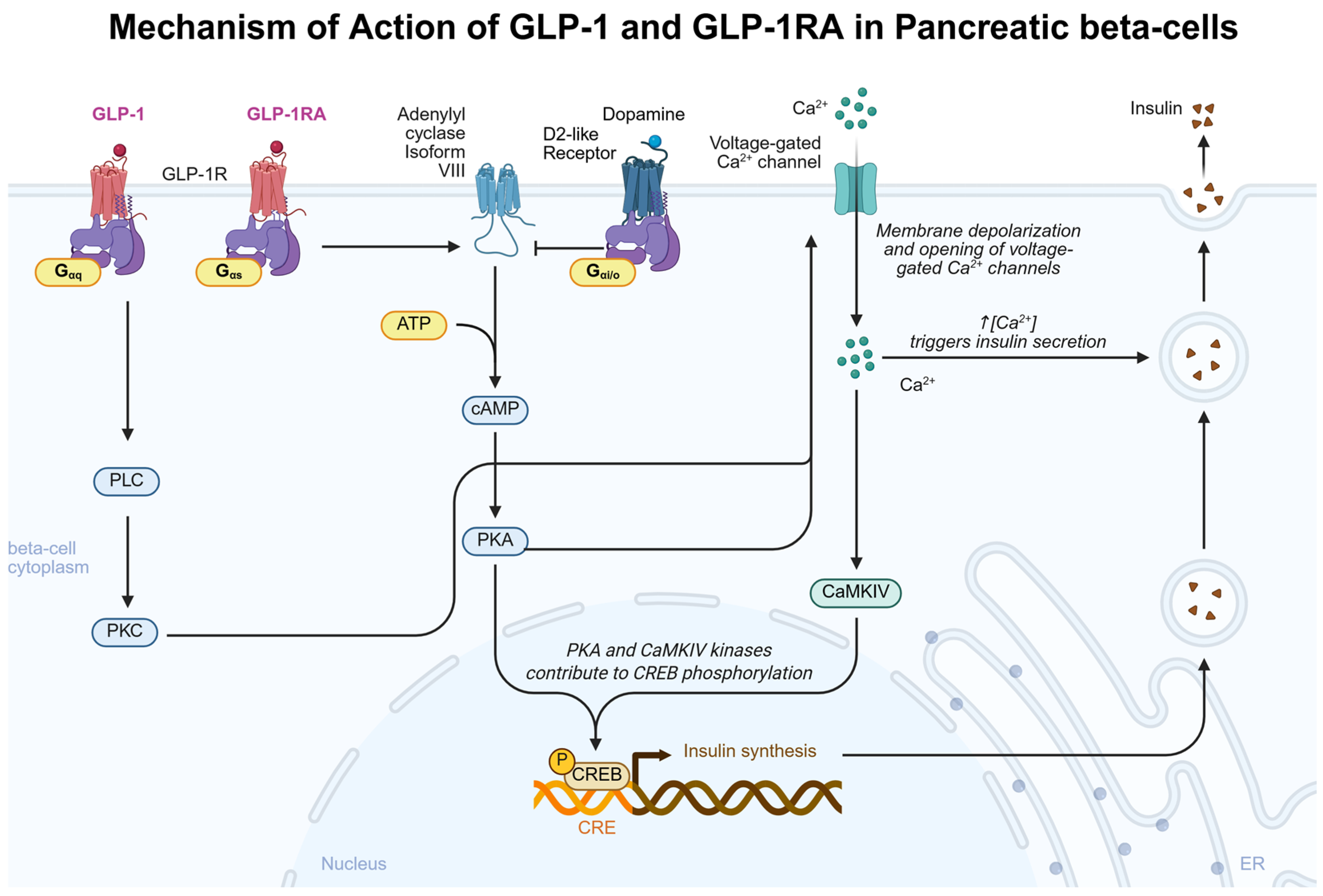

{kind=link}
{kind=link}
{kind=link}
| GLP-1 Intervention | Species | Sex | Regimen | Dose | Administration Route | Outcomes | Reference |
|---|---|---|---|---|---|---|---|
| GLP-1 (7–37) | Rat pancreatic islet cells | N/A | 1-day cell culture incubation | 0.5 µM | N/A | ↑ cAMP ↑ Insulin mRNA ↑ Insulin release | [69] |
| GLP-1 | Rat pancreatic beta cells | Male | 15 min cell culture incubation | 10 nM | N/A | ↑ cAMP ↑ Type VIII adenylate cyclase | [70] |
| GLP-1 | Mice pancreatic islet and beta cells | Female | N/A | 1 µM | N/A | ↑ Insulin ↑ Ca2+ channel activity | [75] |
3.2.2. GLP-1 Effects on the Liver
| GLP-1 Intervention | Species | Sex | Regimen | Dose | Administration Route | Outcomes | Reference |
|---|---|---|---|---|---|---|---|
| GLP-1 | Rat liver cells | Male | N/A | 1nM | GLP-1 added to cell cultures | ↑ GS-a | [84] |
| GLP-1R homozygous knockout | Mice | Male | N/A | N/A | N/A | ↓ Hepatic glycogen content | [85] |
| GLP-1 | Mongrel Dogs | Both | Continuous during experiment | 10 and 20 pmol·kg−1·min−1 | GLP-1 intraportal infusion | ↑ Hepatic glucose uptake | [80] |
| GLP-1 | Mongrel Dogs | Both | Continuous during experiment | 7.5 pmol·kg−1·min−1 | GLP-1-(7–36) peripheral vein infusion | ↑ Hepatic glucose uptake independent of insulin secretion | [87] |
| GLP-1 | PVH zone rat liver cells | Male | Culture incubation | 1000 nM | GLP-1 | Inhibition of 0.1 nM glucagon-induced glycogenolysis | [93] |
3.2.3. GLP-1 Effects on Skeletal Muscle
| GLP-1 Intervention | Species | Sex | Regimen | Dose | Administration Route | Outcomes | Reference |
|---|---|---|---|---|---|---|---|
| Exendin-4 | C2C12 mouse skeletal myoblast cells | N/A | 5 days | 0.5 μM | N/A | ↑ glycogen ↑ membrane Glut-4 | [96] |
| GLP-1 overexpression | Mice | Male | N/A | N/A | GLP-1-AAV | ↑ gene expression of chemokine, AMPK, PI3K/AKT, PLD, cAMP ↑ pAMPK | [96] |
| GLP-1 | Skeletal muscle satellite cells | Male | Cell culture incubation | 100 nM | N/A | ↑ PI3K/AKT | [101] |
| Exendin-4 | Streptozotocin (STZ)-induced rats with diabetes | Male | Culture incubation | 10−9 M Ex-4 | N/A | ↑ PI3K, p70s6K, MAPKs phosphorylation No effect on AKT phosphorylation or glycogen synthase | [102] |
| GLP-1 | Rat skeletal muscle strips | N/A | 10 min culture incubation | 10−10 M | N/A | ↑ PI3K/AKT, p70s6K, p44/42 ↑ glycogen synthase-a activity | [105] |
| GLP-1 | Cultured myocytes from humans with obesity | N/A | 3 min culture incubation | 10−9 M | N/A | ↑ AKT, p70s6K, p44/42 phosphorylation ↑ glycogen synthase activity | [106] |
3.2.4. GLP-1 Effects on Adipose Tissue
| GLP-1 Intervention | Species | Sex | Regimen | Dose | Administration Route | Outcomes | Reference |
|---|---|---|---|---|---|---|---|
| GLP-1 | Mice | Male | 4 days | 0.75 nmol/day | Intracerebroventricular injection | ↑ iBAT thermogenesis | [108] |
| GLP-1 | Rats | Male | Single dose | 0.5 μg | Dorsomedial hypothalamic injection | ↑ iBAT thermogenesis | [109] |
| Liraglutide | Mice | Male | Single dose | 3 μg | Intracerebroventricular (ICV) | ↑ iBAT thermogenesis ↑ UCP1, UCP3, ADRB1, FGF21, PRDM16 ↑ White adipose tissue UCP-1 and PRDM16 | [110] |
| Liraglutide | Mice | Male | 5 days following 6–8 weeks of high-fat diet | 50 μg/kg | Intraperitoneally | ↑ iNKT cell count and IL-10 production ↑ White adipose tissue UCP-1, FGF21, adiponectin PGC1a, and Cidea | [111] |
| Liraglutide | Mice | Male | 2 and 4 weeks | 0.2 mg/kg | Intraperitoneal injection | ↑ UCP1 ↑ AMPK phosphorylation ↓ Fat mass | [112] |
4. Additional Biological Processes Impacted by GLP-1 That Impact Glycogen Metabolism and Energy Homeostasis
4.1. GLP-1 Effects on Gastric Emptying
4.2. GLP-1 Effects on Appetite Regulation
4.3. Physiological Pathways That Impact GLP-1 Activity
4.3.1. TGR5 and Its Physiologic Effects Relevant to GLP-1 and Glycogen Metabolism
4.3.2. PASK Signaling Relevant to GLP-1 and Glycogen Metabolism
4.3.3. Microbiome Relevance to GLP-1 Activity
5. Summary
Funding
Conflicts of Interest
Abbreviations
| GLP-1 | Glucagon-like peptide 1 |
| GLP-1R | Glucagon-like peptide 1 receptor |
| GLP-1RA | Glucagon-like peptide 1 receptor agonist |
| GIP | Gastric inhibitory polypeptide |
| GIP-R | Gastric inhibitory polypeptide receptor |
| T2DM | Type 2 diabetes mellitus |
| BMI | Body mass index |
| DNA | Deoxyribonucleic acid |
| AKT | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| BAT | Brown adipose tissue |
| PASK | PAS kinase |
| UCP1 | Uncoupling protein 1 |
| TGR5 | Takeda G protein-coupled receptor 5 |
| GLUT-4 | Glucose transporter 4 |
| TNF-α | Tumor necrosis factor-alpha |
| GSK-3 | Glycogen synthase kinase-3 |
| IL-6 | Interleukin-6 |
| G6P | Glucose 6-phosphate |
| iNKT | Invariant natural killer T-cell |
| NTS | Nucleus tractus solitarius |
| UDP-ase | UDP-glucose pyrophosphorylase |
| PP-1 | Protein phosphatase-1 |
| PI3K | Phosphoinositide 3-Kinase |
| cAMP | Cyclic adenosine monophosphate |
| LSG | Laparoscopic sleeve gastrectomy |
| ARC | Arcuate nucleus |
| Exn-4 | Exendin-4 |
| PYY | Peptide YY |
| IFN | Intestinofugal neuron |
| HFD | High-fat diet |
| POMC | Pro-opiomelanocortin |
| CART | Cocaine- and amphetamine-regulated transcript |
| NPY | Neuropeptide Y |
| AgRP | Agouti-related peptide |
| GYS | Glycogen synthase |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| SGLT2 | Sodium-glucose-co-transporter-2 |
| SOD | Copper–zinc superoxide dismutase |
| PPargc1a | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| NASH | Non-alcoholic hepatic steatosis |
| CKD | Chronic kidney disease |
| GFR | Glomerular filtration rate |
References
