
Regulatory Genetic Networks by microRNAs: Exploring Genomic Signatures in Cervical Cancer

 ,
,  ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Genomic Signatures of microRNAs in Cervical Cancer
2.1. Genomic Signature of microRNAs in Cervical Tissues
2.2. Genomic Signature of microRNAs Circulating
3. Regulatory Genetic Networks Modulated by MicroRNAs and Their Target Genes in Cervical Cancer
4. MicroRNA-mRNA Regulatory Modules in Cervical Cancer
5. Gene Therapy Clinical Trials with MicroRNAs for Uterine Cancers
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3′-UTRs | 3′-untranslated regions |
| ABCA8 | ATP-binding cassette sub-family A member 8 |
| Akt | Protein kinase B |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| APOBEC3B | Probable DNA dC->dU-editing enzyme APOBEC-3B |
| ATF2 | Activating transcription factor 2 |
| BIRC5 | Baculoviral inhibitor of apoptosis repeat-containing 5 or survivin |
| CCND1 | Cyclin D1 |
| CDC6 | Cell division control protein 6 homolog |
| CeRNA | Competing endogenous RNAs |
| CIN | Cervical intraepithelial neoplasia |
| cGMP | Cyclic guanosine monophosphate |
| cGMP-PKG | dependent protein kinase or protein kinase G |
| c-Myc | MYC proto-oncogene, bHLH transcription factor |
| CXCL8 | IL-8 or chemokine (C-X-C motif) ligand 8 |
| DAXX | Death-associated protein 6 |
| DEGs | Differentially expressed genes |
| DEmiRNAs: | Differentially expressed microRNAs |
| DEmRNAs | Differentially expressed mRNAs |
| DSG2 | Desmoglein-2 |
| DUSP8 | DUSP10: Dual specificity phosphatase 8, 10 |
| E2F7 | Transcription factor E2F7 |
| EC-AIA | Endometrial carcinoma arising in adenomyosis |
| EGFR | Epidermal growth factor receptor |
| EREG | Epiregulin is a member of the epidermal growth factor family |
| EZH2 | Enhancer of zeste homolog 2 |
| FAS | Fas receptor CD95 |
| FASLG | Fas ligand CD95L |
| FFPE | Formalin-fixed paraffin-embedded |
| FGF12 | Fibroblast growth factor 12 |
| FLT1 | Fms-related tyrosine kinase 1 |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GEO | Gene expression omnibus database |
| GO | Gene Ontology |
| GWAS | Genome wide association study |
| HOXA1 | Homeobox protein Hox-A1 |
| HSIL | High-grade squamous intraepithelial lesion |
| HPV | Humanpapilloma virus |
| IL1B | Interleukin-1 beta |
| KEGG | Kyoto encyclopedia of genes and genomes |
| k-Ras | Kirsten rat sarcoma virus |
| LncRNAs | Long non-coding RNAs |
| MAPK | Mitogen-activated protein kinase |
| MEF2C | Myocyte-specific enhancer factor 2C |
| MirDIP | MicroRNA Data Integration Portal |
| MKNK2 | MAP kinase-interacting serine/threonine-protein kinase 2 |
| mTOR | Mammalian target of rapamycin |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NTF3 | Neurotrophin-3 |
| PBMC | Peripheral blood mononuclear cells |
| PI3K | Phosphoinositide 3-kinase |
| PLAGL1 | Zinc finger protein PLAGL1 |
| PPI | Protein-protein interaction |
| PTEN | Phosphatase and tensin homolog |
| RASA1 | RAS p21 protein activator 1 |
| RASGRP1, RASGRP 3 | RAS guanyl-releasing protein 1, 3 |
| RARB | Retinoic acid receptor beta |
| RFCM3 | Relevant and functionally consistent microRNA-mRNA modules |
| RPS6KA3 | Protein S6 kinase, 90kDa, polypeptide 3 |
| SCC | Squamous cell cancer |
| SIG: | Stochastic process model for Identifying differentially co-expressed Gene pair |
| SMAD2 | Mothers against decapentaplegic homolog 2, also known as SMAD family member 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| SVM | Support vector machine |
| TCGA | The Cancer Genome Atlas |
| TGF-B1: TGF-B2 | Transforming growth factor beta 1, 2 |
| TGFBR2 | Transforming growth factor, beta receptor II |
| TP53 | Tumor protein P53 |
| TP63 | Tumor protein P63 |
| Tregs | T regulatory cells |
| TWIST1 | Twist-related protein 1 |
| VEGF | Vascular endothelial growth factor |
| Wnt | Wingless and Int-1 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Loopik, D.L.; Bentley, H.A.; Eijgenraam, M.N.; IntHout, J.; Bekkers, R.L.M.; Bentley, J.R. The natural history of cervical intraepithelial neoplasia grades 1, 2, and 3: A systematic review and meta-analysis. J. Low. Genit. Tract. Dis. 2021, 25, 221–231. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Mackie, A.; Wentzensen, N. The IARC perspective on cervical cancer screening. N. Engl. J. Med. 2022, 386, 607–608. [Google Scholar] [CrossRef]
