
Orchestration of Gut–Liver-Associated Transcription Factors in MAFLD: From Cross-Organ Interactions to Therapeutic Innovation

Abstract
:1. Introduction
2. Metabolism-Related TFs in MAFLD
2.1. PPARs
2.2. SREBP1c
2.3. CHREBP
2.4. CREBH
2.5. FXR
2.6. PXR
2.7. LXR
2.8. AHR
2.9. THR-β
2.10. HNF4α
2.11. FOX
2.12. HIF2α
2.13. MYC
3. Inflammation and Immune-Related TFs in MAFLD
3.1. PPARs
3.2. FXR
3.3. STAT3
3.4. NRF2
3.5. IRF1
3.6. NR4A
3.7. TFEB
4. Microbiota and Metabolite-Related TFs in MAFLD
4.1. PPARs
4.2. FXR
4.3. PXR
4.4. AHR
4.5. HIF2α
4.6. NR1D1
5. Targeted Therapy Strategies of TFs
6. Challenges and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| α-SMA | α-Smooth muscle actin |
| ABCA1 | ATP binding cassette subfamily A member 1 |
| ABCG1 | ATP binding cassette subfamily G member 1 |
| ACC | Acetyl coenzyme A carboxylase |
| ACLY | ATP citrate lyase |
| ACOX1 | Acyl-CoA oxidase 1 |
| AHR | Aryl hydrocarbon receptor |
| AMPK | AMP-activated protein kinase |
| ApoC | Apolipoprotein C |
| ATF3 | Activating transcription factor 3 |
| BA | Bile acid |
| Bcl-xL | BCL2 like 1 |
| BMI | Body mass index |
| CA | Cholic acid |
| Ccl2 | C-C motif chemokine ligand 2 |
| CDCA | Chenodeoxycholic acid |
| cGAS-STING | Cyclic GMP-AMP synthase-stimulator of interferon genes |
| CHREBP | Carbohydrate response element binding protein |
| CPP | Cell-penetrating peptides |
| CPT1A | Carnitine palmitoyltransferase 1A |
| CRC | Colorectal cancer |
| CREBH | cAMP-responsive element-binding protein H |
| CTSB | Cathepsin B |
| CYP7A1 | Cytochrome P450 family 7 subfamily A member 1 |
| CYP8B1 | Cytochrome P450 family 8 subfamily B member 1 |
| Ddit4 | DNA damage inducible transcript 4 |
| DIO | Diet-induced obese |
| DNL | De novo lipogenesis |
| ELOVL6 | Elongation of very long-chain fatty acids-like 6 |
| ER | Endoplasmic reticulum |
| FABP4 | Fatty acid binding protein 4 |
| FAM3A | Family with sequence similarity 3 member A |
| FAO | Fatty acid oxidation |
| FASN | Fatty acid synthase |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FGF19 | Fibroblast growth factor 19 |
| FGF21 | Fibroblast growth factor 21 |
| FMT | Fecal microbiota transplantation |
| FOS | Fructo-oligosaccharides |
| FOXA2 | Forkhead box a2 |
| FOXO1 | Forkhead box o1 |
| FXR | Farnesoid X receptor |
| GLP-1 | Glucagon like peptide 1 |
| GPBAR1 | G protein-coupled bile acid receptor 1 |
| GVB | Gut-vascular barrier |
| HFD | High-fat diet |
| HIF2α | Hypoxia-inducible factor 2α |
| HMGCS2 | 3-Hydroxy-3-methylglutaryl-coa synthase 2 |
| HNF4α | Hepatocyte nuclear factor 4α |
| HO-1 | Heme oxygenase 1 |
| HPC | Hepatic progenitor cells |
| HSC | Hepatic stellate cells |
| iNOS | inducible nitric oxide synthase |
| IRF1 | Interferon regulatory factor 1 |
| KLF10 | Krüppel-like factor 10 |
| LAMP1 | Lysosomal associated membrane protein 1 |
| LDLR | Low-density lipoprotein receptor |
| LPIN1 | Lipin 1 |
| LPS | Lipopolysaccharide |
| LXR | Liver X receptor |
| MAFLD | Metabolic dysfunction-associated fatty liver disease |
| MAP1LC3B | Microtubule associated protein 1 light chain 3 beta |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| MCD | Methionine-choline-deficient |
| Mcl-1 | Myeloid cell leukemia 1 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MRE | Magnetic resonance elastography |
| mTOR | Mechanistic target of rapamycin |
| MTTP | Microsomal triglyceride transfer protein |
| MYC | Myelocytomatosis |
| NAS | NAFLD activity score |
| NLRP3 | NLR family pyrin domain containing 3 |
| NPC1L1 | Niemann–Pick c1-like 1 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| NR1D1 | Nuclear receptor subfamily 1 group d member 1 |
| NR4A | Nuclear receptor subfamily 4 group A |
| NRF2 | Nuclear factor erythroid 2 like 2 |
| Osbpl3 | Oxysterol binding protein like 3 |
| PHD | Prolyl hydroxylase domain enzymes |
| PPAR | Peroxisome proliferator-activated receptor |
| PROTAC | Proteolysis-targeting chimeras |
| PSA | Puromycin-sensitive aminopeptidase |
| PXR | Pregnane X receptor |
| ROS | Reactive oxygen species |
| SCD-1 | Stearoyl coenzyme A desaturase 1 |
| SCFAs | Short-chain fatty acids |
| SLC13A5 | Solute carrier family 13 member 5 |
| SLC27A4 | Solute carrier family 27 member 4 |
| SLUG/SNAI2 | Snail family transcriptional repressor 2 |
| SMAD2 | SMAD family member 2 |
| SOD2 | Superoxide dismutase 2 |
| SREBP1c | Sterol regulatory element binding protein-1c |
| STAT3 | Signal transducer and activator of transcription 3 |
| TCF7L2 | Transcription factor 7-like 2 |
| TF | Transcription factor |
| TFE3 | Transcription factor E3 |
| TFEB | Transcription factor EB |
| TG | Triglyceride |
| TGF-β1 | Transforming growth factor β1 |
| TH | Thyroid hormone |
| THR-β | Thyroid hormone receptor β |
| Th17 | T helper 17 cells |
| TIMP-1 | Tissue inhibitor of metalloproteinases 1 |
| Treg | Regulatory T cells |
| TRIB3 | Tribbles homologue 3 |
| VLDL | Very low-density lipoprotein |
| ZAC1 | Zinc-finger protein regulator of apoptosis and cell-cycle arrest 1 |
| ZEB2 | Zinc finger e-box binding homeobox 2 |
References
- Le, M.H.; Yeo, Y.H.; Li, X.; Li, J.; Zou, B.; Wu, Y.; Ye, Q.; Huang, D.Q.; Zhao, C.; Zhang, J.; et al. 2019 Global NAFLD Prevalence: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 2809–2817.e28. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, F.; Wang, W.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; She, Z.G.; Zhu, L.; Cai, J.; Li, H. Epidemiological Features of NAFLD From 1999 to 2018 in China. Hepatology 2020, 71, 1851–1864. [Google Scholar] [CrossRef]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, L.; Chen, Q.; Wu, L.; He, W.; Tu, D.; Wang, S.; Chen, Y.; Liu, S.; Xie, Z.; et al. Disulfiram ameliorates nonalcoholic steatohepatitis by modulating the gut microbiota and bile acid metabolism. Nat. Commun. 2022, 13, 6862. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, X.; Liu, W.; Wei, H.; Liang, W.; Zhou, Y.; Ding, Y.; Ji, F.; Ho-Kwan Cheung, A.; Wong, N.; et al. Bifidobacterium pseudolongum-generated acetate suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma. J. Hepatol. 2023, 79, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Wang, J.; Li, Y.; Li, M.; Zhao, M.; Ge, K.; Zheng, D.; Cheung, K.C.P.; Liao, B.; Wang, S.; et al. Hyodeoxycholic acid alleviates non-alcoholic fatty liver disease through modulating the gut-liver axis. Cell Metab. 2023, 35, 1752–1766.e8. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Thomas, S.; Sabo, P.J.; Eisen, M.B.; Stamatoyannopoulos, J.A.; Biggin, M.D. The role of chromatin accessibility in directing the widespread, overlapping patterns of Drosophila transcription factor binding. Genome Biol. 2011, 12, R34. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ai, Y.; Hu, C.; Cassim Bawa, F.N.; Xu, Y. Transcription factors, metabolic dysfunction-associated fatty liver disease, and therapeutic implications. Genes Dis. 2025, 12, 101372. [Google Scholar] [CrossRef]
- Gallage, S.; Ali, A.; Barragan Avila, J.E.; Seymen, N.; Ramadori, P.; Joerke, V.; Zizmare, L.; Aicher, D.; Gopalsamy, I.K.; Fong, W.; et al. A 5:2 intermittent fasting regimen ameliorates NASH and fibrosis and blunts HCC development via hepatic PPARα and PCK1. Cell Metab. 2024, 36, 1371–1393.e7. [Google Scholar] [CrossRef]
- Adorini, L.; Trauner, M. FXR agonists in NASH treatment. J. Hepatol. 2023, 79, 1317–1331. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Tacke, F. Metabolic reprogramming in liver fibrosis. Cell Metab. 2024, 36, 1439–1455. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [CrossRef]
- Habibullah, M.; Jemmieh, K.; Ouda, A.; Haider, M.Z.; Malki, M.I.; Elzouki, A.N. Metabolic-associated fatty liver disease: A selective review of pathogenesis, diagnostic approaches, and therapeutic strategies. Front. Med. 2024, 11, 1291501. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.K.S.; Peixoto, C.A. Role of peroxisome proliferator-activated receptors in non-alcoholic fatty liver disease inflammation. Cell. Mol. Life Sci. 2018, 75, 2951–2961. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chen, X.; Sun, Q.; Li, J.; Wang, Q.; Wei, P.; Wang, W.; Li, C.; Wang, Y. Valerenic acid attenuates pathological myocardial hypertrophy by promoting the utilization of multiple substrates in the mitochondrial energy metabolism. J. Adv. Res. 2025, 68, 241–256. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Du, M.; Wang, X.; Yuan, L.; Liu, B.; Mao, X.; Huang, D.; Yang, L.; Huang, K.; Zhang, F.; Wang, Y.; et al. Targeting NFATc4 attenuates non-alcoholic steatohepatitis in mice. J. Hepatol. 2020, 73, 1333–1346. [Google Scholar] [CrossRef]
- Azar, S.; Udi, S.; Drori, A.; Hadar, R.; Nemirovski, A.; Vemuri, K.V.; Miller, M.; Sherill-Rofe, D.; Arad, Y.; Gur-Wahnon, D.; et al. Reversal of diet-induced hepatic steatosis by peripheral CB1 receptor blockade in mice is p53/miRNA-22/SIRT1/PPARα dependent. Mol. Metab. 2020, 42, 101087. [Google Scholar] [CrossRef]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Soufi, N.; Chambers, K.T.; Chen, Z.; Schweitzer, G.G.; McCommis, K.S.; Erion, D.M.; Graham, M.J.; Su, X.; Finck, B.N. Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes 2014, 63, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Lin, S.; Bian, S.; Zheng, W.; Qu, L.; Fan, Y.; Lu, C.; Xiao, M.; Zhou, P. USP7 mediates pathological hepatic de novo lipogenesis through promoting stabilization and transcription of ZNF638. Cell Death Dis. 2020, 11, 843. [Google Scholar] [CrossRef]