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Muzurovic, E.M.; Volcansek, S.; Tomsic, K.Z.; Janez, A.; Mikhailidis, D.P.; Rizzo, M.; Mantzoros, C.S. Glucagon-Like Peptide-1 Receptor Agonists and Dual Glucose-Dependent Insulinotropic Polypeptide/Glucagon-Like Peptide-1 Receptor Agonists in the Treatment of Obesity/Metabolic Syndrome, Prediabetes/Diabetes and Non-Alcoholic Fatty Liver Disease-Current Evidence. J. Cardiovasc. Pharmacol. Ther. 2022, 27, 10742484221146371. [Google Scholar] [CrossRef] [PubMed]
- Grill, H.J. A Role for GLP-1 in Treating Hyperphagia and Obesity. Endocrinology 2020, 161, bqaa093. [Google Scholar] [CrossRef]
- Valverde, I.; Morales, M.; Clemente, F.; Lopez-Delgado, M.I.; Delgado, E.; Perea, A.; Villanueva-Penacarrillo, M.L. Glucagon-like peptide 1: A potent glycogenic hormone. FEBS Lett. 1994, 349, 313–316. [Google Scholar] [CrossRef]
- Popoviciu, M.S.; Paduraru, L.; Yahya, G.; Metwally, K.; Cavalu, S. Emerging Role of GLP-1 Agonists in Obesity: A Comprehensive Review of Randomised Controlled Trials. Int. J. Mol. Sci. 2023, 24, 10449. [Google Scholar] [CrossRef]
- Kuhre, R.E.; Deacon, C.F.; Holst, J.J.; Petersen, N. What Is an L-Cell and How Do We Study the Secretory Mechanisms of the L-Cell? Front. Endocrinol. 2021, 12, 694284. [Google Scholar] [CrossRef]
- Scrocchi, L.A.; Brown, T.J.; MaClusky, N.; Brubaker, P.L.; Auerbach, A.B.; Joyner, A.L.; Drucker, D.J. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat. Med. 1996, 2, 1254–1258. [Google Scholar] [CrossRef]
- Fritsche, A.; Stefan, N.; Hardt, E.; Haring, H.; Stumvoll, M. Characterisation of beta-cell dysfunction of impaired glucose tolerance: Evidence for impairment of incretin-induced insulin secretion. Diabetologia 2000, 43, 852–858. [Google Scholar] [CrossRef]
- Shao, S.; Zhang, X.; Xu, Q.; Pan, R.; Chen, Y. Emerging roles of Glucagon like peptide-1 in the management of autoimmune diseases and diabetes-associated comorbidities. Pharmacol. Ther. 2022, 239, 108270. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, M.; Jiang, D.; Su, Q.; Shi, J. The role of inflammation in autoimmune disease: A therapeutic target. Front. Immunol. 2023, 14, 1267091. [Google Scholar] [CrossRef] [PubMed]
- Maric-Bilkan, C. Obesity and diabetic kidney disease. Med. Clin. N. Am. 2013, 97, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Abasheva, D.; Ortiz, A.; Fernandez-Fernandez, B. GLP-1 receptor agonists in patients with chronic kidney disease and either overweight or obesity. Clin. Kidney J. 2024, 17, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Ferhatbegovic, L.; Mrsic, D.; Macic-Dzankovic, A. The benefits of GLP1 receptors in cardiovascular diseases. Front. Clin. Diabetes Healthc. 2023, 4, 1293926. [Google Scholar] [CrossRef]
- Du, H.; Meng, X.; Yao, Y.; Xu, J. The mechanism and efficacy of GLP-1 receptor agonists in the treatment of Alzheimer’s disease. Front. Endocrinol. 2022, 13, 1033479. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Q.; Qi, X.; Gurney, M.; Perry, G.; Volkow, N.D.; Davis, P.B.; Kaelber, D.C.; Xu, R. Associations of semaglutide with first-time diagnosis of Alzheimer’s disease in patients with type 2 diabetes: Target trial emulation using nationwide real-world data in the US. Alzheimer’s Dement. 2024, 20, 8661–8672. [Google Scholar] [CrossRef]
- Al-Mrabeh, A. Pathogenesis and remission of type 2 diabetes: What has the twin cycle hypothesis taught us? Cardiovasc. Endocrinol. Metab. 2020, 9, 132–142. [Google Scholar] [CrossRef]
- Grant, B.; Sandelson, M.; Agyemang-Prempeh, B.; Zalin, A. Managing obesity in people with type 2 diabetes. Clin. Med. 2021, 21, e327–e331. [Google Scholar] [CrossRef]
- Blum, K.; Thanos, P.K.; Gold, M.S. Dopamine and glucose, obesity, and reward deficiency syndrome. Front. Psychol. 2014, 5, 919. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martin, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Dludla, P.V.; Mabhida, S.E.; Ziqubu, K.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Hanser, S.; Basson, A.K.; Pheiffer, C.; Kengne, A.P. Pancreatic beta-cell dysfunction in type 2 diabetes: Implications of inflammation and oxidative stress. World J. Diabetes 2023, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Himanshu, D.; Ali, W.; Wamique, M. Type 2 diabetes mellitus: Pathogenesis and genetic diagnosis. J. Diabetes Metab. Disord. 2020, 19, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, P.; Weiskirchen, R. The Role of Obesity in Type 2 Diabetes Mellitus-An Overview. Int. J. Mol. Sci. 2024, 25, 1882. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, V.A. Defining and characterizing the progression of type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2), S151–S156. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Freeman, A.M.; Acevedo, L.A.; Pennings, N. Insulin Resistance; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Wondmkun, Y.T. Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications. Diabetes Metab. Syndr. Obes. 2020, 13, 3611–3616. [Google Scholar] [CrossRef]
- Saini, V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J. Diabetes 2010, 1, 68–75. [Google Scholar] [CrossRef]
- He, Q.; Bo, J.; Shen, R.; Li, Y.; Zhang, Y.; Zhang, J.; Yang, J.; Liu, Y. S1P Signaling Pathways in Pathogenesis of Type 2 Diabetes. J. Diabetes Res. 2021, 2021, 1341750. [Google Scholar] [CrossRef]
- Lopez-Pedrosa, J.M.; Camprubi-Robles, M.; Guzman-Rolo, G.; Lopez-Gonzalez, A.; Garcia-Almeida, J.M.; Sanz-Paris, A.; Rueda, R. The Vicious Cycle of Type 2 Diabetes Mellitus and Skeletal Muscle Atrophy: Clinical, Biochemical, and Nutritional Bases. Nutrients 2024, 16, 172. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, G.; Yu, Y.; Zhang, Z. Imaging of Sarcopenia in Type 2 Diabetes Mellitus. Clin. Interv. Aging 2024, 19, 141–151. [Google Scholar] [CrossRef]