- Cox, J.T.; Castle, P.E.; Behrens, C.M.; Sharma, A.; Wright, T.C., Jr.; Cuzick, J.; Athena HPV Study Group. Comparison of cervical cancer screening strategies incorporating different combinations of cytology, HPV testing, and genotyping for HPV 16/18: Results from the ATHENA HPV study. Am. J. Obstet. Gynecol. 2013, 208, 184.e1–184.e11. [Google Scholar] [CrossRef]
- Bahrami, A.; Hasanzadeh, M.; Shahidsales, S.; Farazestanian, M.; Hassanian, S.M.; Moetamani, A.M.; Maftouh, M.; Gharib, M.; Yousefi, Z.; Kadkhodayan, S.; et al. Genetic susceptibility in cervical cancer: From bench to bedside. J. Cell Physiol. 2018, 233, 1929–1939. [Google Scholar] [CrossRef]
- Kang, S.D.; Chatterjee, S.; Alam, S.; Salzberg, A.C.; Milici, J.; van der Burg, S.H.; Meyers, C. Effect of productive human papillomavirus 16 infection on global gene expression in cervical epithelium. J. Virol. 2018, 92, e01261-18. [Google Scholar] [CrossRef] [PubMed]
- Faye, M.D.; Alfieri, J. Advances in radiation oncology for the treatment of cervical cancer. Curr. Oncol. 2022, 29, 928–944. [Google Scholar] [CrossRef] [PubMed]
- Horeweg, N.; Mittal, P.; Gradowska, P.L.; Boere, I.; Nout, R.A.; Chopra, S. A systematic review and meta-analysis of adjuvant chemotherapy after chemoradiation for locally advanced cervical cancer. Crit. Rev. Oncol. Hematol. 2022, 172, 103638. [Google Scholar] [CrossRef]
- Malka, Y.; Steiman-Shimony, A.; Rosenthal, E.; Argaman, L.; Cohen-Daniel, L.; Arbib, E.; Margalit, H.; Kaplan, T.; Berger, M. Post-transcriptional 3′-UTR cleavage of mRNA transcripts generates thousands of stable uncapped autonomous RNA fragments. Nat. Commun. 2017, 8, 2029. [Google Scholar] [CrossRef]
- Galvão-Lima, L.J.; Morais, A.H.F.; Valentim, R.A.M.; Barreto, E.J.S.S. miRNAs as biomarkers for early cancer detection and their application in the development of new diagnostic tools. Biomed. Eng. Online 2021, 20, 21. [Google Scholar] [CrossRef]
- Dragomir, M.P.; Knutsen, E.; Calin, G.A. Classical and noncanonical functions of miRNAs in cancers. Trends Genet. 2022, 38, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Bañuelos-Villegas, E.G.; Pérez-yPérez, M.F.; Alvarez-Salas, L.M. Cervical cancer, papillomavirus, and miRNA dysfunction. Front. Mol. Biosci. 2021, 8, 758337. [Google Scholar] [CrossRef] [PubMed]
- miRTarBase V. Nucleic Acids Research. 2022. Available online: https://tools4mirs.org/software/mirna_databases/mirtarbase/ (accessed on 23 April 2024).
- TargetScan Release 7.1; Institute for Biomedical Research: Cambridge, MA, USA, 2022; Available online: https://www.targetscan.org/vert_71/ (accessed on 23 April 2024).
- Institut Pasteur. miRanda 3.3a. Available online: https://bioweb.pasteur.fr/packages/pack@miRanda@3.3a (accessed on 23 April 2024).
- miRbase. The University of Manchester. 2024. Available online: https://www.mirbase.org/ (accessed on 23 April 2024).
- Medical Faculty Mannheim of the Heidelberg University. MirWalk V2. Available online: http://mirwalk.umm.uni-heidelberg.de/ (accessed on 23 April 2024).
- TriplexRNA. Systems Biology & Bioinformatics, University of Rostock. 2024. Available online: https://triplexrna.org/ (accessed on 23 April 2024).
- miRSearch V3.0. Available online: https://www.mir.com.my/search/ (accessed on 23 April 2024).
- Eclipsebio. Eclipse BioInnovations. 2024. Available online: https://eclipsebio.com/ (accessed on 23 April 2024).
- Harbin Institute of Technology, Indiana University. miR2disease V2.0. 2 April 2008. Available online: http://www.mir2disease.org/ (accessed on 23 April 2024).
- DIANA-miRPath V3. Nucleic Acids Research (2015): gkv403. Available online: https://dianalab.e-ce.uth.gr/html/mirpathv3/index.php?r=mirpath (accessed on 23 April 2024).
- Universidad de Granada. GeneCodis 2007–2021. Available online: https://genecodis.genyo.es/ (accessed on 23 April 2024).
- miRecords: Biolead.org. 2008–2013. Available online: http://c1.accurascience.com/miRecords/ (accessed on 23 April 2024).
- TransmiR v2.0., Django. Available online: http://www.cuilab.cn/transmir (accessed on 23 April 2024).