- Xiao, Z.; Chu, Y.; Qin, W. IGFBP5 modulates lipid metabolism and insulin sensitivity through activating AMPK pathway in non-alcoholic fatty liver disease. Life Sci. 2020, 256, 117997. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, H.; Jiang, L.; Shang, Q.; Yin, L.; Lin, J.D.; Wu, W.S.; Rui, L. Hepatic Slug epigenetically promotes liver lipogenesis, fatty liver disease, and type 2 diabetes. J. Clin. Investig. 2020, 130, 2992–3004. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, J.; Deng, H.; Ma, R.; Liao, J.Y.; Liang, H.; Hu, J.; Li, J.; Guo, Z.; Cai, J.; et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell 2020, 183, 76–93.e22. [Google Scholar] [CrossRef]
- Zhang, S.; Guo, F.; Yu, M.; Yang, X.; Yao, Z.; Li, Q.; Wei, Z.; Feng, K.; Zeng, P.; Zhao, D.; et al. Reduced Nogo expression inhibits diet-induced metabolic disorders by regulating ChREBP and insulin activity. J. Hepatol. 2020, 73, 1482–1495. [Google Scholar] [CrossRef]
- Lei, Y.; Hoogerland, J.A.; Bloks, V.W.; Bos, T.; Bleeker, A.; Wolters, H.; Wolters, J.C.; Hijmans, B.S.; van Dijk, T.H.; Thomas, R.; et al. Hepatic Carbohydrate Response Element Binding Protein Activation Limits Nonalcoholic Fatty Liver Disease Development in a Mouse Model for Glycogen Storage Disease Type 1a. Hepatology 2020, 72, 1638–1653. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Takao, K.; Kato, T.; Horikawa, Y.; Takeda, J. ChREBP Reciprocally Regulates Liver and Plasma Triacylglycerol Levels in Different Manners. Nutrients 2018, 10, 1699. [Google Scholar] [CrossRef]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef]
- Kim, H.; Song, Z.; Zhang, R.; Davies, B.S.J.; Zhang, K. A hepatokine derived from the ER protein CREBH promotes triglyceride metabolism by stimulating lipoprotein lipase activity. Sci. Signal. 2023, 16, eadd6702. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Araki, M.; Nakagawa, Y.; Deisen, L.; Lundsgaard, A.; Kanta, J.M.; Holm, S.; Johann, K.; Brings Jacobsen, J.C.; Jähnert, M.; et al. Dietary medium-chain fatty acids reduce hepatic fat accumulation via activation of a CREBH-FGF21 axis. Mol. Metab. 2024, 87, 101991. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Orihara, K.; Oikawa, F.; Han, S.I.; Kuba, M.; Okuda, K.; Satoh, A.; Osaki, Y.; Takeuchi, Y.; Aita, Y.; et al. Intestinal CREBH overexpression prevents high-cholesterol diet-induced hypercholesterolemia by reducing Npc1l1 expression. Mol. Metab. 2016, 5, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Clifford, B.L.; Sedgeman, L.R.; Williams, K.J.; Morand, P.; Cheng, A.; Jarrett, K.E.; Chan, A.P.; Brearley-Sholto, M.C.; Wahlstrom, A.; Ashby, J.W.; et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021, 33, 1671–1684. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Shen, C.; Pan, Z.; Xie, W.; Zhao, J.; Miao, D.; Zhao, L.; Liu, M.; Zhong, Y.; Zhong, C.; Gonzalez, F.J.; et al. Hepatocyte-specific SLC27A4 deletion ameliorates nonalcoholic fatty liver disease in mice via suppression of phosphatidylcholine-mediated PXR activation. Metabolism 2025, 162, 156054. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, B.; Zhao, J.; Feng, Q.; Peng, J.; Hu, Y.; Zhao, Y. Biological mechanisms and related natural modulators of liver X receptor in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2019, 113, 108778. [Google Scholar] [CrossRef]
- Wang, J.Q.; Li, L.L.; Hu, A.; Deng, G.; Wei, J.; Li, Y.F.; Liu, Y.B.; Lu, X.Y.; Qiu, Z.P.; Shi, X.J.; et al. Inhibition of ASGR1 decreases lipid levels by promoting cholesterol excretion. Nature 2022, 608, 413–420. [Google Scholar] [CrossRef]
- Xu, C.X.; Wang, C.; Zhang, Z.M.; Jaeger, C.D.; Krager, S.L.; Bottum, K.M.; Liu, J.; Liao, D.F.; Tischkau, S.A. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int. J. Obes. 2015, 39, 1300–1309. [Google Scholar] [CrossRef]
- Kannt, A.; Wohlfart, P.; Madsen, A.N.; Veidal, S.S.; Feigh, M.; Schmoll, D. Activation of thyroid hormone receptor-beta improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br. J. Pharmacol. 2021, 178, 2412–2423. [Google Scholar] [CrossRef]
- Thymiakou, E.; Othman, A.; Hornemann, T.; Kardassis, D. Defects in High Density Lipoprotein Metabolism and Hepatic Steatosis In Mice with Liver-Specific Ablation of Hepatocyte Nuclear Factor 4A. Metabolism 2020, 110, 154307. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, M.; Liu, Q.; Xu, T.; Huang, T.; Yao, D.; Wong, C.W.; Liu, J.; Guan, M. 18β-Glycyrrhetinic acid acts through hepatocyte nuclear factor 4 alpha to modulate lipid and carbohydrate metabolism. Pharmacol. Res. 2020, 157, 104840. [Google Scholar] [CrossRef] [PubMed]
- Hosooka, T.; Hosokawa, Y.; Matsugi, K.; Shinohara, M.; Senga, Y.; Tamori, Y.; Aoki, C.; Matsui, S.; Sasaki, T.; Kitamura, T.; et al. The PDK1-FoxO1 signaling in adipocytes controls systemic insulin sensitivity through the 5-lipoxygenase-leukotriene B4 axis. Proc. Natl. Acad. Sci. USA 2020, 117, 11674–11684. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hou, S.; Xiang, R.; Hu, C.; Chen, Z.; Li, N.; Yan, H.; Yu, X.; Li, X.; Chi, Y.; et al. Imipramine activates FAM3A-FOXA2-CPT2 pathway to ameliorate hepatic steatosis. Metabolism 2022, 136, 155292. [Google Scholar] [CrossRef]
- Minamishima, Y.A.; Moslehi, J.; Padera, R.F.; Bronson, R.T.; Liao, R.; Kaelin, W.G., Jr. A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol. Cell. Biol. 2009, 29, 5729–5741. [Google Scholar] [CrossRef]
- Qu, A.; Taylor, M.; Xue, X.; Matsubara, T.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible transcription factor 2alpha promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology 2011, 54, 472–483. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, S.; Wu, X.; Takahashi, S.; Sun, L.; Cai, J.; Krausz, K.W.; Guo, X.; Dias, H.B.; Gavrilova, O.; et al. Intestinal MYC modulates obesity-related metabolic dysfunction. Nat. Metab. 2021, 3, 923–939. [Google Scholar] [CrossRef]
- Baptissart, M.; Bradish, C.M.; Jones, B.S.; Walsh, E.; Tehrani, J.; Marrero-Colon, V.; Mehta, S.; Jima, D.D.; Oh, S.H.; Diehl, A.M.; et al. Zac1 and the Imprinted Gene Network program juvenile NAFLD in response to maternal metabolic syndrome. Hepatology 2022, 76, 1090–1104. [Google Scholar] [CrossRef]
- Sun, N.; Shen, C.; Zhang, L.; Wu, X.; Yu, Y.; Yang, X.; Yang, C.; Zhong, C.; Gao, Z.; Miao, W.; et al. Hepatic Krüppel-like factor 16 (KLF16) targets PPARα to improve steatohepatitis and insulin resistance. Gut 2021, 70, 2183–2195. [Google Scholar] [CrossRef]
- Li, L.; Lin, J.; Huang, C.; Liu, J.; Yuan, Y.; Liu, Z.; Li, Y.; Li, W.; Diao, A. The TFEB activator clomiphene citrate ameliorates lipid metabolic syndrome pathology by activating lipophagy and lipolysis. Biochem. Pharmacol. 2025, 232, 116694. [Google Scholar] [CrossRef]
- Tu, C.; Xiong, H.; Hu, Y.; Wang, W.; Mei, G.; Wang, H.; Li, Y.; Zhou, Z.; Meng, F.; Zhang, P.; et al. Cardiolipin Synthase 1 Ameliorates NASH Through Activating Transcription Factor 3 Transcriptional Inactivation. Hepatology 2020, 72, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Cooreman, M.P.; Vonghia, L.; Francque, S.M. MASLD/MASH and type 2 diabetes: Two sides of the same coin? From single PPAR to pan-PPAR agonists. Diabetes Res. Clin. Pract. 2024, 212, 111688. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C.; Hermans, M.P.; Fruchart-Najib, J.; Kodama, T. Selective Peroxisome Proliferator-Activated Receptor Alpha Modulators (SPPARMα) in the Metabolic Syndrome: Is Pemafibrate Light at the End of the Tunnel? Curr. Atheroscler. Rep. 2021, 23, 3. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Cami, B.; De Boe, V.; Heymans, A.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; et al. Human hepatic in vitro models reveal distinct anti-NASH potencies of PPAR agonists. Cell Biol. Toxicol. 2021, 37, 293–311. [Google Scholar] [CrossRef]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-Lopez, T.; Escola-Gil, J.C.; Cedo, L.; Zali, M.R.; Molaei, M.; et al. Hepatic regulation of VLDL receptor by PPARbeta/delta and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef]
- Lee, C.H.; Chawla, A.; Urbiztondo, N.; Liao, D.; Boisvert, W.A.; Evans, R.M.; Curtiss, L.K. Transcriptional repression of atherogenic inflammation: Modulation by PPARdelta. Science 2003, 302, 453–457. [Google Scholar] [CrossRef]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed]
- Flowers, M.T.; Ntambi, J.M. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr. Opin. Lipidol. 2008, 19, 248–256. [Google Scholar] [CrossRef]
- Esposito, K.; Ciotola, M.; Carleo, D.; Schisano, B.; Saccomanno, F.; Sasso, F.C.; Cozzolino, D.; Assaloni, R.; Merante, D.; Ceriello, A.; et al. Effect of rosiglitazone on endothelial function and inflammatory markers in patients with the metabolic syndrome. Diabetes Care 2006, 29, 1071–1076. [Google Scholar] [CrossRef]