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Bluher, M.; Tschop, M.H.; DiMarchi, R.D. Anti-obesity drug discovery: Advances and challenges. Nat. Rev. Drug Discov. 2022, 21, 201–223. [Google Scholar] [CrossRef] [PubMed]
- Heal, D.J.; Gosden, J.; Smith, S.L. Regulatory challenges for new drugs to treat obesity and comorbid metabolic disorders. Br. J. Clin. Pharmacol. 2009, 68, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Gudzune, K.A.; Kushner, R.F. Medications for Obesity: A Review. JAMA 2024, 332, 571–584. [Google Scholar] [CrossRef]
- Saunders, K.H.; Shukla, A.P.; Igel, L.I.; Kumar, R.B.; Aronne, L.J. Pharmacotherapy for Obesity. Endocrinol. Metab. Clin. North Am. 2016, 45, 521–538. [Google Scholar] [CrossRef]
- Greenway, F.L.; Whitehouse, M.J.; Guttadauria, M.; Anderson, J.W.; Atkinson, R.L.; Fujioka, K.; Gadde, K.M.; Gupta, A.K.; O’Neil, P.; Schumacher, D.; et al. Rational design of a combination medication for the treatment of obesity. Obesity 2009, 17, 30–39. [Google Scholar] [CrossRef]
- Rubino, D.M.; Greenway, F.L.; Khalid, U.; O’Neil, P.M.; Rosenstock, J.; Sorrig, R.; Wadden, T.A.; Wizert, A.; Garvey, W.T.; Investigators, S. Effect of Weekly Subcutaneous Semaglutide vs Daily Liraglutide on Body Weight in Adults With Overweight or Obesity Without Diabetes: The STEP 8 Randomized Clinical Trial. JAMA 2022, 327, 138–150. [Google Scholar] [CrossRef]
- Rodriguez, P.J.; Goodwin Cartwright, B.M.; Gratzl, S.; Brar, R.; Baker, C.; Gluckman, T.J.; Stucky, N.L. Semaglutide vs Tirzepatide for Weight Loss in Adults With Overweight or Obesity. JAMA Intern. Med. 2024, 184, 1056–1064. [Google Scholar] [CrossRef]
- Htike, Z.Z.; Zaccardi, F.; Papamargaritis, D.; Webb, D.R.; Khunti, K.; Davies, M.J. Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: A systematic review and mixed-treatment comparison analysis. Diabetes Obes. Metab. 2017, 19, 524–536. [Google Scholar] [CrossRef]
- Latif, W.; Lambrinos, K.J.; Patel, P.; Rodriguez, R. Compare and Contrast the Glucagon-Like Peptide-1 Receptor Agonists (GLP1RAs); StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes Metab. Res. Rev. 2019, 35, e3070. [Google Scholar] [CrossRef]
- Buse, J.B.; Rosenstock, J.; Sesti, G.; Schmidt, W.E.; Montanya, E.; Brett, J.H.; Zychma, M.; Blonde, L.; Group, L.-S. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: A 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet 2009, 374, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Buse, J.B.; Nauck, M.; Forst, T.; Sheu, W.H.; Shenouda, S.K.; Heilmann, C.R.; Hoogwerf, B.J.; Gao, A.; Boardman, M.K.; Fineman, M.; et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): A randomised, open-label study. Lancet 2013, 381, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ahmann, A.J.; Capehorn, M.; Charpentier, G.; Dotta, F.; Henkel, E.; Lingvay, I.; Holst, A.G.; Annett, M.P.; Aroda, V.R. Efficacy and Safety of Once-Weekly Semaglutide Versus Exenatide ER in Subjects With Type 2 Diabetes (SUSTAIN 3): A 56-Week, Open-Label, Randomized Clinical Trial. Diabetes Care 2018, 41, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Capehorn, M.S.; Catarig, A.M.; Furberg, J.K.; Janez, A.; Price, H.C.; Tadayon, S.; Verges, B.; Marre, M. Efficacy and safety of once-weekly semaglutide 1.0mg vs once-daily liraglutide 1.2mg as add-on to 1-3 oral antidiabetic drugs in subjects with type 2 diabetes (SUSTAIN 10). Diabetes Metab. 2020, 46, 100–109. [Google Scholar] [CrossRef]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Ronne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 2020, 5, e133429. [Google Scholar] [CrossRef]
- Horowitz, M.; Flint, A.; Jones, K.L.; Hindsberger, C.; Rasmussen, M.F.; Kapitza, C.; Doran, S.; Jax, T.; Zdravkovic, M.; Chapman, I.M. Effect of the once-daily human GLP-1 analogue liraglutide on appetite, energy intake, energy expenditure and gastric emptying in type 2 diabetes. Diabetes Res. Clin. Pract. 2012, 97, 258–266. [Google Scholar] [CrossRef]
- van Can, J.; Sloth, B.; Jensen, C.B.; Flint, A.; Blaak, E.E.; Saris, W.H. Effects of the once-daily GLP-1 analog liraglutide on gastric emptying, glycemic parameters, appetite and energy metabolism in obese, non-diabetic adults. Int. J. Obes. 2014, 38, 784–793. [Google Scholar] [CrossRef]
- Rhea, E.M.; Babin, A.; Thomas, P.; Omer, M.; Weaver, R.; Hansen, K.; Banks, W.A.; Talbot, K. Brain uptake pharmacokinetics of albiglutide, dulaglutide, tirzepatide, and DA5-CH in the search for new treatments of Alzheimer’s and Parkinson’s diseases. Tissue Barriers 2024, 12, 2292461. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Guemes, A.; Georgiou, P. Review of the role of the nervous system in glucose homoeostasis and future perspectives towards the management of diabetes. Bioelectron. Med. 2018, 4, 9. [Google Scholar] [CrossRef]
- Sohn, J.W.; Ho, W.K. Cellular and systemic mechanisms for glucose sensing and homeostasis. Pflugers Arch. 2020, 472, 1547–1561. [Google Scholar] [CrossRef] [PubMed]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Annu. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J. Glycogen and its metabolism. Curr. Mol. Med. 2002, 2, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef]
- Stefanovski, D.; Punjabi, N.M.; Boston, R.C.; Watanabe, R.M. Corrigendum: Insulin Action, Glucose Homeostasis and Free Fatty Acid Metabolism: Insights From a Novel Model. Front. Endocrinol. 2021, 12, 789390. [Google Scholar] [CrossRef]
- Berthoud, H.R. Homeostatic and non-homeostatic pathways involved in the control of food intake and energy balance. Obesity 2006, 14 (Suppl. S5), 197S–200S. [Google Scholar] [CrossRef]
- Hill, J.O.; Wyatt, H.R.; Peters, J.C. Energy balance and obesity. Circulation 2012, 126, 126–132. [Google Scholar] [CrossRef]