- Cottrell, K.A.; Chaudhari, H.G.; Cohen, B.A.; Djuranovic, S. PTRE-seq reveals mechanism and interactions of RNA binding proteins and miRNAs. Nat. Commun. 2018, 9, 301. [Google Scholar] [CrossRef]
- Hu, X.; Schwarz, J.K.; Lewis, J.S., Jr.; Huettner, P.C.; Rader, J.S.; Deasy, J.O.; Grigsby, P.W.; Wang, X. A microRNA expression signature for cervical cancer prognosis. Cancer Res. 2010, 70, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.M.; Marques, J.P.; Soares, A.R.; Carreto, L.; Santos, M.A. MicroRNA expression variability in human cervical tissues. PLoS ONE 2010, 5, e11780. [Google Scholar] [CrossRef]
- Cheung, T.H.; Man, K.N.; Yu, M.Y.; Yim, S.F.; Siu, N.S.; Lo, K.W.; Doran, G.; Wong, R.R.; Wang, V.W.; Smith, D.I.; et al. Dysregulated microRNAs in the pathogenesis and progression of cervical neoplasm. Cell Cycle 2012, 11, 2876–2884. [Google Scholar] [CrossRef]
- Zeng, K.; Zheng, W.; Mo, X.; Liu, F.; Li, M.; Liu, Z.; Zhang, W.; Hu, X. Dysregulated microRNAs involved in the progression of cervical neoplasm. Arch. Gynecol. Obstet. 2015, 292, 905–913. [Google Scholar] [CrossRef]
- How, C.; Pintilie, M.; Bruce, J.P.; Hui, A.B.; Clarke, B.A.; Wong, P.; Yin, S.; Yan, R.; Waggott, D.; Boutros, P.C.; et al. Developing a prognostic micro-RNA signature for human cervical carcinoma. PLoS ONE 2015, 10, e0123946. [Google Scholar] [CrossRef]
- Honegger, A.; Schilling, D.; Bastian, S.; Sponagel, J.; Kuryshev, V.; Sültmann, H.; Scheffner, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Dependence of intracellular and exosomal microRNAs on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS Pathog. 2015, 11, e1004712. [Google Scholar] [CrossRef]
- Sun, P.; Shen, Y.; Gong, J.M.; Zhou, L.L.; Sheng, J.H.; Duan, F.J. A new MicroRNA expression signature for cervical cancer. Int. J. Gynecol. Cancer 2017, 27, 339–343. [Google Scholar] [CrossRef]
- Causin, R.L.; da Silva, L.S.; Evangelista, A.F.; Leal, L.F.; Souza, K.C.B.; Pessôa-Pereira, D.; Matsushita, G.M.; Reis, R.M.; Fregnani, J.H.T.G.; Marques, M.M.C. MicroRNA biomarkers of high-grade cervical intraepithelial neoplasia in liquid biopsy. Biomed. Res. Int. 2021, 2021, 6650966. [Google Scholar] [CrossRef]
- Qi, Y.; Lai, Y.L.; Shen, P.C.; Chen, F.H.; Lin, L.J.; Wu, H.H.; Peng, P.H.; Hsu, K.W.; Cheng, W.C. Identification and validation of a miRNA-based prognostic signature for cervical cancer through an integrated bioinformatics approach. Sci. Rep. 2020, 10, 22270. [Google Scholar] [CrossRef]
- Liang, B.; Li, Y.; Wang, T. A three miRNAs signature predicts survival in cervical cancer using bioinformatics analysis. Sci. Rep. 2017, 7, 5624. [Google Scholar] [CrossRef]
- Nair, V.B.; Manasa, V.G.; Sinto, M.S.; Jayasree, K.; James, F.V.; Kannan, S. Differential expression of MicroRNAs in uterine cervical cancer and its implications in carcinogenesis; an integrative approach. Int. J. Gynecol. Cancer 2018, 28, 553–562. [Google Scholar] [CrossRef]
- Liu, S.S.; Chan, K.K.L.; Chu, D.K.H.; Wei, T.N.; Lau, L.S.K.; Ngu, S.F.; Chu, M.M.Y.; Tse, K.Y.; Ip, P.P.C.; Ng, E.K.O.; et al. Oncogenic microRNA signature for early diagnosis of cervical intraepithelial neoplasia and cancer. Mol. Oncol. 2018, 12, 2009–2022. [Google Scholar] [CrossRef]
- Babion, I.; Snoek, B.C.; Novianti, P.W.; Jaspers, A.; van Trommel, N.; Heideman, D.A.M.; Meijer, C.J.L.M.; Snijders, P.J.F.; Steenbergen, R.D.M.; Wilting, S.M. Triage of high-risk HPV-positive women in population-based screening by miRNA expression analysis in cervical scrapes; A feasibility study. Clin. Epigenet. 2018, 10, 76. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, W.; Wu, Q.; Liu, Y.; Wang, C.; Lao, G.; Yang, L.; Liu, P. Identification of a microRNA signature associated with survivability in cervical squamous cell carcinoma. PLoS ONE 2018, 13, e0193625. [Google Scholar] [CrossRef]
- Bayramoglu Tepe, N.; Bozgeyik, E.; Bozdag, Z.; Balat, O.; Ozcan, H.C.; Ugur, M.G. Identification of autophagy-associated miRNA signature for the cervical squamous cell cancer and high-grade cervical intraepithelial lesions. Reprod. Biol. 2021, 21, 100536. [Google Scholar] [CrossRef]
- Shi, C.; Yang, Y.; Zhang, L.; Zhang, T.; Yu, J.; Qin, S.; Gao, Y. Optimal subset of signature miRNAs consisting of 7 miRNAs that can serve as a novel diagnostic and prognostic predictor for the progression of cervical cancer. Oncol. Rep. 2019, 41, 3167–3178. [Google Scholar] [CrossRef]