- Qiu, Y.Y.; Zhang, J.; Zeng, F.Y.; Zhu, Y.Z. Roles of the peroxisome proliferator-activated receptors (PPARs) in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Pharmacol. Res. 2023, 192, 106786. [Google Scholar] [CrossRef]
- Pettinelli, P.; Videla, L.A. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: An additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Nakamuta, M.; Kohjima, M.; Morizono, S.; Kotoh, K.; Yoshimoto, T.; Miyagi, I.; Enjoji, M. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2005, 16, 631–635. [Google Scholar] [PubMed]
- Ezhilarasan, D. Deciphering the molecular pathways of saroglitazar: A dual PPAR α/γ agonist for managing metabolic NAFLD. Metabolism 2024, 155, 155912. [Google Scholar] [CrossRef]
- Cooreman, M.P.; Butler, J.; Giugliano, R.P.; Zannad, F.; Dzen, L.; Huot-Marchand, P.; Baudin, M.; Beard, D.R.; Junien, J.L.; Broqua, P.; et al. The pan-PPAR agonist lanifibranor improves cardiometabolic health in patients with metabolic dysfunction-associated steatohepatitis. Nat. Commun. 2024, 15, 3962. [Google Scholar] [CrossRef]
- Chandrasekaran, P.; Weiskirchen, R. The Role of SCAP/SREBP as Central Regulators of Lipid Metabolism in Hepatic Steatosis. Int. J. Mol. Sci. 2024, 25, 1109. [Google Scholar] [CrossRef]
- Ke, C.; Xiao, C.; Li, J.; Wu, X.; Zhang, Y.; Chen, Y.; Sheng, S.; Fu, Z.; Wang, L.; Ni, C.; et al. FMO2 ameliorates nonalcoholic fatty liver disease by suppressing ER-to-Golgi transport of SREBP1. Hepatology 2025, 81, 181–197. [Google Scholar] [CrossRef]
- Song, K.; Zhang, Y.; Ga, Q.; Bai, Z.; Ge, R.L. High-altitude chronic hypoxia ameliorates obesity-induced non-alcoholic fatty liver disease in mice by regulating mitochondrial and AMPK signaling. Life Sci. 2020, 252, 117633. [Google Scholar] [CrossRef]
- Chang, T.C.; Chiou, W.C.; Lai, W.H.; Huang, H.C.; Huang, Y.L.; Liu, H.K.; Liang, Y.C.; Huang, C. Ugonin J improves metabolic disorder and ameliorates nonalcoholic fatty liver disease by regulating the AMPK/AKT signaling pathway. Pharmacol. Res. 2021, 163, 105298. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.Y.; Yang, X.; Yang, Z.; Li, J.W.; Xu, M.Q.; Qu, Y.X.; Tang, J.J.; Li, Y.F.; Wang, L.; Shao, Y.W.; et al. Discovery of an insulin-induced gene binding compound that ameliorates nonalcoholic steatohepatitis by inhibiting sterol regulatory element-binding protein-mediated lipogenesis. Hepatology 2022, 76, 1466–1481. [Google Scholar] [CrossRef]
- Ju, U.I.; Jeong, D.W.; Seo, J.; Park, J.B.; Park, J.W.; Suh, K.S.; Kim, J.B.; Chun, Y.S. Neddylation of sterol regulatory element-binding protein 1c is a potential therapeutic target for nonalcoholic fatty liver treatment. Cell Death Dis. 2020, 11, 283. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Ide, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Matsuzaka, T.; Yatoh, S.; Kitamine, T.; Okazaki, H.; Tamura, Y.; et al. Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. I. PPARs suppress sterol regulatory element binding protein-1c promoter through inhibition of LXR signaling. Mol. Endocrinol. 2003, 17, 1240–1254. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Yoshikawa, T.; Hasty, A.H.; Tamura, Y.; Osuga, J.; Okazaki, H.; Iizuka, Y.; et al. Dual regulation of mouse Delta(5)- and Delta(6)-desaturase gene expression by SREBP-1 and PPARalpha. J. Lipid Res. 2002, 43, 107–114. [Google Scholar] [CrossRef]
- Gosis, B.S.; Wada, S.; Thorsheim, C.; Li, K.; Jung, S.; Rhoades, J.H.; Yang, Y.; Brandimarto, J.; Li, L.; Uehara, K.; et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science 2022, 376, eabf8271. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef]
- Herman, M.A.; Birnbaum, M.J. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. 2021, 33, 2329–2354. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.; Das, D.; Maity, S.K.; Paul, A.; Rawat, P.; Daniel, P.V.; Das, K.; Mitra, S.; Chakrabarti, P.; Mondal, P. Liver-Derived S100A6 Propels β-Cell Dysfunction in NAFLD. Diabetes 2022, 71, 2284–2296. [Google Scholar] [CrossRef]
- Buziau, A.M.; Oosterveer, M.H.; Wouters, K.; Bos, T.; Tolan, D.R.; Agius, L.; Ford, B.E.; Cassiman, D.; Stehouwer, C.D.A.; Schalkwijk, C.G.; et al. Hepatic glucokinase regulatory protein and carbohydrate response element binding protein attenuation reduce de novo lipogenesis but do not mitigate intrahepatic triglyceride accumulation in Aldob deficiency. Mol. Metab. 2024, 87, 101984. [Google Scholar] [CrossRef]
- Rajas, F.; Dentin, R.; Cannella Miliano, A.; Silva, M.; Raffin, M.; Levavasseur, F.; Gautier-Stein, A.; Postic, C.; Mithieux, G. The absence of hepatic glucose-6 phosphatase/ChREBP couple is incompatible with survival in mice. Mol. Metab. 2021, 43, 101108. [Google Scholar] [CrossRef]
- Scagliola, A.; Miluzio, A.; Ventura, G.; Oliveto, S.; Cordiglieri, C.; Manfrini, N.; Cirino, D.; Ricciardi, S.; Valenti, L.; Baselli, G.; et al. Targeting of eIF6-driven translation induces a metabolic rewiring that reduces NAFLD and the consequent evolution to hepatocellular carcinoma. Nat. Commun. 2021, 12, 4878. [Google Scholar] [CrossRef]
- Meng, J.; Feng, M.; Dong, W.; Zhu, Y.; Li, Y.; Zhang, P.; Wu, L.; Li, M.; Lu, Y.; Chen, H.; et al. Identification of HNF-4alpha as a key transcription factor to promote ChREBP expression in response to glucose. Sci. Rep. 2016, 6, 23944. [Google Scholar] [CrossRef]
- Lee, D.S.; An, T.H.; Kim, H.; Jung, E.; Kim, G.; Oh, S.Y.; Kim, J.S.; Chun, H.J.; Jung, J.; Lee, E.W.; et al. Tcf7l2 in hepatocytes regulates de novo lipogenesis in diet-induced non-alcoholic fatty liver disease in mice. Diabetologia 2023, 66, 931–954. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Imai, J.; Watanabe, M.; Komatsu, T.; Suzuki, Y.; Kataoka, K.; Watanabe, S.; Tanigami, A.; Sugano, S. CREB-H: A novel mammalian transcription factor belonging to the CREB/ATF family and functioning via the box-B element with a liver-specific expression. Nucleic Acids Res. 2001, 29, 2154–2162. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 2012, 55, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Song, K.H.; Li, T.; Owsley, E.; Strom, S.; Chiang, J.Y. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009, 49, 297–305. [Google Scholar] [CrossRef]
- Xiao, J.; Hou, Y.; Luo, X.; Zhu, Y.; Li, W.; Li, B.; Zhou, L.; Chen, X.; Guo, Y.; Zhang, X.; et al. Clostridium Scindens Protects Against Vancomycin-Induced Cholestasis and Liver Fibrosis by Activating Intestinal FXR-FGF15/19 Signaling. Adv. Sci. 2025, 12, e2406445. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Caron, S.; Huaman Samanez, C.; Dehondt, H.; Ploton, M.; Briand, O.; Lien, F.; Dorchies, E.; Dumont, J.; Postic, C.; Cariou, B.; et al. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol. Cell. Biol. 2013, 33, 2202–2211. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Tang, Y.; Fan, Y.; Wang, Y.; Wang, D.; Huang, Q.; Chen, T.; Cao, X.; Wen, C.; Shen, X.; Li, J.; et al. A Current Understanding of FXR in NAFLD: The Multifaceted Regulatory Role of FXR and Novel Lead Discovery for Drug Development. Biomed. Pharmacother. 2024, 175, 116658. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Garzel, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 2015, 87, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhai, Y.; Mu, Y.; Gong, H.; Uppal, H.; Toma, D.; Ren, S.; Evans, R.M.; Xie, W. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 2006, 281, 15013–15020. [Google Scholar] [CrossRef]
- He, J.; Gao, J.; Xu, M.; Ren, S.; Stefanovic-Racic, M.; O’Doherty, R.M.; Xie, W. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes 2013, 62, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Merrell, M.D.; Cherrington, N.J. Drug metabolism alterations in nonalcoholic fatty liver disease. Drug Metab. Rev. 2011, 43, 317–334. [Google Scholar] [CrossRef]
- Zhao, L.Y.; Xu, J.Y.; Shi, Z.; Englert, N.A.; Zhang, S.Y. Pregnane X receptor (PXR) deficiency improves high fat diet-induced obesity via induction of fibroblast growth factor 15 (FGF15) expression. Biochem. Pharmacol. 2017, 142, 194–203. [Google Scholar] [CrossRef]
- Nakamura, K.; Moore, R.; Negishi, M.; Sueyoshi, T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J. Biol. Chem. 2007, 282, 9768–9776. [Google Scholar] [CrossRef]
- Griffett, K.; Burris, T.P. Development of LXR inverse agonists to treat MAFLD, NASH, and other metabolic diseases. Front. Med. 2023, 10, 1102469. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.G. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef]
- Graelmann, F.J.; Gondorf, F.; Majlesain, Y.; Niemann, B.; Klepac, K.; Gosejacob, D.; Gottschalk, M.; Mayer, M.; Iriady, I.; Hatzfeld, P.; et al. Differential cell type-specific function of the aryl hydrocarbon receptor and its repressor in diet-induced obesity and fibrosis. Mol. Metab. 2024, 85, 101963. [Google Scholar] [CrossRef]
- Angrish, M.M.; Jones, A.D.; Harkema, J.R.; Zacharewski, T.R. Aryl hydrocarbon receptor-mediated induction of Stearoyl-CoA desaturase 1 alters hepatic fatty acid composition in TCDD-elicited steatosis. Toxicol. Sci. 2011, 124, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wada, T.; Febbraio, M.; He, J.; Matsubara, T.; Lee, M.J.; Gonzalez, F.J.; Xie, W. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 2010, 139, 653–663. [Google Scholar] [CrossRef]