- Hopkins, M.; Blundell, J.E. Energy balance, body composition, sedentariness and appetite regulation: Pathways to obesity. Clin. Sci. 2016, 130, 1615–1628. [Google Scholar] [CrossRef]
- von Loeffelholz, C.; Birkenfeld, A.L. Non-Exercise Activity Thermogenesis in Human Energy Homeostasis; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Fenzl, A.; Kiefer, F.W. Brown adipose tissue and thermogenesis. Horm. Mol. Biol. Clin. Investig. 2014, 19, 25–37. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, J.; Dai, H.; Duan, Y.; An, Y.; Shi, L.; Lv, Y.; Li, H.; Wang, C.; Ma, Q.; et al. Brown and beige adipose tissue: A novel therapeutic strategy for obesity and type 2 diabetes mellitus. Adipocyte 2021, 10, 48–65. [Google Scholar] [CrossRef]
- Roesler, A.; Kazak, L. UCP1-independent thermogenesis. Biochem. J. 2020, 477, 709–725. [Google Scholar] [CrossRef]
- Hropot, T.; Herman, R.; Janez, A.; Lezaic, L.; Jensterle, M. Brown Adipose Tissue: A New Potential Target for Glucagon-like Peptide 1 Receptor Agonists in the Treatment of Obesity. Int. J. Mol. Sci. 2023, 24, 8592. [Google Scholar] [CrossRef]
- Jones, B.; Bloom, S.R.; Buenaventura, T.; Tomas, A.; Rutter, G.A. Control of insulin secretion by GLP-1. Peptides 2018, 100, 75–84. [Google Scholar] [CrossRef]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef]
- Knauf, C.; Cani, P.D.; Perrin, C.; Iglesias, M.A.; Maury, J.F.; Bernard, E.; Benhamed, F.; Gremeaux, T.; Drucker, D.J.; Kahn, C.R.; et al. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J. Clin. Investig. 2005, 115, 3554–3563. [Google Scholar] [CrossRef]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, W.L.; Habener, J.F. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 3434–3438. [Google Scholar] [CrossRef]
- Delmeire, D.; Flamez, D.; Hinke, S.A.; Cali, J.J.; Pipeleers, D.; Schuit, F. Type VIII adenylyl cyclase in rat beta cells: Coincidence signal detector/generator for glucose and GLP-1. Diabetologia 2003, 46, 1383–1393. [Google Scholar] [CrossRef]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef]
- Moens, K.; Heimberg, H.; Flamez, D.; Huypens, P.; Quartier, E.; Ling, Z.; Pipeleers, D.; Gremlich, S.; Thorens, B.; Schuit, F. Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 1996, 45, 257–261. [Google Scholar] [CrossRef]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Shigeto, M.; Cha, C.Y.; Rorsman, P.; Kaku, K. A role of PLC/PKC-dependent pathway in GLP-1-stimulated insulin secretion. J. Mol. Med. 2017, 95, 361–368. [Google Scholar] [CrossRef]
- Shigeto, M.; Ramracheya, R.; Tarasov, A.I.; Cha, C.Y.; Chibalina, M.V.; Hastoy, B.; Philippaert, K.; Reinbothe, T.; Rorsman, N.; Salehi, A.; et al. GLP-1 stimulates insulin secretion by PKC-dependent TRPM4 and TRPM5 activation. J. Clin. Investig. 2015, 125, 4714–4728. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schaffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Overgaard, R.V.; Hertz, C.L.; Ingwersen, S.H.; Navarria, A.; Drucker, D.J. Levels of circulating semaglutide determine reductions in HbA1c and body weight in people with type 2 diabetes. Cell Rep. Med. 2021, 2, 100387. [Google Scholar] [CrossRef]
- Rubi, B.; Ljubicic, S.; Pournourmohammadi, S.; Carobbio, S.; Armanet, M.; Bartley, C.; Maechler, P. Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J. Biol. Chem. 2005, 280, 36824–36832. [Google Scholar] [CrossRef]
- D’Alessio, D.; Vahl, T.; Prigeon, R. Effects of glucagon-like peptide 1 on the hepatic glucose metabolism. Horm. Metab. Res. 2004, 36, 837–841. [Google Scholar] [CrossRef]
- Nishizawa, M.; Moore, M.C.; Shiota, M.; Gustavson, S.M.; Snead, W.L.; Neal, D.W.; Cherrington, A.D. Effect of intraportal glucagon-like peptide-1 on glucose metabolism in conscious dogs. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1027–E1036. [Google Scholar] [CrossRef]
- Johnson, K.M.; Edgerton, D.S.; Rodewald, T.; Scott, M.; Farmer, B.; Neal, D.; Cherrington, A.D. Intraportal GLP-1 infusion increases nonhepatic glucose utilization without changing pancreatic hormone levels. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1085–E1091. [Google Scholar] [CrossRef]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef]
- Pyke, C.; Heller, R.S.; Kirk, R.K.; Orskov, C.; Reedtz-Runge, S.; Kaastrup, P.; Hvelplund, A.; Bardram, L.; Calatayud, D.; Knudsen, L.B. GLP-1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 2014, 155, 1280–1290. [Google Scholar] [CrossRef]
- Lopez-Delgado, M.I.; Morales, M.; Villanueva-Penacarrillo, M.L.; Malaisse, W.J.; Valverde, I. Effects of glucagon-like peptide 1 on the kinetics of glycogen synthase a in hepatocytes from normal and diabetic rats. Endocrinology 1998, 139, 2811–2817. [Google Scholar] [CrossRef]
- Ayala, J.E.; Bracy, D.P.; James, F.D.; Burmeister, M.A.; Wasserman, D.H.; Drucker, D.J. Glucagon-like peptide-1 receptor knockout mice are protected from high-fat diet-induced insulin resistance. Endocrinology 2010, 151, 4678–4687. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef]
- Dardevet, D.; Moore, M.C.; Neal, D.; DiCostanzo, C.A.; Snead, W.; Cherrington, A.D. Insulin-independent effects of GLP-1 on canine liver glucose metabolism: Duration of infusion and involvement of hepatoportal region. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E75–E81. [Google Scholar] [CrossRef]
- Redondo, A.; Trigo, M.V.; Acitores, A.; Valverde, I.; Villanueva-Penacarrillo, M.L. Cell signalling of the GLP-1 action in rat liver. Mol. Cell Endocrinol. 2003, 204, 43–50. [Google Scholar] [CrossRef]
- Janzen, N.R.; Whitfield, J.; Murray-Segal, L.; Kemp, B.E.; Hawley, J.A.; Hoffman, N.J. Mice with Whole-Body Disruption of AMPK-Glycogen Binding Have Increased Adiposity, Reduced Fat Oxidation and Altered Tissue Glycogen Dynamics. Int. J. Mol. Sci. 2021, 22, 9616. [Google Scholar] [CrossRef]