- Bozgeyik, E.; Tepe, N.B.; Bozdag, Z. Identification of microRNA expression signature for the diagnosis and prognosis of cervical squamous cell carcinoma. Pathol. Res. Pract. 2020, 216, 153159. [Google Scholar] [CrossRef]
- Causin, R.L.; Freitas, A.J.A.; Trovo Hidalgo Filho, C.M.; Reis, R.D.; Reis, R.M.; Marques, M.M.C. A systematic review of micrornas involved in cervical cancer progression. Cells 2021, 10, 668. [Google Scholar] [CrossRef]
- Pedroza-Torres, A.; Fernández-Retana, J.; Peralta-Zaragoza, O.; Jacobo-Herrera, N.; Cantú de Leon, D.; Cerna-Cortés, J.F.; Lopez-Camarillo, C.; Pérez-Plasencia, C. A microRNA expression signature for clinical response in locally advanced cervical cancer. Gynecol. Oncol. 2016, 142, 557–565. [Google Scholar] [CrossRef]
- Jia, W.; Wu, Y.; Zhang, Q.; Gao, G.E.; Zhang, C.; Xiang, Y. Expression profile of circulating microRNAs as a promising fingerprint for cervical cancer diagnosis and monitoring. Mol. Clin. Oncol. 2015, 3, 851–858. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, D.; Wang, F.; Xu, D.; Guo, Y.; Cui, W. Serum miRNAs panel (miR-16-2*, miR-195, miR-2861, miR-497) as novel non-invasive biomarkers for detection of cervical cancer. Sci. Rep. 2015, 5, 17942. [Google Scholar] [CrossRef]
- Xin, F.; Liu, P.; Ma, C.F. A circulating serum miRNA panel as early detection biomarkers of cervical intraepithelial neoplasia. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4846–4851. [Google Scholar] [PubMed]
- Palatnik, A.; Ye, S.; Kendziorski, C.; Iden, M.; Zigman, J.S.; Hessner, M.J.; Rader, J.S. Correction: Identification of a serum-induced transcriptional signature associated with metastatic cervical cancer. PLoS ONE 2018, 13, e0193687, Erratum in PLoS ONE 2017, 12, e0181242. [Google Scholar] [CrossRef]
- Aftab, M.; Poojary, S.S.; Seshan, V.; Kumar, S.; Agarwal, P.; Tandon, S.; Zutshi, V.; Das, B.C. Urine miRNA signature as a potential non-invasive diagnostic and prognostic biomarker in cervical cancer. Sci. Rep. 2021, 11, 10323. [Google Scholar] [CrossRef]
- Ferrero, G.; Cordero, F.; Tarallo, S.; Arigoni, M.; Riccardo, F.; Gallo, G.; Ronco, G.; Allasia, M.; Kulkarni, N.; Matullo, G.; et al. Small non-coding RNA profiling in human biofluids and surrogate tissues from healthy individuals: Description of the diverse and most represented species. Oncotarget 2017, 9, 3097–3111. [Google Scholar] [CrossRef]
- Ma, G.; Song, G.; Zou, X.; Shan, X.; Liu, Q.; Xia, T.; Zhou, X.; Zhu, W. Circulating plasma microRNA signature for the diagnosis of cervical cancer. Cancer Biomark. 2019, 26, 491–500. [Google Scholar] [CrossRef]
- Qiu, H.; Liang, D.; Liu, L.; Xiang, Q.; Yi, Z.; Ji, Y. A novel circulating MiRNA-based signature for the diagnosis and prognosis prediction of early-stage cervical cancer. Technol. Cancer Res. Treat. 2020, 19, 1533033820970667. [Google Scholar] [CrossRef]
- Cao, M.M.; Liu, Q.X.; Zou, X.; Fan, X.C.; Liu, C.; Zhang, S.Y.; Wang, T.S.; Li, C.Y.; Zhang, C.; Geng, X.N.; et al. Identification of a serum three-microRNA signature for cervical cancer diagnosis. Chin. Med. J. 2021, 134, 1756–1758. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, Y.Y.; Guo, Y.L.; Li, Z.J.; Geng, L. Profiling of microRNA-mRNA reveals roles of microRNAs in cervical cancer. Chin. Med. J. 2012, 125, 4270–4276. [Google Scholar] [PubMed]
- Sharma, G.; Agarwal, S.M. Identification of critical microRNA gene targets in cervical cancer using network properties. Microrna 2014, 3, 37–44. [Google Scholar] [CrossRef]
- Wang, N.; Xu, Z.; Wang, K.; Zhu, M.; Li, Y. Construction and analysis of regulatory genetic networks in cervical cancer based on involved microRNAs, target genes, transcription factors and host genes. Oncol. Lett. 2014, 7, 1279–1283. [Google Scholar] [CrossRef]
- Mo, W.; Tong, C.; Zhang, Y.; Lu, H. microRNAs’ differential regulations mediate the progress of Human Papillomavirus (HPV)-induced Cervical Intraepithelial Neoplasia (CIN). BMC Syst. Biol. 2015, 9, 4. [Google Scholar] [CrossRef]
- He, Y.; Lin, J.; Ding, Y.; Liu, G.; Luo, Y.; Huang, M.; Xu, C.; Kim, T.K.; Etheridge, A.; Lin, M.; et al. A systematic study on dysregulated microRNAs in cervical cancer development. Int. J. Cancer 2016, 138, 1312–1327. [Google Scholar] [CrossRef]
