
Patients with Papillary Renal Cancer and Germline Duplication of MET Exons 5-21

 , , ,
, , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Anamnesis
2.2. Genetic Laboratory Testing
2.3. Bioinformatic Analysis
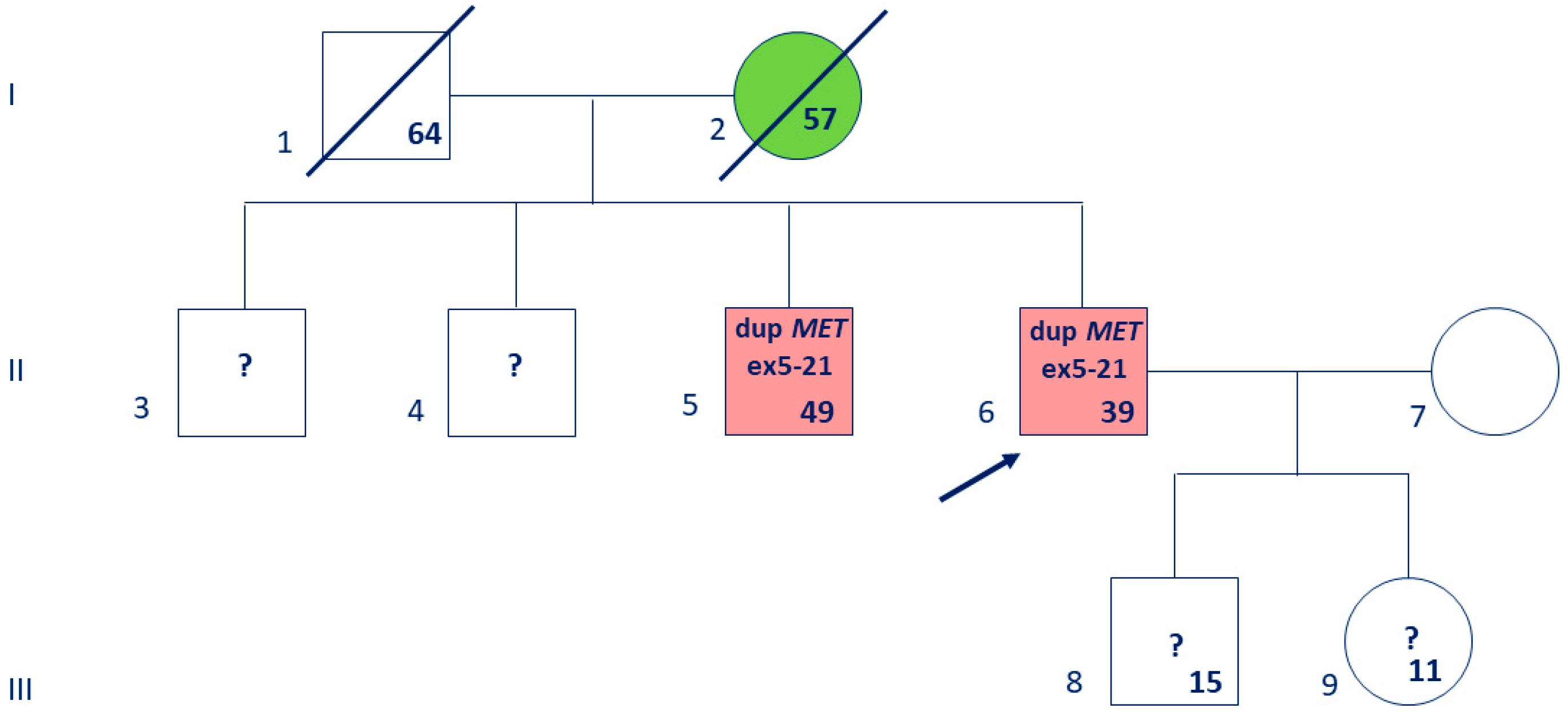
3. Results
3.1. Absence of MET Gene Alteration in Routine Testing
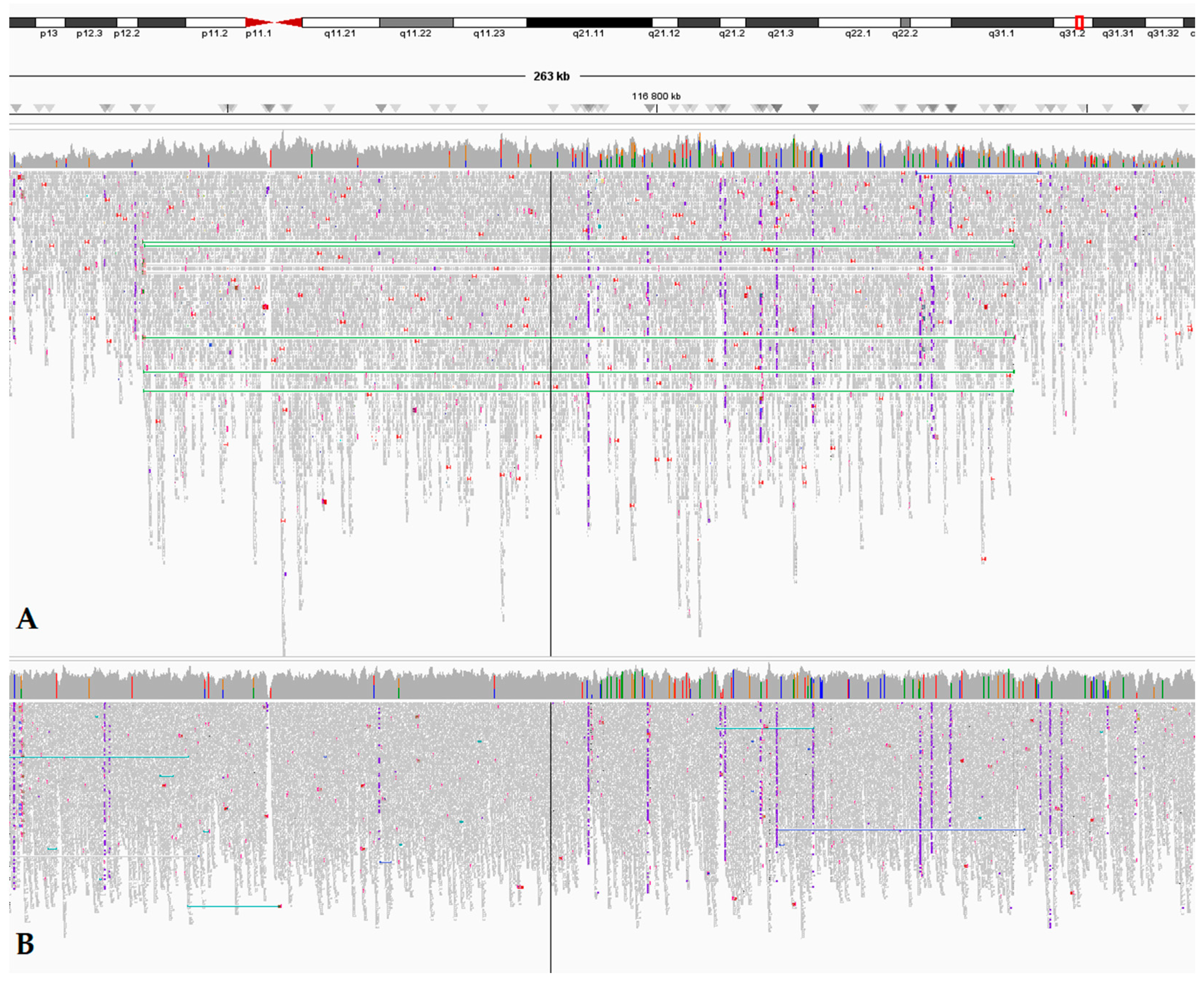
3.2. Whole-Genome Sequencing Reveals a Large MET Duplication
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HPRC | Hereditary Papillary Renal Carcinoma |
| ACMG | American College of Medical Genetics and Genomics |
| VUS | Variant of Uncertain Significance |
| RC | Renal Cancer |
| WGS | Whole-Genome Sequencing |
| WHO | World Health Organization |
| ISUP | International Society of Urological Pathology |
| P/LP | Pathogenic/Likely Pathogenic |
| MRI | Magnetic Resonance Imaging |
| USA | The United States of America |
| PCR | Polymerase Chain Reaction |
| MLPA | Multiplex Ligation-dependent Probe Amplification |
| DNA | DeoxyriboNucleic Acid |
| NGS | Next Generation Sequencing |
| HGVS | Human Genome Variation Society |
| CNV | Copy Number Variation |
| OMIM | Online Mendelian Inheritance in Man |
| FIP | Federal Institute of Industrial Property (Russia) |
| IGV | Integrative Genomics Viewer |
| HRR | Homologous Recombination Repair |
| AMP | Association for Molecular Pathology |
| ASCO | American Society of Clinical Oncology |
| CAP | College of American Pathologists |
| NCCN | The National Comprehensive Cancer Network |
| CGC | Cancer Genomics Consortium |
| VICC | Variant Interpretation for Cancer Consortium |
References
- ECIS—European Cancer Information System. Available online: https://ecis.jrc.ec.europa.eu (accessed on 10 December 2024).