- Ayala, J.E.; Bracy, D.P.; James, F.D.; Julien, B.M.; Wasserman, D.H.; Drucker, D.J. The glucagon-like peptide-1 receptor regulates endogenous glucose production and muscle glucose uptake independent of its incretin action. Endocrinology 2009, 150, 1155–1164. [Google Scholar] [CrossRef]
- Lingohr, M.K.; Bull, R.J.; Kato-Weinstein, J.; Thrall, B.D. Dichloroacetate stimulates glycogen accumulation in primary hepatocytes through an insulin-independent mechanism. Toxicol. Sci. 2002, 68, 508–515. [Google Scholar] [CrossRef]
- Sepulveda-Quinenao, C.; Rodriguez, J.M.; Diaz-Castro, F.; Del Campo, A.; Bravo-Sagua, R.; Troncoso, R. Glucocorticoid Receptor beta Overexpression Has Agonist-Independent Insulin-Mimetic Effects on HepG2 Glucose Metabolism. Int. J. Mol. Sci. 2022, 23, 5582. [Google Scholar] [CrossRef]
- Ikezawa, Y.; Yamatani, K.; Ohnuma, H.; Daimon, M.; Manaka, H.; Sasaki, H. Glucagon-like peptide-1 inhibits glucagon-induced glycogenolysis in perivenous hepatocytes specifically. Regul. Pept. 2003, 111, 207–210. [Google Scholar] [CrossRef]
- Irimia, J.M.; Meyer, C.M.; Segvich, D.M.; Surendran, S.; DePaoli-Roach, A.A.; Morral, N.; Roach, P.J. Lack of liver glycogen causes hepatic insulin resistance and steatosis in mice. J. Biol. Chem. 2017, 292, 10455–10464. [Google Scholar] [CrossRef]
- Lopez-Soldado, I.; Guinovart, J.J.; Duran, J. Active Glycogen Synthase in the Liver Prevents High-Fat Diet-Induced Glucose Intolerance, Decreases Food Intake, and Lowers Body Weight. Int. J. Mol. Sci. 2023, 24, 2574. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, M.; Li, T.; Dong, N.; Yi, L.; Zhang, Q.; Mi, M. GLP-1 regulates exercise endurance and skeletal muscle remodeling via GLP-1R/AMPK pathway. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119300. [Google Scholar] [CrossRef]
- Holmes, B.F.; Sparling, D.P.; Olson, A.L.; Winder, W.W.; Dohm, G.L. Regulation of muscle GLUT4 enhancer factor and myocyte enhancer factor 2 by AMP-activated protein kinase. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E1071–E1076. [Google Scholar] [CrossRef]
- Hoffman, N.J.; Whitfield, J.; Janzen, N.R.; Belhaj, M.R.; Galic, S.; Murray-Segal, L.; Smiles, W.J.; Ling, N.X.Y.; Dite, T.A.; Scott, J.W.; et al. Genetic loss of AMPK-glycogen binding destabilises AMPK and disrupts metabolism. Mol. Metab. 2020, 41, 101048. [Google Scholar] [CrossRef]
- Luque, M.A.; Gonzalez, N.; Marquez, L.; Acitores, A.; Redondo, A.; Morales, M.; Valverde, I.; Villanueva-Penacarrillo, M.L. Glucagon-like peptide-1 (GLP-1) and glucose metabolism in human myocytes. J. Endocrinol. 2002, 173, 465–473. [Google Scholar] [CrossRef]
- Taheri, R.; Mokhtari, Y.; Yousefi, A.M.; Bashash, D. The PI3K/Akt signaling axis and type 2 diabetes mellitus (T2DM): From mechanistic insights into possible therapeutic targets. Cell Biol. Int. 2024, 48, 1049–1068. [Google Scholar] [CrossRef]
- Green, C.J.; Henriksen, T.I.; Pedersen, B.K.; Solomon, T.P. Glucagon like peptide-1-induced glucose metabolism in differentiated human muscle satellite cells is attenuated by hyperglycemia. PLoS ONE 2012, 7, e44284. [Google Scholar] [CrossRef]
- Arnes, L.; Gonzalez, N.; Tornero-Esteban, P.; Sancho, V.; Acitores, A.; Valverde, I.; Delgado, E.; Villanueva-Penacarrillo, M.L. Characteristics of GLP-1 and exendins action upon glucose transport and metabolism in type 2 diabetic rat skeletal muscle. Int. J. Mol. Med. 2008, 22, 127–132. [Google Scholar] [CrossRef]
- Lai, Y.C.; Liu, Y.; Jacobs, R.; Rider, M.H. A novel PKB/Akt inhibitor, MK-2206, effectively inhibits insulin-stimulated glucose metabolism and protein synthesis in isolated rat skeletal muscle. Biochem. J. 2012, 447, 137–147. [Google Scholar] [CrossRef]
- Luo, J.; Sobkiw, C.L.; Hirshman, M.F.; Logsdon, M.N.; Li, T.Q.; Goodyear, L.J.; Cantley, L.C. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006, 3, 355–366. [Google Scholar] [CrossRef]
- Acitores, A.; Gonzalez, N.; Sancho, V.; Valverde, I.; Villanueva-Penacarrillo, M.L. Cell signalling of glucagon-like peptide-1 action in rat skeletal muscle. J. Endocrinol. 2004, 180, 389–398. [Google Scholar] [CrossRef]
- Villanueva-Penacarrillo, M.L.; Martin-Duce, A.; Ramos-Alvarez, I.; Gutierrez-Rojas, I.; Moreno, P.; Nuche-Berenguer, B.; Acitores, A.; Sancho, V.; Valverde, I.; Gonzalez, N. Characteristic of GLP-1 effects on glucose metabolism in human skeletal muscle from obese patients. Regul. Pept. 2011, 168, 39–44. [Google Scholar] [CrossRef]
- Huang, S.; Ma, S.; Ning, M.; Yang, W.; Ye, Y.; Zhang, L.; Shen, J.; Leng, Y. TGR5 agonist ameliorates insulin resistance in the skeletal muscles and improves glucose homeostasis in diabetic mice. Metabolism 2019, 99, 45–56. [Google Scholar] [CrossRef]
- Lockie, S.H.; Heppner, K.M.; Chaudhary, N.; Chabenne, J.R.; Morgan, D.A.; Veyrat-Durebex, C.; Ananthakrishnan, G.; Rohner-Jeanrenaud, F.; Drucker, D.J.; DiMarchi, R.; et al. Direct control of brown adipose tissue thermogenesis by central nervous system glucagon-like peptide-1 receptor signaling. Diabetes 2012, 61, 2753–2762. [Google Scholar] [CrossRef]
- Lee, S.J.; Sanchez-Watts, G.; Krieger, J.P.; Pignalosa, A.; Norell, P.N.; Cortella, A.; Pettersen, K.G.; Vrdoljak, D.; Hayes, M.R.; Kanoski, S.E.; et al. Loss of dorsomedial hypothalamic GLP-1 signaling reduces BAT thermogenesis and increases adiposity. Mol. Metab. 2018, 11, 33–46. [Google Scholar] [CrossRef]
- Beiroa, D.; Imbernon, M.; Gallego, R.; Senra, A.; Herranz, D.; Villarroya, F.; Serrano, M.; Ferno, J.; Salvador, J.; Escalada, J.; et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 2014, 63, 3346–3358. [Google Scholar] [CrossRef]