- Kori, M.; Yalcin Arga, K. Potential biomarkers and therapeutic targets in cervical cancer: Insights from the meta-analysis of transcriptomics data within network biomedicine perspective. PLoS ONE 2018, 13, e0200717. [Google Scholar] [CrossRef]
- Paul, S.; Madhumita. RFCM3: Computational method for identification of miRNA-mRNA regulatory modules in cervical cancer. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1729–1740. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J.; Gao, F.; Li, S.; Nie, S.; Meng, H.; Sun, R.; Wan, Y.; Jiang, Y.; Ma, X.; et al. A microRNA-messenger RNA regulatory network and its prognostic value in cervical cancer. DNA Cell Biol. 2020, 39, 1328–1346. [Google Scholar] [CrossRef]
- Zong, S.; Liu, X.; Zhou, N.; Yue, Y. E2F7, EREG, miR-451a and miR-106b-5p are associated with the cervical cancer development. Arch. Gynecol. Obstet. 2019, 299, 1089–1098. [Google Scholar] [CrossRef]
- Mei, Y.; Jiang, P.; Shen, N.; Fu, S.; Zhang, J. Identification of miRNA-mRNA regulatory network and construction of prognostic signature in cervical cancer. DNA Cell Biol. 2020, 39, 1023–1040. [Google Scholar] [CrossRef]
- Masud Karim, S.M.; Liu, L.; Le, T.D.; Li, J. Identification of miRNA-mRNA regulatory modules by exploring collective group relationships. BMC Genomics 2016, 17 (Suppl. 1), 7. [Google Scholar] [CrossRef]
- Huang, J.C.; Morris, Q.D.; Frey, B.J. Bayesian inference of microRNA targets from sequence and expression data. J. Comput. Biol. 2007, 14, 550–563. [Google Scholar] [CrossRef]
- Stingo, F.C.; Chen, Y.A.; Vannucci, M.; Barrier, M.; Mirkes, P.E. A bayesian graphical modeling approach to microRNA regulatory network inference. Ann. Appl. Stat. 2010, 4, 2024–2048. [Google Scholar] [CrossRef]
- Liu, B.; Li, J.; Tsykin, A.; Liu, L.; Gaur, A.B.; Goodall, G.J. Exploring complex miRNA-mRNA interactions with Bayesian networks by splitting-averaging strategy. BMC Bioinform. 2009, 10, 408. [Google Scholar] [CrossRef]
- Dogan, H.; Hakguder, Z.; Madadjim, R.; Scott, S.; Pierobon, M.; Cui, J. Elucidation of dynamic microRNA regulations in cancer progression using integrative machine learning. Brief. Bioinform. 2021, 22, bbab270. [Google Scholar] [CrossRef]
- Nersisyan, S.; Galatenko, A.; Galatenko, V.; Shkurnikov, M.; Tonevitsky, A. miRGTF-net: Integrative miRNA-gene-TF network analysis reveals key drivers of breast cancer recurrence. PLoS ONE 2021, 16, e0249424. [Google Scholar] [CrossRef]
- Joung, J.G.; Hwang, K.B.; Nam, J.W.; Kim, S.J.; Zhang, B.T. Discovery of microRNA-mRNA modules via population-based probabilistic learning. Bioinformatics 2007, 23, 1141–1147. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Q.; Liu, J.; Zhou, X.J. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. Bioinformatics 2011, 27, i401–i409. [Google Scholar] [CrossRef]
- Tran, D.H.; Satou, K.; Ho, T.B. Finding microRNA regulatory modules in human genome using rule induction. BMC Bioinformatics 2008, 9 (Suppl. 12), S5. [Google Scholar] [CrossRef]
- Gamberger, D.; Lavrac, N.; Zelezný, F.; Tolar, J. Induction of comprehensible models for gene expression datasets by subgroup discovery methodology. J. Biomed. Inform. 2004, 37, 269–284. [Google Scholar] [CrossRef]
- Yousef, M.; Goy, G.; Bakir-Gungor, B. miRModuleNet: Detecting miRNA-mRNA regulatory modules. Front. Genet. 2022, 13, 767455. [Google Scholar] [CrossRef]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef]
- Aziz, M.A.; Islam, M.S. MAP3K1 rs889312 polymorphism and cancer prognosis: A systematic review and meta-analysis. Cancer Rep. 2023, 6, e1773. [Google Scholar] [CrossRef]
- Peralta-Zaragoza, O.; Deas, J.; Gómez-Cerón, C.; García-Suastegui, W.A.; Fierros-Zárate, G.d.S.; Jacobo-Herrera, N.J. HPV-Based screening, triage, treatment, and followup strategies in the management of cervical intraepithelial neoplasia. Obstet. Gynecol. Int. 2013, 2013, 912780. [Google Scholar] [CrossRef]
- Drakopoulou, E.; Anagnou, N.P.; Pappa, K.I. Gene Therapy for malignant and benign gynaecological disorders: A systematic review of an emerging success story. Cancers 2022, 14, 3238. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Pineros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020; Available online: http://gco.iarc.fr/today/home (accessed on 23 April 2024).