- Xia, H.; Zhu, X.; Zhang, X.; Jiang, H.; Li, B.; Wang, Z.; Li, D.; Jin, Y. Alpha-naphthoflavone attenuates non-alcoholic fatty liver disease in oleic acid-treated HepG2 hepatocytes and in high fat diet-fed mice. Biomed. Pharmacother. 2019, 118, 109287. [Google Scholar] [CrossRef]
- Girer, N.G.; Carter, D.; Bhattarai, N.; Mustafa, M.; Denner, L.; Porter, C.; Elferink, C.J. Inducible Loss of the Aryl Hydrocarbon Receptor Activates Perigonadal White Fat Respiration and Brown Fat Thermogenesis via Fibroblast Growth Factor 21. Int. J. Mol. Sci. 2019, 20, 950. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Targher, G. Hepatic thyroid hormone receptor-beta signalling: Mechanisms and recent advancements in the treatment of metabolic dysfunction-associated steatohepatitis. Diabetes Obes. Metab. 2025, 27, 1635–1647. [Google Scholar] [CrossRef]
- Krause, C.; Grohs, M.; El Gammal, A.T.; Wolter, S.; Lehnert, H.; Mann, O.; Mittag, J.; Kirchner, H. Reduced expression of thyroid hormone receptor beta in human nonalcoholic steatohepatitis. Endocr. Connect. 2018, 7, 1448–1456. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, Y.; Hu, S.; Xu, Y.; Stroup, D.; Pan, X.; Bawa, F.C.; Chen, S.; Gopoju, R.; Yin, L.; et al. Hepatocyte Nuclear Factor 4α Prevents the Steatosis-to-NASH Progression by Regulating p53 and Bile Acid Signaling (In Mice). Hepatology 2021, 73, 2251–2265. [Google Scholar] [CrossRef]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4alpha pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef] [PubMed]
- Thymiakou, E.; Tzardi, M.; Kardassis, D. Impaired hepatic glucose metabolism and liver-alpha-cell axis in mice with liver-specific ablation of the Hepatocyte Nuclear Factor 4alpha (Hnf4a) gene. Metabolism 2023, 139, 155371. [Google Scholar] [CrossRef]
- Yu, D.; Chen, G.; Pan, M.; Zhang, J.; He, W.; Liu, Y.; Nian, X.; Sheng, L.; Xu, B. High fat diet-induced oxidative stress blocks hepatocyte nuclear factor 4alpha and leads to hepatic steatosis in mice. J. Cell. Physiol. 2018, 233, 4770–4782. [Google Scholar] [CrossRef]
- Pan, X.; Hu, S.; Xu, Y.; Gopoju, R.; Zhu, Y.; Cassim Bawa, F.N.; Wang, H.; Wang, J.; Batayneh, Z.; Clark, A.; et al. Krüppel-like factor 10 protects against metabolic dysfunction-associated steatohepatitis by regulating HNF4α-mediated metabolic pathways. Metabolism 2024, 155, 155909. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Altomonte, J.; Perdomo, G.; He, J.; Fan, Y.; Kamagate, A.; Meseck, M.; Dong, H.H. Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology 2006, 147, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Su, D.; Altomonte, J.; Kamagate, A.; He, J.; Perdomo, G.; Tse, T.; Jiang, Y.; Dong, H.H. PPARalpha mediates the hypolipidemic action of fibrates by antagonizing FoxO1. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E421–E434. [Google Scholar] [CrossRef]
- Gouw, A.M.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.K.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019, 30, 556–572.e5. [Google Scholar] [CrossRef]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsboll, T. Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 2018, 14, 390–403. [Google Scholar] [CrossRef]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Bronneke, H.S.; et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef]
- Xie, C.; Jiang, C.; Shi, J.; Gao, X.; Sun, D.; Sun, L.; Wang, T.; Takahashi, S.; Anitha, M.; Krausz, K.W.; et al. An Intestinal Farnesoid X Receptor-Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 2017, 66, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Martin-Mateos, R.; Albillos, A. The Role of the Gut-Liver Axis in Metabolic Dysfunction-Associated Fatty Liver Disease. Front. Immunol. 2021, 12, 660179. [Google Scholar] [CrossRef]
- Jiang, S.; Uddin, M.J.; Yu, X.; Piao, L.; Dorotea, D.; Oh, G.T.; Ha, H. Peroxisomal Fitness: A Potential Protective Mechanism of Fenofibrate Against High Fat Diet-Induced Non-Alcoholic Fatty Liver Disease In Mice. Diabetes Metab. J. 2022, 46, 829–842. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Radun, R.; Dixon, E.D.; Mlitz, V.; Timelthaler, G.; Halilbasic, E.; Herac, M.; Jonker, J.W.; Ronda, O.; Tardelli, M.; et al. Hepatocyte-specific deletion of adipose triglyceride lipase (adipose triglyceride lipase/patatin-like phospholipase domain containing 2) ameliorates dietary induced steatohepatitis in mice. Hepatology 2022, 75, 125–139. [Google Scholar] [CrossRef]
- Zhang, M.; Barroso, E.; Ruart, M.; Peña, L.; Peyman, M.; Aguilar-Recarte, D.; Montori-Grau, M.; Rada, P.; Cugat, C.; Montironi, C.; et al. Elafibranor upregulates the EMT-inducer S100A4 via PPARβ/δ. Biomed. Pharmacother. 2023, 167, 115623. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Vandoros, G.P.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papavassiliou, A.G. NF-kappaB/PPAR gamma and/or AP-1/PPAR gamma ‘on/off’ switches and induction of CBP in colon adenocarcinomas: Correlation with COX-2 expression. Int. J. Color. Dis. 2007, 22, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Danzeng, A.; Liu, Q.; Zeng, C.; Xu, L.; Mo, J.; Pingcuo, C.; Wang, X.; Wang, C.; Zhang, B.; et al. The Role of Nuclear Receptors in the Pathogenesis and Treatment of Non-alcoholic Fatty Liver Disease. Int. J. Biol. Sci. 2024, 20, 113–126. [Google Scholar] [CrossRef]
- Zhou, L.; Qiu, X.; Meng, Z.; Liu, T.; Chen, Z.; Zhang, P.; Kuang, H.; Pan, T.; Lu, Y.; Qi, L.; et al. Hepatic danger signaling triggers TREM2+ macrophage induction and drives steatohepatitis via MS4A7-dependent inflammasome activation. Sci. Transl. Med. 2024, 16, eadk1866. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, J.; Yang, X.; Shen, C.; Huang, J.; Zhang, D.; Liu, N.; Liu, C.; Zhong, Y.; Chen, Y.; et al. Hepatic Zbtb18 (Zinc Finger and BTB Domain Containing 18) Alleviates Hepatic Steatohepatitis via FXR (Farnesoid X Receptor). Signal Transduct. Target Ther. 2024, 9, 20. [Google Scholar] [CrossRef]
- Qin, D.; Pan, P.; Lyu, B.; Chen, W.; Gao, Y. Lupeol improves bile acid metabolism and metabolic dysfunction-associated steatotic liver disease in mice via FXR signaling pathway and gut-liver axis. Biomed. Pharmacother. 2024, 177, 116942. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, W.; Ding, D.; Tan, K.; Ding, W.; Wang, Z.; Fu, S.; Hou, G.; Zhou, W.P.; Gu, F. IL-17a promotes hepatocellular carcinoma by increasing FAP expression in hepatic stellate cells via activation of the STAT3 signaling pathway. Cell Death Discov. 2024, 10, 230. [Google Scholar] [CrossRef]
- Kui, L.; Kim, A.D.; Onyuru, J.; Hoffman, H.M.; Feldstein, A.E. BRP39 Regulates Neutrophil Recruitment in NLRP3 Inflammasome-Induced Liver Inflammation. Cell. Mol. Gastroenterol. Hepatol. 2024, 17, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, J.; Lee, A.; Tsaur, I.; Ohira, M.; Duong, V.; Vo, N.; Watari, K.; Su, H.; Kim, J.Y.; et al. IL-22 resolves MASLD via enterocyte STAT3 restoration of diet-perturbed intestinal homeostasis. Cell Metab. 2024, 36, 2341–2354.e6. [Google Scholar] [CrossRef]
- Huang, B.; Xiong, X.; Zhang, L.; Liu, X.; Wang, Y.; Gong, X.; Sang, Q.; Lu, Y.; Qu, H.; Zheng, H.; et al. PSA controls hepatic lipid metabolism by regulating the NRF2 signaling pathway. J. Mol. Cell Biol. 2021, 13, 527–539. [Google Scholar] [CrossRef]
- Sun, X.; Yang, Z.; Min, L.; Gong, S.; Miao, X.; Wang, B.; Kong, X.; Zhu, Q. Interferon regulatory factor 1 contributes to metabolic dysfunction associated steatotic liver disease. Life Sci. 2025, 370, 123575. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, G.; Fang, Z.; Gao, W.; Song, Y.; Lei, L.; Du, X.; Li, X. Buddleoside alleviates nonalcoholic steatohepatitis by targeting the AMPK-TFEB signaling pathway. Autophagy 2025, 21, 1316–1334. [Google Scholar] [CrossRef]
- Aki, D.; Hayakawa, T.; Srirat, T.; Shichino, S.; Ito, M.; Saitoh, S.I.; Mise-Omata, S.; Yoshimura, A. The Nr4a family regulates intrahepatic Treg proliferation and liver fibrosis in MASLD models. J. Clin. Investig. 2024, 134, e175305. [Google Scholar] [CrossRef] [PubMed]
- Jeelani, I.; Moon, J.S.; da Cunha, F.F.; Nasamran, C.A.; Jeon, S.; Zhang, X.; Bandyopadhyay, G.K.; Dobaczewska, K.; Mikulski, Z.; Hosseini, M.; et al. HIF-2α drives hepatic Kupffer cell death and proinflammatory recruited macrophage activation in nonalcoholic steatohepatitis. Sci. Transl. Med. 2024, 16, eadi0284. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Jo, S.H.; Lee, E.S.; Ha, K.B.; Park, N.W.; Kong, D.H.; Park, S.I.; Park, J.S.; Chung, C.H. DWN12088, A Prolyl-tRNA Synthetase Inhibitor, Alleviates Hepatic Injury in Nonalcoholic Steatohepatitis. Diabetes Metab. J. 2024, 48, 97–111. [Google Scholar] [CrossRef]
- Tian, S.Y.; Chen, S.M.; Pan, C.X.; Li, Y. FXR: Structures, biology, and drug development for NASH and fibrosis diseases. Acta Pharmacol. Sin. 2022, 43, 1120–1132. [Google Scholar] [CrossRef]