- Lynch, L.; Hogan, A.E.; Duquette, D.; Lester, C.; Banks, A.; LeClair, K.; Cohen, D.E.; Ghosh, A.; Lu, B.; Corrigan, M.; et al. iNKT Cells Induce FGF21 for Thermogenesis and Are Required for Maximal Weight Loss in GLP1 Therapy. Cell Metab. 2016, 24, 510–519. [Google Scholar] [CrossRef]
- Gutierrez, A.D.; Gao, Z.; Hamidi, V.; Zhu, L.; Saint Andre, K.B.; Riggs, K.; Ruscheinsky, M.; Wang, H.; Yu, Y.; Miller, C., III; et al. Anti-diabetic effects of GLP1 analogs are mediated by thermogenic interleukin-6 signaling in adipocytes. Cell Rep. Med. 2022, 3, 100813. [Google Scholar] [CrossRef]
- Keinan, O.; Valentine, J.M.; Xiao, H.; Mahata, S.K.; Reilly, S.M.; Abu-Odeh, M.; Deluca, J.H.; Dadpey, B.; Cho, L.; Pan, A.; et al. Glycogen metabolism links glucose homeostasis to thermogenesis in adipocytes. Nature 2021, 599, 296–301. [Google Scholar] [CrossRef]
- Zhuo, S.; Bai, M.; Wang, Z.; Chen, L.; Li, Z.; Zhu, X.; Chen, J.; Ye, X.; Guo, C.; Chen, Y. Glycogen synthesis is required for adaptive thermogenesis in beige adipose tissue and affects diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2024, 326, E696–E708. [Google Scholar] [CrossRef]
- Tavares, G.; Rosendo-Silva, D.; Simoes, F.; Eickhoff, H.; Marques, D.; Sacramento, J.F.; Capucho, A.M.; Seica, R.; Conde, S.V.; Matafome, P. Circulating Dopamine Is Regulated by Dietary Glucose and Controls Glucagon-like 1 Peptide Action in White Adipose Tissue. Int. J. Mol. Sci. 2023, 24, 2464. [Google Scholar] [CrossRef]
- van Zuylen, M.L.; Siegelaar, S.E.; Plummer, M.P.; Deane, A.M.; Hermanides, J.; Hulst, A.H. Perioperative management of long-acting glucagon-like peptide-1 (GLP-1) receptor agonists: Concerns for delayed gastric emptying and pulmonary aspiration. Br. J. Anaesth. 2024, 132, 644–648. [Google Scholar] [CrossRef]
- Urva, S.; Coskun, T.; Loghin, C.; Cui, X.; Beebe, E.; O’Farrell, L.; Briere, D.A.; Benson, C.; Nauck, M.A.; Haupt, A. The novel dual glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 (GLP-1) receptor agonist tirzepatide transiently delays gastric emptying similarly to selective long-acting GLP-1 receptor agonists. Diabetes Obes. Metab. 2020, 22, 1886–1891. [Google Scholar] [CrossRef]
- Jalleh, R.J.; Plummer, M.P.; Marathe, C.S.; Umapathysivam, M.M.; Quast, D.R.; Rayner, C.K.; Jones, K.L.; Wu, T.; Horowitz, M.; Nauck, M.A. Clinical Consequences of Delayed Gastric Emptying With GLP-1 Receptor Agonists and Tirzepatide. J. Clin. Endocrinol. Metab. 2024, 110, 1–15. [Google Scholar] [CrossRef]
- Kobori, T.; Onishi, Y.; Yoshida, Y.; Tahara, T.; Kikuchi, T.; Kubota, T.; Iwamoto, M.; Sawada, T.; Kobayashi, R.; Fujiwara, H.; et al. Association of glucagon-like peptide-1 receptor agonist treatment with gastric residue in an esophagogastroduodenoscopy. J. Diabetes Investig. 2023, 14, 767–773. [Google Scholar] [CrossRef]
- Xie, C.; Huang, W.; Watson, L.E.; Soenen, S.; Young, R.L.; Jones, K.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Plasma GLP-1 Response to Oral and Intraduodenal Nutrients in Health and Type 2 Diabetes-Impact on Gastric Emptying. J. Clin. Endocrinol. Metab. 2022, 107, e1643–e1652. [Google Scholar] [CrossRef]
- Naslund, E.; Bogefors, J.; Gryback, P.; Bjellerup, P.; Jacobsson, H.; Holst, J.J.; Hellstrom, P.M. GLP-1 inhibits gastric emptying of water but does not influence plasma. Scand. J. Gastroenterol. 2001, 36, 156–162. [Google Scholar] [CrossRef]
- Quitadamo, P.; Zenzeri, L.; Mozzillo, E.; Giorgio, V.; Rocco, A.; Franzese, A.; Nardone, G.; Staiano, A. Plasma dosage of ghrelin, IGF-1, GLP- 1 and leptin related to gastric emptying and esophageal pH-impedance in children with obesity. J. Endocrinol. Investig. 2021, 44, 1275–1281. [Google Scholar] [CrossRef]
- Murphy, C.F.; Elliott, J.A.; Docherty, N.G.; Mohamed, A.A.; Vincent, R.P.; Ravi, N.; Reynolds, J.V.; Le Roux, C.W. Exaggerated postprandial GLP-1 secretion following esophagectomy is not associated with gastric emptying and intestinal transit. Dis. Esophagus 2021, 34, doaa098. [Google Scholar] [CrossRef]
- Sista, F.; Abruzzese, V.; Clementi, M.; Carandina, S.; Cecilia, M.; Amicucci, G. The effect of sleeve gastrectomy on GLP-1 secretion and gastric emptying: A prospective study. Surg. Obes. Relat. Dis. 2017, 13, 7–14. [Google Scholar] [CrossRef]
- Kittah, E.; Camilleri, M.; Jensen, M.D.; Vella, A. A Pilot Study Examining the Effects of GLP-1 Receptor Blockade Using Exendin-(9,39) on Gastric Emptying and Caloric Intake in Subjects With and Without Bariatric Surgery. Metab. Syndr. Relat. Disord. 2020, 18, 406–412. [Google Scholar] [CrossRef]
- Lopez-Ferreras, L.; Richard, J.E.; Noble, E.E.; Eerola, K.; Anderberg, R.H.; Olandersson, K.; Taing, L.; Kanoski, S.E.; Hayes, M.R.; Skibicka, K.P. Lateral hypothalamic GLP-1 receptors are critical for the control of food reinforcement, ingestive behavior and body weight. Mol. Psychiatry 2018, 23, 1157–1168. [Google Scholar] [CrossRef]
- Psilopanagioti, A.; Nikou, S.; Logotheti, S.; Arbi, M.; Chartoumpekis, D.V.; Papadaki, H. Glucagon-like Peptide-1 Receptor in the Human Hypothalamus Is Associated with Body Mass Index and Colocalizes with the Anorexigenic Neuropeptide Nucleobindin-2/Nesfatin-1. Int. J. Mol. Sci. 2022, 23, 14899. [Google Scholar] [CrossRef]
- Stice, E.; Yokum, S.; Blum, K.; Bohon, C. Weight gain is associated with reduced striatal response to palatable food. J. Neurosci. 2010, 30, 13105–13109. [Google Scholar] [CrossRef]
- Ard, J.; Fitch, A.; Fruh, S.; Herman, L. Weight Loss and Maintenance Related to the Mechanism of Action of Glucagon-Like Peptide 1 Receptor Agonists. Adv. Ther. 2021, 38, 2821–2839. [Google Scholar] [CrossRef]