- Schubert, M.; Bauerschlag, D.O.; Muallem, M.Z.; Maass, N.; Alkatout, I. Challenges in the diagnosis and individualized treatment of cervical cancer. Medicina 2023, 59, 925. [Google Scholar] [CrossRef]
- Sagae, S.; Toita, T.; Matsuura, M.; Saito, M.; Matsuda, T.; Sato, N.; Shimizu, A.; Endo, T.; Fujii, M.; Gaffney, D.K.; et al. Improvement in radiation techniques for locally advanced cervical cancer during the last two decades. Int. J. Gynecol. Cancer 2023, 33, 1295–1303. [Google Scholar] [CrossRef]
- Ma, X.; Fang, J.; Zhang, L.; Huang, Y.; Shen, H.; Ma, X.; Zhang, S.; Zhang, B. Efficacy and safety of adjuvant chemotherapy for locally advanced cervical cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2023, 184, 103953. [Google Scholar] [CrossRef]
- US National Institutes of Health. Database of Clinical Trials. Revision: v2.9.6. Available online: http://clinicaltrials.gov/ct2/home (accessed on 23 April 2024).
- Ravegnini, G.; De Leo, A.; Coada, C.; Gorini, F.; de Biase, D.; Ceccarelli, C.; Dondi, G.; Tesei, M.; De Crescenzo, E.; Santini, D.; et al. Identification of miR-499a-5p as a potential novel biomarker for risk stratification in endometrial cancer. Front. Oncol. 2021, 11, 757678. [Google Scholar] [CrossRef]
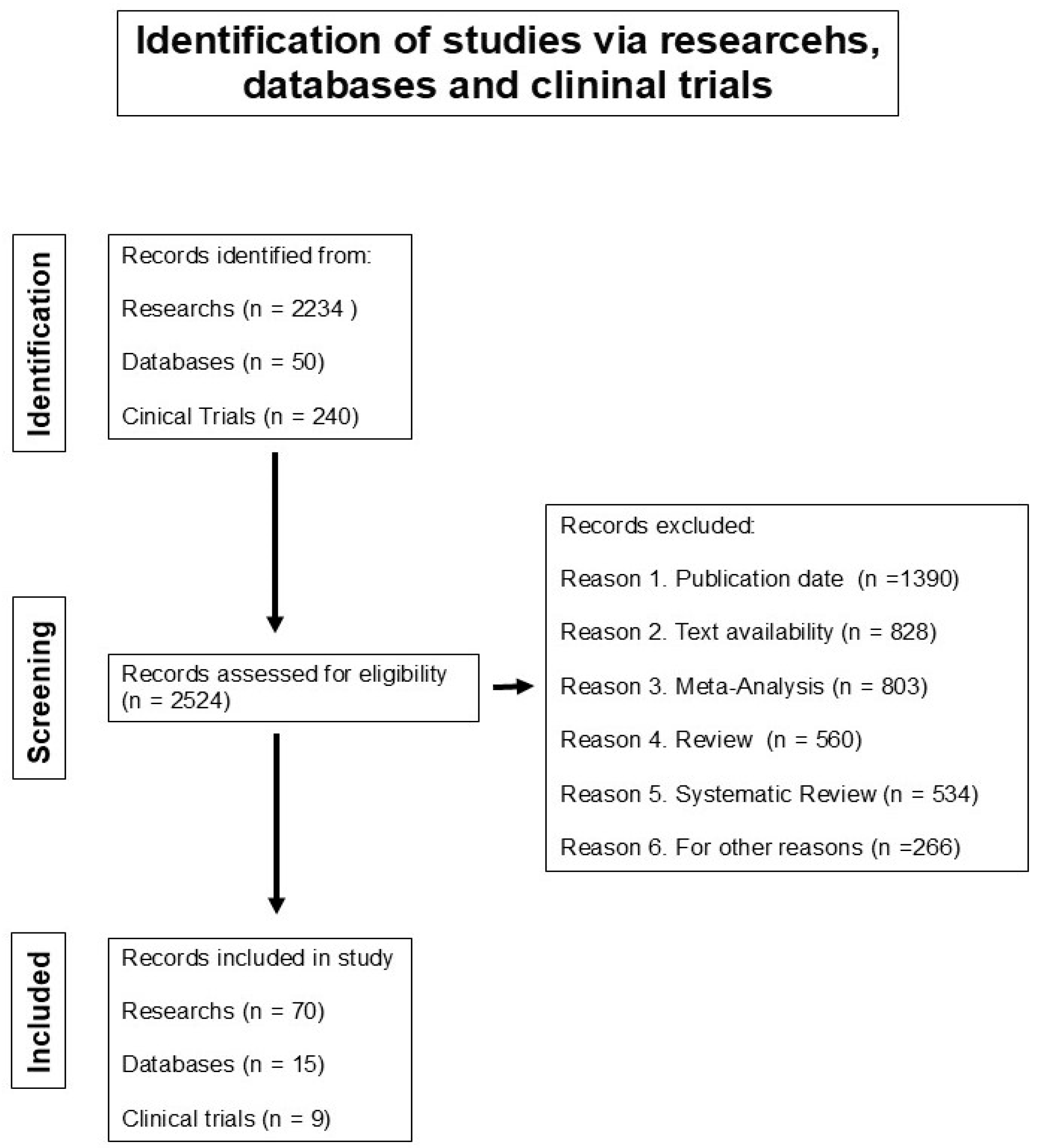


{kind=link}
{kind=link}
| Biospecimen | Genomic Signature of Up-Expressed microRNAs | Genomic Signature of Down-Expressed microRNAs | Reference |
|---|---|---|---|
| Cervical tissues | miR-9, miR-21, miR-200a, miR-203, and miR-218. | [27] | |
| Cervical tissues | miR-26a, miR-29a, miR-99a, miR-143, miR-145, miR-199a, miR-203 and miR-513. | miR-16, miR-27a, miR-106a, miR-142-5p, miR-197 and miR-205. | [28] |
| Cervical tissues | miR-9, miR-10a, miR-20b, miR-34b, miR-34c, miR-193b, miR-203, miR-338, miR-345, miR-424, miR-512-5p, and miR-518a | [29] | |
| Cervical tissues | miR-15b, miR-16, miR-21, miR-21* miR-188-5p, miR-483-5p, miR-663, miR-765, miR-1300 | miR-142-3, miR-149, miR-152, miR-218, miR-374b, miR-376c | [30] |
| Cervical tissues | miR-1, miR-21 | [31] | |
| Cervical tissues | miR-466 | [33] | |
| Cervical tissues | let-7f-5p, miR-128-2-5p, miR-130a-3p, miR-202-3p, miR-205-5p, miR-323a-5p, miR-3136-3p | miR-381-3p, miR-4531 | [34] |
| Cervical tissues | miR-216b-5p, miR-585-5p, and miR-7641 | [35] | |
| Cervical tissues | miR-145, miR-200c, miR218-1 | [36] | |
| Cervical tissues | miR-21-5p, miR-135b-5p, miR-363-3p, and miR-429 | miR-136-5p, miR-218-5p, miR-377-3p, miR-497-5p, miR-1184, miR-3196, miR-4687-3p, miR-5587-3p, and miR-5572 | [37] |
| Cervical tissues | miR-20a, miR-92a, miR-141, miR-183*, miR-210, and miR-944 | [38] | |
| Cervical tissues | miR-9-5p, miR-15b-5p, and miR-28-5p | miR-100-5p, miR-125b-5p, miR-149-5p, miR-203a-3p, and miR-375 | [39] |
| Cervical tissues | miR-378c and miR-642a | [40] | |
| Cervical tissues | miR-130a | miR-30c, miR-143, miR-372, and miR-375 | [41] |
| Cervical tissues | miR-144, miR-147b, miR-218-2, miR-425, miR-451, miR-483, and miR-486. | [42] | |
| Cervical tissues | miR-34a, miR-200a, and miR-455 | [43] | |
| Cervical tissues | miR-31, miR-92a, miR-93, miR-96-5p, miR-199b-5p, miR-200a, and miR-224 | miR-1, miR-107, miR-132, miR-139-3p, miR-143, miR-195, miR-335-5p, miR-337-3p, miR-361-5p, miR-383-5p, miR-411, miR-424-5p, miR-433, miR-545, miR-573, miR-874, miR-1284, miR-2861, and miR-3941 | [44] |
| Cervical tissues | miR-31-3p, miR-100-5p, miR-125a-5p, miR-125b-5p, miR-200a-5p, miR-342, and miR-3676 | [45] | |