- Zheng, C.; Wang, L.; Zou, T.; Lian, S.; Luo, J.; Lu, Y.; Hao, H.; Xu, Y.; Xiang, Y.; Zhang, X.; et al. Ileitis promotes MASLD progression via bile acid modulation and enhanced TGR5 signaling in ileal CD8+ T cells. J. Hepatol. 2024, 80, 764–777. [Google Scholar] [CrossRef]
- Jiao, J.; Sanchez, J.I.; Saldarriaga, O.A.; Solis, L.M.; Tweardy, D.J.; Maru, D.M.; Stevenson, H.L.; Beretta, L. Spatial molecular and cellular determinants of STAT3 activation in liver fibrosis progression in non-alcoholic fatty liver disease. JHEP Rep. Innov. Hepatol. 2023, 5, 100628. [Google Scholar] [CrossRef]
- Inoue, H.; Ogawa, W.; Ozaki, M.; Haga, S.; Matsumoto, M.; Furukawa, K.; Hashimoto, N.; Kido, Y.; Mori, T.; Sakaue, H.; et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat. Med. 2004, 10, 168–174. [Google Scholar] [CrossRef]
- Hakeem, A.N.; Kamal, M.M.; Tawfiq, R.A.; Abdelrahman, B.A.; Hammam, O.A.; Elmazar, M.M.; El-Khatib, A.S.; Attia, Y.M. Elafibranor modulates ileal macrophage polarization to restore intestinal integrity in NASH: Potential crosstalk between ileal IL-10/STAT3 and hepatic TLR4/NF-kappaB axes. Biomed. Pharmacother. 2023, 157, 114050. [Google Scholar] [CrossRef]
- Lei, Z.; Yu, J.; Wu, Y.; Shen, J.; Lin, S.; Xue, W.; Mao, C.; Tang, R.; Sun, H.; Qi, X.; et al. CD1d protects against hepatocyte apoptosis in non-alcoholic steatohepatitis. J. Hepatol. 2024, 80, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Anandhan, A.; Chen, J.; Shakya, A.; Dodson, M.; Ooi, A.; Chapman, E.; White, E.; Garcia, J.G.; Zhang, D.D. Decreased autophagosome biogenesis, reduced NRF2, and enhanced ferroptotic cell death are underlying molecular mechanisms of non-alcoholic fatty liver disease. Redox Biol. 2023, 59, 102570. [Google Scholar] [CrossRef]
- Vanani, A.R.; Kalantari, H.; Mahdavinia, M.; Rashno, M.; Khorsandi, L.; Khodayar, M.J. Dimethyl fumarate reduces oxidative stress, inflammation and fat deposition by modulation of Nrf2, SREBP-1c and NF-κB signaling in HFD fed mice. Life Sci. 2021, 283, 119852. [Google Scholar] [CrossRef]
- Fernández-Ginés, R.; Encinar, J.A.; Escoll, M.; Carnicero-Senabre, D.; Jiménez-Villegas, J.; García-Yagüe, Á.J.; González-Rodríguez, Á.; Garcia-Martinez, I.; Valverde, Á.M.; Rojo, A.I.; et al. Specific targeting of the NRF2/β-TrCP axis promotes beneficial effects in NASH. Redox Biol. 2024, 69, 103027. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Chen, Y.; Chen, M.T.; Fu, T.T.; Liu, J.; Liu, F.F.; Xu, C.J.; Li, W.S.; Li, B.L.; Jiang, Z.P.; et al. Natural linoleic acid from marine fungus Eutypella sp. F0219 blocks KEAP1/NRF2 interaction and ameliorates MASLD by targeting FABP4. Free. Radic. Biol. Med. 2024, 224, 630–643. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, Q.; Fang, X.; Li, L.; Yang, G.; Xu, X.; Yang, M. Nuclear factor erythroid 2-related factor 2 ameliorates disordered glucose and lipid metabolism in liver: Involvement of gasdermin D in regulating pyroptosis. Clin. Transl. Med. 2025, 15, e70233. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cao, Z.; Yang, Z.; Chen, Y.; Yao, H.; Zhou, D.; Ou, P.; Huang, W.; Jiao, S.; Chen, S.; et al. Design, synthesis, and biological studies of novel sulfonamide derivatives as farnesoid X receptor agonists. Eur. J. Med. Chem. 2023, 258, 115614. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, J.; Liu, H.; Guan, L.; Xu, S.; Wang, Z.; Qiu, Y.; Liu, H.; Peng, L.; Men, X. Ajugol enhances TFEB-mediated lysosome biogenesis and lipophagy to alleviate non-alcoholic fatty liver disease. Pharmacol. Res. 2021, 174, 105964. [Google Scholar] [CrossRef]
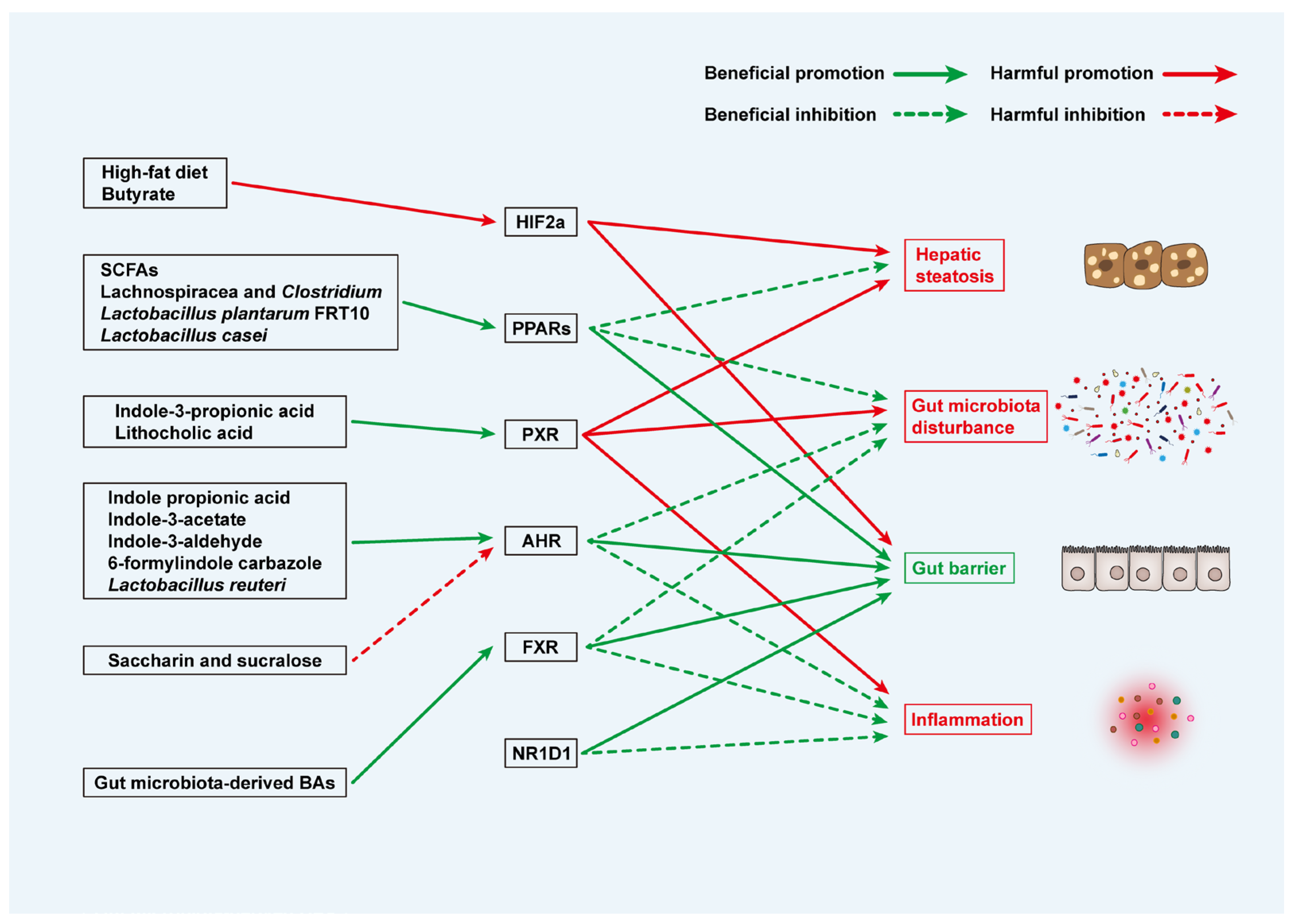
- Cai, W.; Qiu, T.; Hu, W.; Fang, T. Changes in the intestinal microbiota of individuals with non-alcoholic fatty liver disease based on sequencing: An updated systematic review and meta-analysis. PLoS ONE 2024, 19, e0299946. [Google Scholar] [CrossRef]
- Saeed, H.; Diaz, L.A.; Gil-Gomez, A.; Burton, J.; Bajaj, J.S.; Romero-Gomez, M.; Arrese, M.; Arab, J.P.; Khan, M.Q. Microbiome-centered therapies for the management of metabolic dysfunction-associated steatotic liver disease. Clin. Mol. Hepatol. 2025, 31, S94–S111. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Birchenough, G.M.H.; Stahlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Backhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40.e7. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-Invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062. [Google Scholar] [CrossRef]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Stahl, C.; Schroder, M.; Vetter, W.; Bischoff, S.C.; Bergheim, I. Lactobacillus casei Shirota protects from fructose-induced liver steatosis: A mouse model. J. Nutr. Biochem. 2013, 24, 531–538. [Google Scholar] [CrossRef]
- Cai, H.; Wen, Z.; Li, X.; Meng, K.; Yang, P. Lactobacillus plantarum FRT10 alleviated high-fat diet-induced obesity in mice through regulating the PPARalpha signal pathway and gut microbiota. Appl. Microbiol. Biotechnol. 2020, 104, 5959–5972. [Google Scholar] [CrossRef]
- Sun, S.S.; Wang, K.; Ma, K.; Bao, L.; Liu, H.W. An insoluble polysaccharide from the sclerotium of Poria cocos improves hyperglycemia, hyperlipidemia and hepatic steatosis in ob/ob mice via modulation of gut microbiota. Chin. J. Nat. Med. 2019, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Nihei, N.; Okamoto, H.; Furune, T.; Ikuta, N.; Sasaki, K.; Rimbach, G.; Yoshikawa, Y.; Terao, K. Dietary alpha-cyclodextrin modifies gut microbiota and reduces fat accumulation in high-fat-diet-fed obese mice. Biofactors 2018, 44, 336–347. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Kim, S.; Choi, S.; Dutta, M.; Asubonteng, J.O.; Polunas, M.; Goedken, M.; Gonzalez, F.J.; Cui, J.Y.; Gyamfi, M.A. Pregnane X receptor exacerbates nonalcoholic fatty liver disease accompanied by obesity- and inflammation-prone gut microbiome signature. Biochem. Pharmacol. 2021, 193, 114698. [Google Scholar] [CrossRef]
- Shi, Z.; Lei, H.; Chen, G.; Yuan, P.; Cao, Z.; Ser, H.L.; Zhu, X.; Wu, F.; Liu, C.; Dong, M.; et al. Impaired Intestinal Akkermansia muciniphila and Aryl Hydrocarbon Receptor Ligands Contribute to Nonalcoholic Fatty Liver Disease in Mice. mSystems 2021, 6, e00985-20. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zhao, Y.; Ma, L.; Wang, Z.; Ni, L.; Hu, L.; Fu, Z. Pharmacological activation of REV-ERBalpha improves nonalcoholic steatohepatitis by regulating intestinal permeability. Metabolism 2021, 114, 154409. [Google Scholar] [CrossRef]
- Kondo, T.; Kishi, M.; Fushimi, T.; Kaga, T. Acetic acid upregulates the expression of genes for fatty acid oxidation enzymes in liver to suppress body fat accumulation. J. Agric. Food Chem. 2009, 57, 5982–5986. [Google Scholar] [CrossRef]
- Moreau, F.; Brunao, B.B.; Liu, X.Y.; Tremblay, F.; Fitzgerald, K.; Avila-Pacheco, J.; Clish, C.; Kahn, R.C.; Softic, S. Liver-specific FGFR4 knockdown in mice on an HFD increases bile acid synthesis and improves hepatic steatosis. J. Lipid Res. 2023, 64, 100324. [Google Scholar] [CrossRef]
- Verbeke, L.; Farre, R.; Verbinnen, B.; Covens, K.; Vanuytsel, T.; Verhaegen, J.; Komuta, M.; Roskams, T.; Chatterjee, S.; Annaert, P.; et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am. J. Pathol. 2015, 185, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Henry, Z.; Meadows, V.; Guo, G.L. FXR and NASH: An avenue for tissue-specific regulation. Hepatol. Commun. 2023, 7, e0127. [Google Scholar] [CrossRef]
- Jarret, A.; Jackson, R.; Duizer, C.; Healy, M.E.; Zhao, J.; Rone, J.M.; Bielecki, P.; Sefik, E.; Roulis, M.; Rice, T.; et al. Enteric Nervous System-Derived IL-18 Orchestrates Mucosal Barrier Immunity. Cell 2020, 180, 50–63.e12. [Google Scholar] [CrossRef]
- Sun, D.; Bai, R.; Zhou, W.; Yao, Z.; Liu, Y.; Tang, S.; Ge, X.; Luo, L.; Luo, C.; Hu, G.F.; et al. Angiogenin maintains gut microbe homeostasis by balancing alpha-Proteobacteria and Lachnospiraceae. Gut 2021, 70, 666–676. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Cui, J.Y. Review: Mechanisms of How the Intestinal Microbiota Alters the Effects of Drugs and Bile Acids. Drug Metab. Dispos. 2015, 43, 1505–1521. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Dempsey, J.L.; Wang, D.; Siginir, G.; Fei, Q.; Raftery, D.; Gu, H.; Yue Cui, J. Pharmacological Activation of PXR and CAR Downregulates Distinct Bile Acid-Metabolizing Intestinal Bacteria and Alters Bile Acid Homeostasis. Toxicol. Sci. 2019, 168, 40–60. [Google Scholar] [CrossRef]
- Li, C.Y.; Dempsey, J.L.; Wang, D.; Lee, S.; Weigel, K.M.; Fei, Q.; Bhatt, D.K.; Prasad, B.; Raftery, D.; Gu, H.; et al. PBDEs Altered Gut Microbiome and Bile Acid Homeostasis in Male C57BL/6 Mice. Drug Metab. Dispos. 2018, 46, 1226–1240. [Google Scholar] [CrossRef]
- Niu, B.; Pan, T.; Xiao, Y.; Wang, H.; Zhu, J.; Tian, F.; Lu, W.; Chen, W. The therapeutic potential of dietary intervention: Based on the mechanism of a tryptophan derivative-indole propionic acid on metabolic disorders. Crit. Rev. Food Sci. Nutr. 2025, 65, 1729–1748. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Lee, S.O.; Sridharan, G.; Lee, K.; Davidson, L.A.; Jayaraman, A.; Chapkin, R.S.; Alaniz, R.; Safe, S. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharmacol. 2014, 85, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Cella, M.; Colonna, M. Aryl hydrocarbon receptor: Linking environment to immunity. Semin. Immunol. 2015, 27, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk Between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef]