- Kadouh, H.; Chedid, V.; Halawi, H.; Burton, D.D.; Clark, M.M.; Khemani, D.; Vella, A.; Acosta, A.; Camilleri, M. GLP-1 Analog Modulates Appetite, Taste Preference, Gut Hormones, and Regional Body Fat Stores in Adults with Obesity. J. Clin. Endocrinol. Metab. 2020, 105, 1552–1563. [Google Scholar] [CrossRef]
- Friedrichsen, M.; Breitschaft, A.; Tadayon, S.; Wizert, A.; Skovgaard, D. The effect of semaglutide 2.4 mg once weekly on energy intake, appetite, control of eating, and gastric emptying in adults with obesity. Diabetes Obes. Metab. 2021, 23, 754–762. [Google Scholar] [CrossRef]
- Singh, I.; Wang, L.; Xia, B.; Liu, J.; Tahiri, A.; El Ouaamari, A.; Wheeler, M.B.; Pang, Z.P. Activation of arcuate nucleus glucagon-like peptide-1 receptor-expressing neurons suppresses food intake. Cell Biosci. 2022, 12, 178. [Google Scholar] [CrossRef]
- Quast, D.R.; Nauck, M.A.; Schenker, N.; Menge, B.A.; Kapitza, C.; Meier, J.J. Macronutrient intake, appetite, food preferences and exocrine pancreas function after treatment with short- and long-acting glucagon-like peptide-1 receptor agonists in type 2 diabetes. Diabetes Obes. Metab. 2021, 23, 2344–2353. [Google Scholar] [CrossRef]
- Badulescu, S.; Tabassum, A.; Le, G.H.; Wong, S.; Phan, L.; Gill, H.; Llach, C.D.; McIntyre, R.S.; Rosenblat, J.; Mansur, R. Glucagon-like peptide 1 agonist and effects on reward behaviour: A systematic review. Physiol. Behav. 2024, 283, 114622. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.J.; Hibberd, T.J. GLP-1 appetite control via intestinofugal neurons. Cell Res. 2022, 32, 711–712. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, C.; Blundell, J.; Tetens Hoff, S.; Dahl, K.; Bauer, R.; Baekdal, T. Effects of oral semaglutide on energy intake, food preference, appetite, control of eating and body weight in subjects with type 2 diabetes. Diabetes Obes. Metab. 2021, 23, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Spezani, R.; Marinho, T.S.; Reis, T.S.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Cotadutide (GLP-1/Glucagon dual receptor agonist) modulates hypothalamic orexigenic and anorexigenic neuropeptides in obese mice. Peptides 2024, 173, 171138. [Google Scholar] [CrossRef]
- Costa, A.; Ai, M.; Nunn, N.; Culotta, I.; Hunter, J.; Boudjadja, M.B.; Valencia-Torres, L.; Aviello, G.; Hodson, D.J.; Snider, B.M.; et al. Anorectic and aversive effects of GLP-1 receptor agonism are mediated by brainstem cholecystokinin neurons, and modulated by GIP receptor activation. Mol. Metab. 2022, 55, 101407. [Google Scholar] [CrossRef]
- Holter, M.M.; Chirikjian, M.K.; Govani, V.N.; Cummings, B.P. TGR5 Signaling in Hepatic Metabolic Health. Nutrients 2020, 12, 2598. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Potts, A.; He, T.; Duarte, J.A.; Taussig, R.; Mangelsdorf, D.J.; Kliewer, S.A.; Burgess, S.C. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2013, 304, G371–G380. [Google Scholar] [CrossRef]
- Rutter, J.; Michnoff, C.H.; Harper, S.M.; Gardner, K.H.; McKnight, S.L. PAS kinase: An evolutionarily conserved PAS domain-regulated serine/threonine kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 8991–8996. [Google Scholar] [CrossRef]
- Perez-Garcia, A.; Hurtado-Carneiro, V.; Herrero-De-Dios, C.; Dongil, P.; Garcia-Maurino, J.E.; Sanchez, M.D.; Sanz, C.; Alvarez, E. Storage and Utilization of Glycogen by Mouse Liver during Adaptation to Nutritional Changes Are GLP-1 and PASK Dependent. Nutrients 2021, 13, 2552. [Google Scholar] [CrossRef]
- Engin, A. Protein Kinases in Obesity, and the Kinase-Targeted Therapy. Adv. Exp. Med. Biol. 2024, 1460, 199–229. [Google Scholar] [CrossRef]
- Hurtado-Carneiro, V.; Dongil, P.; Perez-Garcia, A.; Alvarez, E.; Sanz, C. Preventing Oxidative Stress in the Liver: An Opportunity for GLP-1 and/or PASK. Antioxidants 2021, 10, 2028. [Google Scholar] [CrossRef] [PubMed]
- Dicter, N.; Madar, Z.; Tirosh, O. Alpha-lipoic acid inhibits glycogen synthesis in rat soleus muscle via its oxidative activity and the uncoupling of mitochondria. J. Nutr. 2002, 132, 3001–3006. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lin, H.; Shen, C.; Zhang, M.; Wang, X.; Yuan, M.; Yuan, M.; Jia, S.; Cao, Z.; Wu, C.; et al. Gut microbiota regulates postprandial GLP-1 response via ileal bile acid-TGR5 signaling. Gut. Microbes 2023, 15, 2274124. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, J.; Xia, L.; Wei, T.; Cui, X.; Wang, D.; Jin, Z.; Lin, X.; Li, F.; Yang, K.; et al. Gut Microbiota-Tryptophan Metabolism-GLP-1 Axis Participates in beta-Cell Regeneration Induced by Dapagliflozin. Diabetes 2024, 73, 926–940. [Google Scholar] [CrossRef]
- Yoon, H.S.; Cho, C.H.; Yun, M.S.; Jang, S.J.; You, H.J.; Kim, J.H.; Han, D.; Cha, K.H.; Moon, S.H.; Lee, K.; et al. Akkermansia muciniphila secretes a glucagon-like peptide-1-inducing protein that improves glucose homeostasis and ameliorates metabolic disease in mice. Nat. Microbiol. 2021, 6, 563–573. [Google Scholar] [CrossRef]
- Chen, C.; Liu, L.; Zhong, Y.; Wang, M.; Ai, Y.; Hou, Y.; Chen, H.; Lin, X.; Zhang, Y.; Ding, M.; et al. Gut microbiota-bile acids-glucagon like peptide-1 axis contributes the resistance to high fat diet-induced obesity in mice. J. Nutr. Biochem. 2023, 117, 109358. [Google Scholar] [CrossRef]
- Drucker, D.J. GLP-1 physiology informs the pharmacotherapy of obesity. Mol. Metab. 2022, 57, 101351. [Google Scholar] [CrossRef]
- van Eyk, H.J.; Paiman, E.H.M.; Bizino, M.B.; SL, I.J.; Kleiburg, F.; Boers, T.G.W.; Rappel, E.J.; Burakiewicz, J.; Kan, H.E.; Smit, J.W.A.; et al. Liraglutide decreases energy expenditure and does not affect the fat fraction of supraclavicular brown adipose tissue in patients with type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 616–624. [Google Scholar] [CrossRef]