| Circulating | miR-21, miR-25, miR-26b, miR-29a, miR-29c, miR-101, miR-140-3p, miR-146b-5p, miR-191, miR-200a, miR-423-3p, and miR-486-5p | [46] | |
| Circulating | miR-16-2* and miR-497 | miR-195 and miR-2861 | [47] |
| Circulating | miR-9, miR-10a, miR-20a, and miR-196a | [48] | |
| Circulating | miR-23a-3p, miR-23b-3p, miR-193b-5p, and miR-944 | [49] | |
| Circulating | miR-21, miR-199a, and miR-155-5p | [50] | |
| Circulating | miR-320a, miR-589-5p, miR-636, miR-1273a, miR-3960, miR-4419a, miR-4497, miR-4709-5p, miR-4792, miR-7641-1, and miR-7641-2 | [51] | |
| Circulating | miR-21-5p, miR-146a-5p, miR-151a-3p, and miR-2110 | [52] | |
| Circulating | miR-21 | miR-125b and miR-370 | [53] |
| Circulating | miR-20a-5p and miR-122-5p | miR-133a-3p | [54] |
| ClinicalTrials.gov Identifier | Study Title/Country | Condition Diseases and Biospecimen | Study Type, Study Design and Status | Study Population | Current Primary Outcome | First Submitted Date/ Last Update Posted Date |
|---|---|---|---|---|---|---|
| NCT04087785 | MicroRNA profile associated with positive lymph node metastasis in early-stage cervical cancer. México | Cervical cancer Samples from formalin-fixed paraffin-embedded tissue blocks. | Observational. Observational, retrospective Completed. | Patients diagnosed with cervical cancer between January 2006 and December 2013 at the Department of Oncologic Gynecology of the National Cancer Institute (Mexico City). | Prognostic miRNAs | 11 September 2019 13 September 2019 |
| NCT03824613 | Urinary microRNA expression in endometrial cancer patients—a feasibility study United Kingdom | Endometrial Cancer. Urine samples with DNA or microRNA | Observational. Observational, prospective Completed | Endometrial cancer patients | Accuracy of predictive value of miRNA test in detecting endometrial cancer | 5 December 2018 1 March 2021 |
| NCT04845425 | Identification of miRNAs in endometrial cancer as novel diagnostic and prognostic biomarkers Italy | Endometrial Cancer. Biospecimen is not provided. | Observational. Cohort, retrospective Recruiting | Women with endometrial cancer | Evaluate miRNA expression based on the 4 molecular groups recently identified | 10 April 2021 21 July 2022 |
| NCT03776630 | Exploring the Potential of Novel Biomarkers Based on Plasma microRNAs for a Better Management of Pelvic Gynecologic Tumors France | Ovarian cancer Endometrial cancer Blood sample | Interventional. Allocation: Non-Randomized Intervention Model: Parallel Assignment Masking: None (open label) Primary purpose: Diagnostic Active, not recruiting | Patients’ diagnosis with ovarian cancer and endometrial cancer | It will aim to investigate the links of the 5-miR index with classical predictors of lymph node involvement in the context of endometrial cancer. It also will aim to validate multiplexed homogenous miR detection based on RCA-FRET compared to conventional RT-qPCR in plasma samples. | 24 September 2018 30 June 2022 |
| NCT01119573 | MicroRNAs associated with lymph node metastasis in endometrial cancer. United State | Endometrial cancer. Tumor tissue samples from endometrial cancer patients. | Observational. Unknown status | Not provided | Association between microRNA expression and lymph node metastasis | 6 May 2010 22 June 2010 |