- Xie, C.; Yagai, T.; Luo, Y.; Liang, X.; Chen, T.; Wang, Q.; Sun, D.; Zhao, J.; Ramakrishnan, S.K.; Sun, L.; et al. Activation of intestinal hypoxia-inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat. Med. 2017, 23, 1298–1308. [Google Scholar] [CrossRef]
- Glover, L.E.; Bowers, B.E.; Saeedi, B.; Ehrentraut, S.F.; Campbell, E.L.; Bayless, A.J.; Dobrinskikh, E.; Kendrick, A.A.; Kelly, C.J.; Burgess, A.; et al. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 19820–19825. [Google Scholar] [CrossRef]
- Xie, L.; Xue, X.; Taylor, M.; Ramakrishnan, S.K.; Nagaoka, K.; Hao, C.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible factor/MAZ-dependent induction of caveolin-1 regulates colon permeability through suppression of occludin, leading to hypoxia-induced inflammation. Mol. Cell. Biol. 2014, 34, 3013–3023. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Artis, D.; Becker, C. The intestinal barrier: A pivotal role in health, inflammation, and cancer. Lancet Gastroenterol. Hepatol. 2025, 10, 573–592. [Google Scholar] [CrossRef]
- Yang, K.; Song, M. New Insights into the Pathogenesis of Metabolic-Associated Fatty Liver Disease (MAFLD): Gut-Liver-Heart Crosstalk. Nutrients 2023, 15, 3970. [Google Scholar] [CrossRef] [PubMed]
- Oduro, P.K.; Zheng, X.; Wei, J.; Yang, Y.; Wang, Y.; Zhang, H.; Liu, E.; Gao, X.; Du, M.; Wang, Q. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm. Sin. B 2022, 12, 50–75. [Google Scholar] [CrossRef]
- Xie, W.; Gan, J.; Zhou, X.; Tian, H.; Pan, X.; Liu, W.; Li, X.; Du, J.; Xu, A.; Zheng, M.; et al. Myocardial infarction accelerates the progression of MASH by triggering immunoinflammatory response and induction of periosti. Cell Metab. 2024, 36, 1269–1286.e9. [Google Scholar] [CrossRef]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef]
- Kim, E.R.; Park, J.S.; Kim, J.H.; Oh, J.Y.; Oh, I.J.; Choi, D.H.; Lee, Y.S.; Park, I.S.; Kim, S.; Lee, D.H.; et al. A GLP-1/GLP-2 receptor dual agonist to treat NASH: Targeting the gut-liver axis and microbiome. Hepatology 2022, 75, 1523–1538. [Google Scholar] [CrossRef]
- Higarza, S.G.; Arboleya, S.; Arias, J.L.; Gueimonde, M.; Arias, N. Akkermansia muciniphila and environmental enrichment reverse cognitive impairment associated with high-fat high-cholesterol consumption in rats. Gut Microbes 2021, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H. Multiple organs involved in the pathogenesis of non-alcoholic fatty liver disease. Cell Biosci. 2020, 10, 140. [Google Scholar] [CrossRef]
- Zhou, J.; Tripathi, M.; Ho, J.P.; Widjaja, A.A.; Shekeran, S.G.; Camat, M.D.; James, A.; Wu, Y.; Ching, J.; Kovalik, J.P.; et al. Thyroid Hormone Decreases Hepatic Steatosis, Inflammation, and Fibrosis in a Dietary Mouse Model of Nonalcoholic Steatohepatitis. Thyroid 2022, 32, 725–738. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, L.; Sun, C.; Ireland, N.; Shah, Y.M.; Liu, Y.; Rui, L. Insulin/Snail1 axis ameliorates fatty liver disease by epigenetically suppressing lipogenesis. Nat. Commun. 2018, 9, 2751. [Google Scholar] [CrossRef] [PubMed]
- Collins, S. A heart-adipose tissue connection in the regulation of energy metabolism. Nat. Rev. Endocrinol. 2014, 10, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Crudele, L.; De Matteis, C.; Novielli, F.; Petruzzelli, S.; Di Buduo, E.; Graziano, G.; Cariello, M.; Piccinin, E.; Gadaleta, R.M.; Moschetta, A. Fasting hyperglycaemia and fatty liver drive colorectal cancer: A retrospective analysis in 1145 patients. Intern. Emerg. Med. 2024, 19, 1267–1277. [Google Scholar] [CrossRef]
- Crudele, L.; Piccinin, E.; Moschetta, A. Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis. Nutrients 2021, 13, 2101. [Google Scholar] [CrossRef]
- Lee, H.; Lee, H.W.; Kim, S.U.; Chang Kim, H. Metabolic Dysfunction-Associated Fatty Liver Disease Increases Colon Cancer Risk: A Nationwide Cohort Study. Clin. Transl. Gastroenterol. 2022, 13, e00435. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Jeong, I.K.; Ahn, K.J.; Chung, H.Y.; Hwang, Y.C. Fenofibrate, a PPARalpha agonist, reduces hepatic fat accumulation through the upregulation of TFEB-mediated lipophagy. Metabolism 2021, 120, 154798. [Google Scholar] [CrossRef]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients with Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-Alpha/Gamma Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Bhandari, B.R.; Ruane, P.J.; Kohli, A.; Harting, E.; Ding, D.; Chuang, J.C.; Huss, R.S.; Chung, C.; Myers, R.P.; et al. Fenofibrate Mitigates Hypertriglyceridemia in Nonalcoholic Steatohepatitis Patients Treated with Cilofexor/Firsocostat. Clin. Gastroenterol. Hepatol. 2023, 21, 143–152.e3. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.; Kleiner, D.E.; Goodman, Z.; Brunt, E.; Avigan, M.I.; Regev, A.; Hayashi, P.H.; Lewis, J.H.; Mehta, R.; Harrison, S.A.; et al. Liver biopsy for assessment of suspected drug-induced liver injury in metabolic dysfunction-associated steatohepatitis clinical trials: Expert consensus from the Liver Forum. Aliment. Pharmacol. Ther. 2024, 59, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Rastogi, A.; Dunbar, R.L.; Thacker, H.P.; Bhatt, J.; Parmar, K.; Parmar, D.V. Abrogation of postprandial triglyceridemia with dual PPAR alpha/gamma agonist in type 2 diabetes mellitus: A randomized, placebo-controlled study. Acta Diabetol. 2020, 57, 809–818. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Idowu, M.O.; Parmar, D.; Borg, B.B.; Denham, D.; Loo, N.M.; Lazas, D.; Younes, Z.; Sanyal, A.J. A Phase 2 Double Blinded, Randomized Controlled Trial of Saroglitazar in Patients with Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2021, 19, 2670–2672. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-Alpha and -Delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrene, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages☆. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef]
- Kim, W.; Kim, B.G.; Lee, J.S.; Lee, C.K.; Yeon, J.E.; Chang, M.S.; Kim, J.H.; Kim, H.; Yi, S.; Lee, J.; et al. Randomised clinical trial: The efficacy and safety of oltipraz, a liver X receptor alpha-inhibitory dithiolethione in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 45, 1073–1083. [Google Scholar] [CrossRef]
- Alkhouri, N.; Herring, R.; Kabler, H.; Kayali, Z.; Hassanein, T.; Kohli, A.; Huss, R.S.; Zhu, Y.; Billin, A.N.; Damgaard, L.H.; et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. J. Hepatol. 2022, 77, 607–618. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Tully, D.C.; Rucker, P.V.; Chianelli, D.; Williams, J.; Vidal, A.; Alper, P.B.; Mutnick, D.; Bursulaya, B.; Schmeits, J.; Wu, X.; et al. Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J. Med. Chem. 2017, 60, 9960–9973. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, Y.; Liu, Y.; Wang, W.; Tian, X.; Chen, S.; Lu, Y.; Du, J.; Cai, W. A nonbile acid farnesoid X receptor agonist tropifexor potently inhibits cholestatic liver injury and fibrosis by modulating the gut-liver axis. Liver Int. 2021, 41, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; McKilligin, E.; Pei, L.; Watson, M.A.; Collins, A.R.; Laffitte, B.A.; Chen, M.; Noh, G.; Goodman, J.; Hagger, G.N.; et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7604–7609. [Google Scholar] [CrossRef]
- Griffett, K.; Solt, L.A.; El-Gendy Bel, D.; Kamenecka, T.M.; Burris, T.P. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem. Biol. 2013, 8, 559–567. [Google Scholar] [CrossRef]
- Yasuda, T.; Grillot, D.; Billheimer, J.T.; Briand, F.; Delerive, P.; Huet, S.; Rader, D.J. Tissue-specific liver X receptor activation promotes macrophage reverse cholesterol transport in vivo. Arter. Thromb. Vasc. Biol. 2010, 30, 781–786. [Google Scholar] [CrossRef]
- Katz, A.; Udata, C.; Ott, E.; Hickey, L.; Burczynski, M.E.; Burghart, P.; Vesterqvist, O.; Meng, X. Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J. Clin. Pharmacol. 2009, 49, 643–649. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.; Moussa, S.E.; McCarty, K.; Pablo Frias, J.; Taub, R.; Alkhouri, N. Effects of Resmetirom on Noninvasive Endpoints in a 36-Week Phase 2 Active Treatment Extension Study in Patients with NASH. Hepatol. Commun. 2021, 5, 573–588. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Zucchi, R. Thyroid Hormone Analogues: An Update. Thyroid 2020, 30, 1099–1105. [Google Scholar] [CrossRef]