- Wang, X.F.; Liu, J.J.; Xia, J.; Liu, J.; Mirabella, V.; Pang, Z.P. Endogenous Glucagon-like Peptide-1 Suppresses High-Fat Food Intake by Reducing Synaptic Drive onto Mesolimbic Dopamine Neurons. Cell Rep. 2015, 12, 726–733. [Google Scholar] [CrossRef]
- Nestler, E.J.; Carlezon, W.A., Jr. The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 2006, 59, 1151–1159. [Google Scholar] [CrossRef]
- Jensen, M.E.; Galli, A.; Thomsen, M.; Jensen, K.L.; Thomsen, G.K.; Klausen, M.K.; Vilsboll, T.; Christensen, M.B.; Holst, J.J.; Owens, A.; et al. Glucagon-like peptide-1 receptor regulation of basal dopamine transporter activity is species-dependent. Neurochem. Int. 2020, 138, 104772. [Google Scholar] [CrossRef] [PubMed]
- Kornelius, E.; Huang, J.Y.; Lo, S.C.; Huang, C.N.; Yang, Y.S. The risk of depression, anxiety, and suicidal behavior in patients with obesity on glucagon like peptide-1 receptor agonist therapy. Sci. Rep. 2024, 14, 24433. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Lu, Y.; Donahoo, W.T.; Westen, S.C.; Chen, Y.; Bian, J.; Guo, J. Glucagon-Like Peptide-1 Receptor Agonists and Risk for Depression in Older Adults With Type 2 Diabetes: A Target Trial Emulation Study. Ann. Intern. Med. 2025, 178, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Thomas, N.; Singleton, A.; Piggott, M.; Lloyd, S.; Perry, E.K.; Morris, C.M.; Perry, R.H.; Ferrier, I.N.; Court, J.A. D2 dopamine receptor gene (DRD2) Taq1 A polymorphism: Reduced dopamine D2 receptor binding in the human striatum associated with the A1 allele. Pharmacogenetics 1997, 7, 479–484. [Google Scholar] [CrossRef]
- Modestino, E.J.; Bowirrat, A.; Baron, D.; Thanos, P.K.; Hanna, C.; Bagchi, D.; Braverman, E.R.; Dennen, C.A.; Badgaiyan, R.D.; Pollack, A.R.; et al. Is There a Natural, Non-addictive, and Non-anti-reward, Safe, Gene-based Solution to Treat Reward Deficiency Syndrome? KB220 Variants vs GLP-1 Analogs. J. Addict Psychiatry 2024, 8, 34–49. [Google Scholar]
- Cardel, M.I.; Lemas, D.J.; Lee, A.M.; Miller, D.R.; Huo, T.; Klimentidis, Y.C.; Fernandez, J.R. Taq1a polymorphism (rs1800497) is associated with obesity-related outcomes and dietary intake in a multi-ethnic sample of children. Pediatr. Obes. 2019, 14, e12470. [Google Scholar] [CrossRef]
- Blum, K.; Dennen, C.A.; Lewandrowski, K.-U.; Sharafshah, A.; Pinhasov, A.; Bowirrat, A.; Elman, I.; Cadet, J.L.; Braverman, E.R.; Thanos, P.K.; et al. Hypothesizing glucagon-like peptide 1 (GLP-1), agonists promote hypodopaminergia, resulting in heightened addictive reward-seeking and altered mood: Breaking the bubble and adding salt to a wound. Med. Hypotheses 2025, 198, 111612. [Google Scholar] [CrossRef]
- Sharafshah, A.; Lewandrowski, K.U.; Gold, M.S.; Fuehrlein, B.; Ashford, J.W.; Thanos, P.K.; Wang, G.J.; Hanna, C.; Cadet, J.L.; Gardner, E.L.; et al. In Silico Pharmacogenomic Assessment of Glucagon-like Peptide-1 (GLP1) Agonists and the Genetic Addiction Risk Score (GARS) Related Pathways: Implications for Suicide Ideation and Substance Use Disorder. Curr. Neuropharmacol. 2025, 23, 974–995. [Google Scholar] [CrossRef]
- Wang, M.; Tan, Y.; Shi, Y.; Wang, X.; Liao, Z.; Wei, P. Diabetes and Sarcopenic Obesity: Pathogenesis, Diagnosis, and Treatments. Front. Endocrinol. 2020, 11, 568. [Google Scholar] [CrossRef]
- Prado, C.M.; Phillips, S.M.; Gonzalez, M.C.; Heymsfield, S.B. Muscle matters: The effects of medically induced weight loss on skeletal muscle. Lancet Diabetes Endocrinol. 2024, 12, 785–787. [Google Scholar] [CrossRef]
- Yuan, S.; Larsson, S.C. Epidemiology of sarcopenia: Prevalence, risk factors, and consequences. Metabolism 2023, 144, 155533. [Google Scholar] [CrossRef] [PubMed]
- Blomstrand, E.; Saltin, B. Effect of muscle glycogen on glucose, lactate and amino acid metabolism during exercise and recovery in human subjects. J. Physiol. 1999, 514 Pt 1, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Linge, J.; Birkenfeld, A.L. Changes in lean body mass with glucagon-like peptide-1-based therapies and mitigation strategies. Diabetes Obes. Metab. 2024, 26 (Suppl. S4), 16–27. [Google Scholar] [CrossRef] [PubMed]
- Mastaitis, J.W.; Gomez, D.; Raya, J.G.; Li, D.; Min, S.; Stec, M.; Kleiner, S.; McWilliams, T.; Altarejos, J.Y.; Murphy, A.J.; et al. GDF8 and activin A blockade protects against GLP-1-induced muscle loss while enhancing fat loss in obese male mice and non-human primates. Nat. Commun. 2025, 16, 4377. [Google Scholar] [CrossRef]
- Yang, H.; Wang, S.; Ye, Y.; Xie, M.; Li, Y.; Jin, H.; Li, J.; Gao, L. GLP-1 preserves beta cell function via improvement on islet insulin signaling in high fat diet feeding mice. Neuropeptides 2021, 85, 102110. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lotosky, J.; Jean, X.; Altankhuyag, A.; Khan, S.; Bernotas, A.; Sharafshah, A.; Blum, K.; Posner, A.; Thanos, P.K. GLP-1 and Its Role in Glycogen Production: A Narrative Review. Biomedicines 2025, 13, 1610. https://doi.org/10.3390/biomedicines13071610
Lotosky J, Jean X, Altankhuyag A, Khan S, Bernotas A, Sharafshah A, Blum K, Posner A, Thanos PK. GLP-1 and Its Role in Glycogen Production: A Narrative Review. Biomedicines. 2025; 13(7):1610. https://doi.org/10.3390/biomedicines13071610
Chicago/Turabian StyleLotosky, Joseph, Xavier Jean, Anungoo Altankhuyag, Saqib Khan, Ashley Bernotas, Alireza Sharafshah, Kenneth Blum, Alan Posner, and Panayotis K. Thanos. 2025. "GLP-1 and Its Role in Glycogen Production: A Narrative Review" Biomedicines 13, no. 7: 1610. https://doi.org/10.3390/biomedicines13071610
APA StyleLotosky, J., Jean, X., Altankhuyag, A., Khan, S., Bernotas, A., Sharafshah, A., Blum, K., Posner, A., & Thanos, P. K. (2025). GLP-1 and Its Role in Glycogen Production: A Narrative Review. Biomedicines, 13(7), 1610. https://doi.org/10.3390/biomedicines13071610