| NCT04010487 | A multi-omics study on the pathogenesis of malignant transformation of adenomyosis. China | Adenomyosis, endometrial cancer, ectopic endometrial tissue, eutopic endometrium | Observational, case-control, retrospective. Recruiting | Group 1. Patients pathologically conformed endometrial carcinoma arising in adenomyosis. Group 2. Patients pathologically diagnosed with adenomyosis | Frequencies of somatic driving mutations. Frequencies of alteration of RNA expression. | 3 July 2019 18 July 2019 |
| NCT02983279 | Caloric restriction for oncology research: pre-operative caloric restriction prior to definitive oncologic surgery. United State | Breast carcinoma, endometrial carcinoma, prostate carcinosarcoma | Interventional, Single group assignment, Completed | Patients diagnosed with prostate, endometrial or breast cancer. | Change in miR-21 expression assessed in serum will be evaluated by a two-sided paired t-test. | 14 November 2016 6 January 2023 |
| NCT05292573 | MicroRNAs as biomarkers of predicting future endometrial malignancy and longitudinal follow-up with randomized intervention in women with endometrial hyperplasia without atypia. Taiwan | Endometrial tissues and sera | Interventional, phase 3. Randomized, parallel assignment. Recruiting. | This study will prospectively enroll a total of 1000 patients with simple hyperplasia/complex hyperplasia without atypia to endometrial cancer of the 1989–2011 cohort | To evaluate the ROC (operating characteristic curve) of the prediction microRNA panel of 3 microRNAs. | 8 April 2021 23 March 2022 |
| NCT03742869 | Multi-omics Study on the Human Papillomavirus Integration and Tumorigenesis of Uterine Cervical Adenocarcinoma. China | Samples with DNA from peripheral venous blood and 50 μL cancer tissue and tissue adjacent to cancer will be collected from eligible patients. | Observational Prospective case-control Unknown status | All patients confirmed primary adenocarcinoma of the uterine cervix | Alteration of mRNA, miRNA, and lncRNA pattern expression in patients with and without HPV integration | 11 November 2018 23 November 2021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Plasencia, C.; Orbe-Orihuela, Y.C.; Méndez-Herrera, A.; Deas, J.; Gómez-Cerón, C.; Jiménez-Wences, H.; Ortiz-Ortiz, J.; Fernández-Tilapa, G.; Clemente-Soto, A.F.; Parra-Unda, J.R.; et al. Regulatory Genetic Networks by microRNAs: Exploring Genomic Signatures in Cervical Cancer. Biomedicines 2025, 13, 1457. https://doi.org/10.3390/biomedicines13061457
Pérez-Plasencia C, Orbe-Orihuela YC, Méndez-Herrera A, Deas J, Gómez-Cerón C, Jiménez-Wences H, Ortiz-Ortiz J, Fernández-Tilapa G, Clemente-Soto AF, Parra-Unda JR, et al. Regulatory Genetic Networks by microRNAs: Exploring Genomic Signatures in Cervical Cancer. Biomedicines. 2025; 13(6):1457. https://doi.org/10.3390/biomedicines13061457
Chicago/Turabian StylePérez-Plasencia, Carlos, Yaneth Citlalli Orbe-Orihuela, Armando Méndez-Herrera, Jessica Deas, Claudia Gómez-Cerón, Hilda Jiménez-Wences, Julio Ortiz-Ortiz, Gloria Fernández-Tilapa, Aldo Francisco Clemente-Soto, Jesús Ricardo Parra-Unda, and et al. 2025. "Regulatory Genetic Networks by microRNAs: Exploring Genomic Signatures in Cervical Cancer" Biomedicines 13, no. 6: 1457. https://doi.org/10.3390/biomedicines13061457
APA StylePérez-Plasencia, C., Orbe-Orihuela, Y. C., Méndez-Herrera, A., Deas, J., Gómez-Cerón, C., Jiménez-Wences, H., Ortiz-Ortiz, J., Fernández-Tilapa, G., Clemente-Soto, A. F., Parra-Unda, J. R., Velarde-Felix, J. S., Rodríguez-Dorantes, M., & Peralta-Zaragoza, O. (2025). Regulatory Genetic Networks by microRNAs: Exploring Genomic Signatures in Cervical Cancer. Biomedicines, 13(6), 1457. https://doi.org/10.3390/biomedicines13061457