- Zhou, J.; Waskowicz, L.R.; Lim, A.; Liao, X.H.; Lian, B.; Masamune, H.; Refetoff, S.; Tran, B.; Koeberl, D.D.; Yen, P.M. A Liver-Specific Thyromimetic, VK2809, Decreases Hepatosteatosis in Glycogen Storage Disease Type Ia. Thyroid 2019, 29, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Sepe, V.; Biagioli, M.; Fiorillo, B.; Rapacciuolo, P.; Distrutti, E.; Zampella, A. Development of bile acid activated receptors hybrid molecules for the treatment of inflammatory and metabolic disorders. Biochem. Pharmacol. 2023, 216, 115776. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.C.; Jiang, N.; Chen, L.L.; Liu, F.; Liu, S.Q.; Ding, C.H.; Wu, S.H.; Wang, K.Q.; Luo, Y.Y.; Peng, Y.; et al. TRIB3-TRIM8 complex drives NAFLD progression by regulating HNF4alpha stability. J. Hepatol. 2024, 80, 778–791. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, G.; Wang, Y.; Yin, J.; Wang, J.; Xia, B.; Li, T.; Yang, X.; Hou, P.; Hu, S.; et al. Fucoidan A2 from the Brown Seaweed Ascophyllum nodosum Lowers Lipid by Improving Reverse Cholesterol Transport in C57BL/6J Mice Fed a High-Fat Diet. J. Agric. Food Chem. 2019, 67, 5782–5791. [Google Scholar] [CrossRef]
- Bao, T.; He, F.; Zhang, X.; Zhu, L.; Wang, Z.; Lu, H.; Wang, T.; Li, Y.; Yang, S.; Wang, H. Inulin Exerts Beneficial Effects on Non-Alcoholic Fatty Liver Disease via Modulating gut Microbiome and Suppressing the Lipopolysaccharide-Toll-Like Receptor 4-Mpsi-Nuclear Factor-kappaB-Nod-Like Receptor Protein 3 Pathway via Gut-Liver Axis In Mice. Front. Pharmacol. 2020, 11, 558525. [Google Scholar] [CrossRef]
- Daubioul, C.A.; Horsmans, Y.; Lambert, P.; Danse, E.; Delzenne, N.M. Effects of oligofructose on glucose and lipid metabolism in patients with nonalcoholic steatohepatitis: Results of a pilot study. Eur. J. Clin. Nutr. 2005, 59, 723–726. [Google Scholar] [CrossRef]
- Bomhof, M.R.; Parnell, J.A.; Ramay, H.R.; Crotty, P.; Rioux, K.P.; Probert, C.S.; Jayakumar, S.; Raman, M.; Reimer, R.A. Histological improvement of non-alcoholic steatohepatitis with a prebiotic: A pilot clinical trial. Eur. J. Nutr. 2019, 58, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Paone, P.; Latousakis, D.; Terrasi, R.; Vertommen, D.; Jian, C.; Borlandelli, V.; Suriano, F.; Johansson, M.E.V.; Puel, A.; Bouzin, C.; et al. Human milk oligosaccharide 2′-fucosyllactose protects against high-fat diet-induced obesity by changing intestinal mucus production, composition and degradation linked to changes in gut microbiota and faecal proteome profiles in mice. Gut 2024, 73, 1632–1649. [Google Scholar] [CrossRef]
- Li, H.; Wang, X.K.; Tang, M.; Lei, L.; Li, J.R.; Sun, H.; Jiang, J.; Dong, B.; Li, H.Y.; Jiang, J.D.; et al. Bacteroides thetaiotaomicron ameliorates mouse hepatic steatosis through regulating gut microbial composition, gut-liver folate and unsaturated fatty acids metabolism. Gut Microbes 2024, 16, 2304159. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, L.; Wu, J.; Hu, J.; Wan, M.; Bie, J.; Li, J.; Pan, D.; Sun, G.; Yang, C. Optimal probiotic combinations for treating nonalcoholic fatty liver disease: A systematic review and network meta-analysis. Clin. Nutr. 2024, 43, 1224–1239. [Google Scholar] [CrossRef]
- Silva-Sperb, A.S.; Moraes, H.A.; Barcelos, S.T.A.; de Moura, B.C.; Longo, L.; Michalczuk, M.T.; Cerski, C.T.S.; Uribe-Cruz, C.; da Silveira, T.R.; Alvares-da-Silva, M.R.; et al. Probiotic supplementation for 24 weeks in patients with non-alcoholic steatohepatitis: The PROBILIVER randomized clinical trial. Front. Nutr. 2024, 11, 1362694. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.X.; Ding, W.J.; Chen, Y.W.; Fan, J.G. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 2017, 7, 1529. [Google Scholar] [CrossRef] [PubMed]
- Burz, S.D.; Monnoye, M.; Philippe, C.; Farin, W.; Ratziu, V.; Strozzi, F.; Paillarse, J.M.; Chene, L.; Blottiere, H.M.; Gerard, P. Fecal Microbiota Transplant from Human to Mice Gives Insights into the Role of the Gut Microbiota in Non-Alcoholic Fatty Liver Disease (NAFLD). Microorganisms 2021, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.J.; Smits, L.P.; Pekmez, C.T.; Prodan, A.; Meijnikman, A.S.; Troelstra, M.A.; Bouter, K.E.C.; Herrema, H.; Levin, E.; Holleboom, A.G.; et al. Donor Fecal Microbiota Transplantation Alters Gut Microbiota and Metabolites in Obese Individuals with Steatohepatitis. Hepatol. Commun. 2020, 4, 1578–1590. [Google Scholar] [CrossRef]
- Xue, L.; Deng, Z.; Luo, W.; He, X.; Chen, Y. Effect of Fecal Microbiota Transplantation on Non-Alcoholic Fatty Liver Disease: A Randomized Clinical Trial. Front. Cell. Infect. Microbiol. 2022, 12, 759306. [Google Scholar] [CrossRef]
- Kang, Y.; Ren, P.; Shen, X.; Kuang, X.; Yang, X.; Liu, H.; Yan, H.; Yang, H.; Kang, X.; Ding, Z.; et al. A Newly Synbiotic Combination Alleviates Obesity by Modulating the Gut Microbiota-Fat Axis and Inhibiting the Hepatic TLR4/NF-KappaB Signaling Pathway. Mol. Nutr. Food Res. 2023, 67, e2300141. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Transcription Factor | Mechanism | Core Functions | Role in MAFLD |
|---|---|---|---|
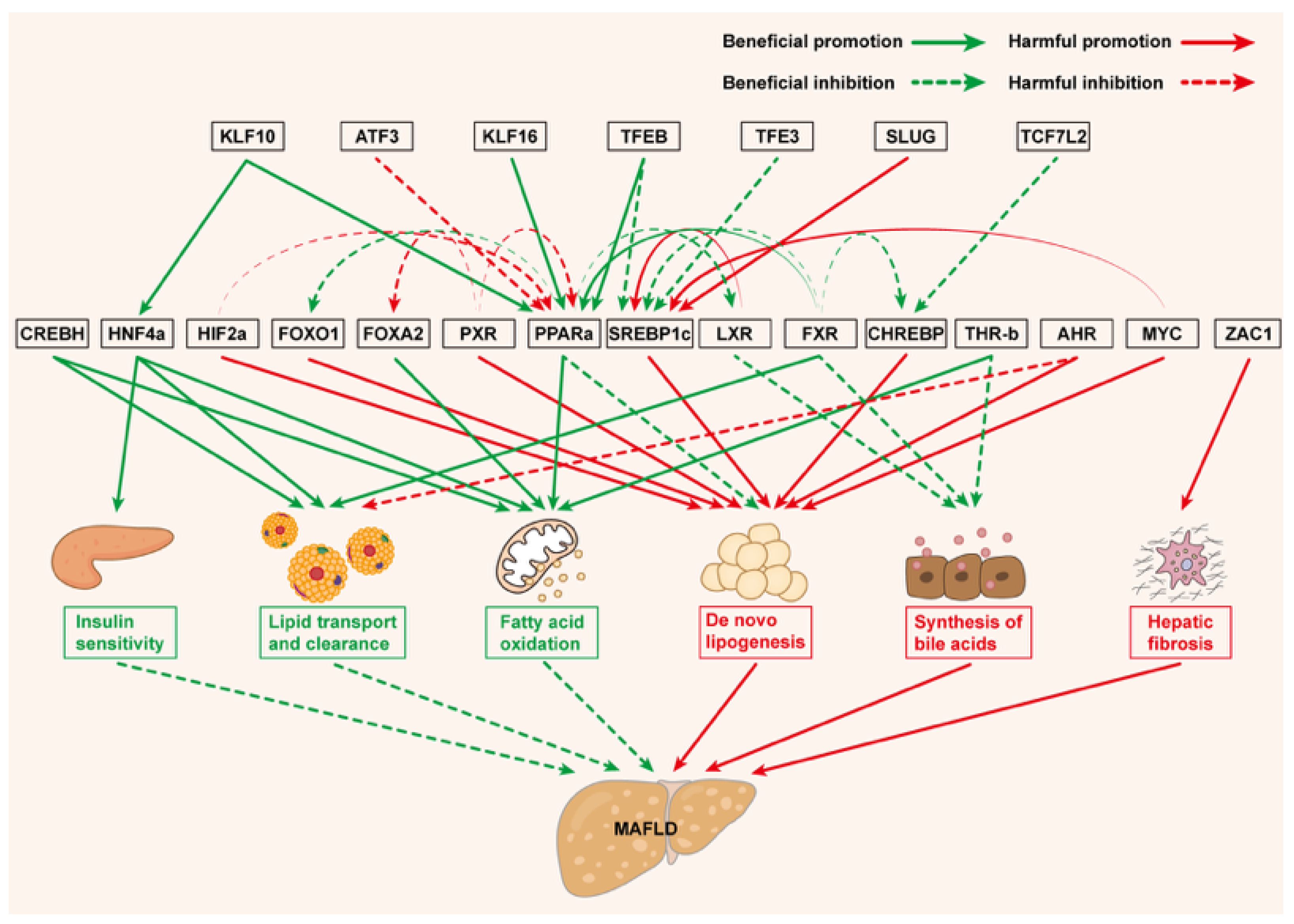
| PPARα | Activates β-oxidation genes (CPT1A, ACOX1); inhibits de novo lipogenesis; reduces lipid uptake and VLDL secretion via transcriptional regulation of CD36 and MTTP; antagonizes SREBP1c transcription | Liver: Lipid oxidation Gut: Modulates intestinal lipid absorption | Attenuates MAFLD: Attenuates steatosis via enhanced fatty acid utilization [18,19] |
| PPARβ/δ | Lowers apoC-III and VLDL receptor and increases apoA-II | Liver: Enhances fatty acid oxidation and energy uncoupling | Attenuates MAFLD: Ameliorates hepatic steatosis [20] |
| PPARγ | Regulates adipocyte differentiation, lipid uptake and energy storage | Liver: Promote lipid redistribution | Attenuates MAFLD: Improves lipid disorders [21] Exacerbates MAFLD: PPARγ-deficient exhibits resistance to diet-induced MASH [22] |
| SREBP1c | Drives ACLY, FASN, and ELOVL6 expression; phosphorylation of SREBP1c increases its expression | Liver: Lipogenesis and cholesterol biosynthesis | Exacerbates MAFLD: Exacerbates hepatic lipid accumulation [23,24,25] |
| CHREBP | Upregulates lipogenic genes (FASN, SCD-1, LPIN1 and ACLY); interacts with PPARγ to amplify lipid storage | Liver: Carbohydrate-to-lipid conversion Gut: Regulates dietary sugar metabolism | Exacerbates MAFLD: Promotes DNL under high-carbohydrate diets [26,27,28]; Induces hepatic steatosis [29,30] |
| CREBH | Cleaved C-terminal fragment activates lipoprotein lipase; induces apoC2-5 and FGF21; suppresses Niemann-Pick C1-like 1 | Liver: Promotes fatty acid oxidation Gut: Inhibits dietary cholesterol uptake; enhances intestinal cholesterol efflux | Attenuates MAFLD: Reduces HFD-induced steatosis [31,32]; mitigates hypercholesterolemia [33] |
| FXR | Stimulates FGF19/FGFR4 signaling to inhibit bile acid synthase CYP7A1 and CYP8B1; alters the ratio of cholic acid to taurocholic acid | Liver: Inhibits bile acid production; suppresses the generation of monounsaturated fatty acids; Gut: Inhibits the absorption of polyunsaturated fatty acids | Attenuates MAFLD: Reduces hepatic TG accumulation [34]; improves fibrosis in MASH patients [35] |
| PXR | Targets CD36 and PPARγ; Induces S14, LPIN1 and SLC13A5 to promote lipogenesis; upregulates SLC27A4 to increase fatty acid uptake; activates c-Jun and LPIN1 to decrease mitochondrial β-oxidation; downregulates PPARα, FGF15, and FOXA2 signaling pathways | Liver: Lipid uptake and synthesis | Exacerbates MAFLD: Exacerbates steatosis [36] |
| LXR | Increases SREBP1c, FASN, SCD-1, and ACC; Targets ABCA1 and ABCG1; | Liver: Promotes lipid deposition; Gut: Increases fecal excretion of bile acids | Exacerbates MAFLD: Promotes the deterioration of MASH [37] Attenuates MAFLD: Increases insulin sensitivity [38] |
| AHR | Promotes SCD-1 expression to enhance monounsaturated fatty acid synthesis; promotes the expression of fatty acid transport-related genes CD36, LDLR, VLDLR, and FABP4; | Liver: Facilitates hepatic fatty acid uptake and accumulation | Exacerbates MAFLD: Promotes hepatic steatosis [39] |
| THR-β | Upregulates CPT1A, medium-chain acyl-coenzyme A dehydrogenase and LDL-R; downregulates SCD-1 and glycerol-3-phosphate acyltransferase-3; stimulates FASN, ACC-α, SREBP1c, and carbohydrate-responsive element-binding protein; | Liver: Promotes mitochondrial fatty acid uptake and β-oxidation; decreases circulating cholesterol | Attenuates MAFLD: Reduces hepatic steatosis and inflammation [40] |
| HNF4α | Regulates lipid transport genes apoB and MTTP, and gluconeogenic genes glucose-6-phosphatase catalytic and phosphoenolpyruvate carboxykinase | Liver: VLDL and HDL secretion; glucose homeostasis | Attenuates MAFLD: Loss promotes steatosis; antagonism reduces VLDL output [41,42] |
| FOXO1 | Induces insulin resistance and gluconeogenesis; regulates autophagy and glycophagy | Liver: Glucose production | Exacerbates MAFLD: Exacerbates MAFLD progression; links adipocyte dysfunction to hepatic insulin resistance [43] |
| FOXA2 | Upregulates CPT2; regulated by insulin and CaM signaling | Liver: Enhances mitochondrial oxidation | Attenuates MAFLD: Restores lipid catabolism [44] |
| HIF2α | Upregulates sterol regulatory element binding transcription factor 1, FASN, and CD36; downregulates PPARα and ACOX1 | Liver: Promotes fatty acid synthesis and uptake; downregulates fatty acid β-oxidation | Exacerbates MAFLD: Exacerbates hepatic steatosis [45,46] |
| MYC | Induces SREBP1; reduces GLP-1 secretion; activates de novo ceramide synthesis | Liver: Drives DNL; Gut: Reduces GLP-1 secretion | Exacerbates MAFLD: Aggravates hepatic ceramide accumulation and hepatic steatosis [47] |
| ZAC1 | Regulates imprinted genes; activates TGF-β1/Collagen Type VI α2 | Liver: Drives hepatic fibrosis | Exacerbates MAFLD: Drives juvenile MAFLD fibrosis [48] |
| Transcription Factor | Main Mechanisms | Core Functions | Association with MAFLD |
|---|---|---|---|
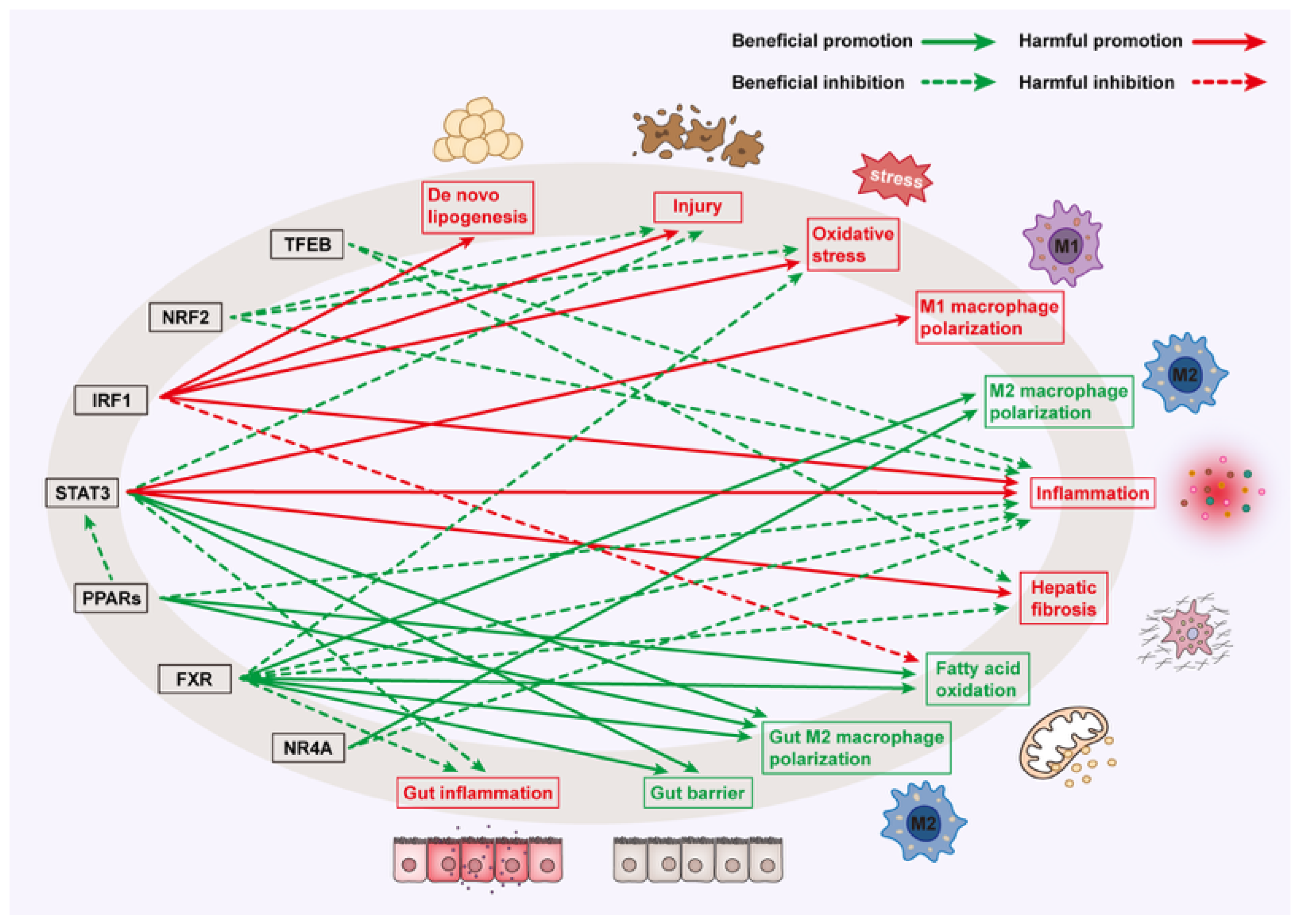
| PPARs | PPARα: reduces lipid accumulation and oxidative stress; PPARδ: reduces IL-1β, F4/80, and NLRP3 inflammasome activity; macrophage polarization to M2 phenotype; PPARγ: Transcriptional repression of NF-κB and STAT; suppresses TGF-β1 | Liver: Inhibits inflammation; | Attenuates MAFLD: Improves NAFLD activity score (NAS) [124]; suppresses inflammation and fibrosis [120] |
| FXR | Inhibits NLRP3 and NF-κB signaling, reducing pro-inflammatory cytokines (TNF-α, IL-6) and chemokines (MCP-1); promotes macrophage M2 polarization; suppresses TGF-β/SMAD2/3 signaling, and reduces HSC activation; prevents ileal CD8+ T cell infiltration; improves gut barrier function via GPBAR1-dependent IL-10 production | Liver: Suppresses inflammation; Gut: Enhances barrier function | Attenuates MAFLD: Attenuates steatohepatitis and fibrosis [86,125,126] |
| STAT3 | Promotes IL-1β, fibrotic genes (Timp-1, α-SMA and Zeb2), TGF-β1, IL-17 secretion, HPC expansion, and HSC activation; improves insulin sensitivity and resolves steatosis and inflammation; promotes M2 polarization; activates anti-apoptotic pathways in hepatocytes | Liver: Promotes pro-inflammatory and fibrogenic signaling; suppresses apoptosis; Gut: Improves insulin sensitivity and intestinal permeability; resolves inflammation | Exacerbates MAFLD: Promotes inflammation, HSC activation and fibrosis [127,128]; Attenuates MAFLD: Reduces hepatocyte and enterocytes death [129] |
| NRF2 | Induces antioxidant genes (SOD2, HO-1 and NQO1); restores glutathione levels; inhibits gasdermin D expression | Liver: Reduces ROS and suppresses lipogenic pathways | Attenuates MAFLD: Attenuates oxidative stress and fibrosis [130] |
| IRF1 | Upregulates Osbpl3, Ddit4, and Ccl2; inhibits AMPK-TFEB autophagy; drives macrophage ferroptosis; | Liver: Promotes lipogenesis and oxidative stress; | Exacerbates MAFLD: Aggravates steatosis and inflammation [131,132] |
| NR4A | Modulates Treg/Th17 balance; inhibits NF-κB, TNF-α and IL-6 in macrophages | Liver: Suppresses Kupffer cell apoptosis | Attenuates MAFLD: Deficiency exacerbates fibrosis and inflammation; agonists reduce inflammation [133] |
| TFEB | Induces lysosomal/autophagy genes; clears lipid droplets, damaged mitochondria, and ROS | Liver: Enhances lipid degradation and autophagy | Attenuates MAFLD: Restores hepatocyte function; reduces hepatic TG and fibrosis [134] |
| Transcription Factor | Main Mechanisms | Core Functions | Association with MAFLD |
|---|---|---|---|
| PPARs | Restores the expression of tight junction proteins; Lactobacillus casei, Lactobacillus plantarum FRT10, Lachnospiraceae, Clostridium, and SCFAs enhance PPARs activity | Liver: Reduces hepatic steatosis; Gut: Activated by bacteria; improves intestinal barrier | Attenuates MAFLD: Prevents hepatic steatosis and flora disturbance [158] |
| FXR | Suppresses hepatic bile acid synthesis; enhances intestinal tight junction; activates Wnt/β-catenin signaling to ameliorate disrupted GVB; upregulates the expression of iNOS, IL-18, and angiopoietin 1 | Gut: Improves intestinal barrier; strengthens antimicrobial defense | Attenuates MAFLD: Ameliorates steatosis and fibrosis [159] |
| PXR | Decreases bile acid-metabolizing bacteria, Allobaculum, Bifidobacterium; increases Akkermansia muciniphila, Allobaculum spp., specific pro-inflammatory Lactobacillus, and Firmicutes/Bacteroidetes ratio; elevates deoxycholic acid | Gut: Drives microbial dysbiosis and inflammation | Exacerbates MAFLD: Promotes hepatic injury and lipid metabolism disorders in MAFLD [160] |
| AHR | Activated by indole propionic acid to reduce TNF-α, IFN-γ, IL-1β; promotes mucin production and intestinal homeostasis | Gut: Protects the intestinal mucosa and intestinal homeostasis; inhibits inflammation | Attenuates MAFLD: Decreased AHR ligands and colonic AHR expression is related to MAFLD [161]; suppresses the intestinal inflammation and prevents bacterial translocation |
| HIF2α | Butyrate consumes oxygen to activate HIF signaling; activates neuraminidase 3 to increase ceramide; acute activation preserves barrier integrity while chronic activation disrupts tight junctions | Gut: Increases intestinal and serum ceramide | Exacerbates MAFLD: Positively correlates with body mass index and hepatic enzymes; HFD promotes HIF2α activation; promotes hepatic steatosis and obesity [162] |
| NR1D1 | Binds to promoters of Claudin 1, Claudin 3, and Zona occludens 3; agonist SR9009 promotes goblet cell populations and mucin secretion | Gut: Restores intestinal barrier integrity | Attenuates MAFLD: Ameliorates hepatic steatosis, insulin resistance, and inflammation in MASH mice [163] |
| Transcription Factor | Drug | Progress | Challenges |
|---|---|---|---|
| PPARα | Fenofibrate | Improved dyslipidemia and inflammation [201] | Minimal effect on insulin sensitivity or liver histology [196] |
| Pemafibrate | Decreased MRE-based liver stiffness | Did not decrease liver fat [197] | |
| PPARβ/δ | Seladelpar | Improvement in insulin sensitivity, liver enzymes, and hepatic steatosis | Termination due to worrisome histology results [202] |
| Endurobol/GW501516 | Increases HDL-C and reduces LDL-C and TG in obese monkeys with insulin resistance [57] | ||
| PPARγ | Pioglitazone | Reduced histological liver fat, inflammation, and fibrosis [203] | Increased risk of fluid retention, edema, congestive heart failure, and bladder cancer [203,204] |
| Rosiglitazone | Improve insulin sensitivity [203] | Does not appear to be as effective as pioglitazone in ameliorating MAFLD, and might cause pro-inflammatory changes [203] | |
| PPARα and γ | Saroglitazar | Improved postprandial triglycerides in people with diabetic dyslipidemia [205]; improved ALT and hepatic fat [199] | No improvement in delta change of NAS from baseline to week 24 biopsy [206] |
| PPARα and δ | Elafibranor | Resolved NASH without fibrosis worsening in patients [207] | Not met the primary endpoint (NASH resolution) in the large phase 3 trial [207] |
| PPARα, δ and γ | Lanifibranor | Decreased at least 2 points in the activity part of steatosis, activity, fibrosis scoring system of MASH patients [200]; decreased MAFLD in rodents [208] | Increased the risk of diarrhea, nausea, peripheral edema, anemia, and weight gain [200] |
| SREBP1c | Oltipraz | Decreased liver fat and BMI [209] | No difference in insulin resistance, liver enzymes, lipids, and cytokines |
| FXR | Cilofexor | Decreased fat accumulation, serum bile acids, and fibrosis in MASH patients [210] | |
| Obeticholic acid | Decreased liver enzymes; improved liver histology in MASH patients [35] | No statistically significant effect [211]; side effect: Pruritus [35] | |
| Tropifexor | Decreased liver fat [212]; ameliorates liver injury, fibrosis, intestinal barrier injury in piglets [213] | ||
| LXR | GW3965 | Alters the VLDL-C and LDL-C levels in hamsters and cynomolgus monkeys; inhibits atherosclerosis in mice [214] | Unsuitable for clinical development due to their pleotropic effects |
| SR9238 | Inhibition of LXR-driven lipogenesis [215] | Decreased liver lipid content and inflammation in mice [215] | |
| GW6340 | Promotes excretion of (3)H-sterol in feces [216] | ||
| LXR-623 | Regulates lipid levels in monkeys [217] | Termination due to unexpected adverse neurological events | |
| THR-β | Resmetirom | Reduced liver fat content, LDL, and apoB of MASH patients [218] | Higher incidence of transient mild diarrhea and nausea [219] |
| VK2809 | Reduced liver fat content [220]; decreased hepatic mass and TG in mice [221] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, A.; Huang, M.; Xi, Y.; Deng, X.; Xu, K. Orchestration of Gut–Liver-Associated Transcription Factors in MAFLD: From Cross-Organ Interactions to Therapeutic Innovation. Biomedicines 2025, 13, 1422. https://doi.org/10.3390/biomedicines13061422
Liu A, Huang M, Xi Y, Deng X, Xu K. Orchestration of Gut–Liver-Associated Transcription Factors in MAFLD: From Cross-Organ Interactions to Therapeutic Innovation. Biomedicines. 2025; 13(6):1422. https://doi.org/10.3390/biomedicines13061422
Chicago/Turabian StyleLiu, Ao, Mengting Huang, Yuwen Xi, Xiaoling Deng, and Keshu Xu. 2025. "Orchestration of Gut–Liver-Associated Transcription Factors in MAFLD: From Cross-Organ Interactions to Therapeutic Innovation" Biomedicines 13, no. 6: 1422. https://doi.org/10.3390/biomedicines13061422
APA StyleLiu, A., Huang, M., Xi, Y., Deng, X., & Xu, K. (2025). Orchestration of Gut–Liver-Associated Transcription Factors in MAFLD: From Cross-Organ Interactions to Therapeutic Innovation. Biomedicines, 13(6), 1422. https://doi.org/10.3390/biomedicines13061422