- Yanus, G.A.; Kuligina, E.S.; Imyanitov, E.N. Hereditary renal cancer syndromes. Med. Sci. 2024, 12, 12. [Google Scholar] [CrossRef]
- Zhang, H.; Andreou, A.; Bhatt, R.; Whitworth, J.; Yngvadottir, B.; Maher, E.R. Characteristics, aetiology and implications for management of multiple primary renal tumours: A systematic review. Eur. J. Hum. Genet. 2024, 32, 887–894. [Google Scholar] [CrossRef]
- Mikhaylenko, D.S.; Klimov, A.V.; Matveev, V.B.; Samoylova, S.I.; Strelnikov, V.V.; Zaletaev, D.V.; Lubchenko, L.N.; Alekseev, B.Y.; Nemtsova, M.V. Case of hereditary papillary renal cell carcinoma type I in a patient with a germline MET mutation in Russia. Front. Oncol. 2020, 9, 1566. [Google Scholar] [CrossRef]
- Fujino, T.; Suda, K.; Mitsudomi, T. Lung cancer with MET exon 14 skipping mutation: Genetic feature, current treatments, and future challenges. Lung Cancer 2021, 12, 35–50. [Google Scholar] [CrossRef]
- Webster, B.R.; Gopal, N.; Ball, M.W. Tumorigenesis mechanisms found in hereditary renal cell carcinoma: A review. Genes 2022, 13, 2122. [Google Scholar] [CrossRef]
- Villacis, R.A.; Basso, T.R.; Canto, L.M.; Nobrega, A.F.; Achatz, M.I.; Rogatto, S.R. Germline large genomic alterations on 7q in patients with multiple primary cancers. Sci. Rep. 2017, 7, 41677. [Google Scholar] [CrossRef]
- Yang, Y.; Ricketts, C.J.; Vocke, C.D.; Killian, J.K.; Padilla-Nash, H.M.; Lang, M.; Wei, D.; Lee, Y.H.; Wangsa, D.; Sourbier, C.; et al. Characterization of genetically defined sporadic and hereditary type 1 papillary renal cell carcinoma cell lines. Genes Chromosomes Cancer 2021, 60, 434–446. [Google Scholar] [CrossRef]
- Filippova, M.G.; Mikhaylenko, D.S.; Samoylenko, I.V.; Sergeev, Y.S.; Kozlov, N.A.; Fainstein, I.A.; Alekseeva, E.A. Hereditary leiomyomatosis and renal cell cancer: A case report. Cancer Urol. 2022, 18, 211–216. (In Russian) [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Garrett, A.; Durkie, M.; Callaway, A.; Burghel, G.J.; Robinson, R.; Drummond, J.; Torr, B.; Cubuk, C.; Berry, I.R.; Wallace, A.J.; et al. Combining evidence for and against pathogenicity for variants in cancer susceptibility genes: CanVIG-UK consensus recommendations. J. Med. Genet. 2021, 58, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Griffith, M.; Danos, A.M.; Pitel, B.A.; Madhavan, S.; Liu, X.; Chow, C.; Williams, H.; Carmody, L.; Barrow-Laing, L.; et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet. Med. 2022, 24, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Amin, M.B.; Berney, D.M.; Comperat, E.M.; Gill, A.J.; Hartmann, A.; Menon, S.; Raspollini, M.R.; Rubin, M.A.; Srigley, J.R.; et al. The 2022 World Health Organization classification of tumours of the urinary system and male genital organs—Part A: Renal, penile, and testicular tumours. Eur. Urol. 2022, 82, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Lee, K.; Abul-Husn, N.S.; Amendola, L.M.; Brothers, K.; Chung, W.K.; Gollob, M.H.; Gordon, A.S.; Harrison, S.M.; Hershberger, R.E.; et al. ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2023, 25, 100866. [Google Scholar] [CrossRef]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, W.H.; Zhou, S.H.; Gu, J.L.; Yu, Q.; Zhu, Y.Z.; Yan, Y.J.; Zhu, Z.; Shang, J.B. Molecular genetics, therapeutics and RET inhibitor resistance for medullary thyroid carcinoma and future perspectives. Cell Commun. Signal. 2024, 22, 460. [Google Scholar] [CrossRef]
- Aarabi, M.; Darabi, H.; Bashar, A.; Bellissimo, D.; Rajkovic, A.; Yatsenko, S.A. Copy-number variants in the ACMG secondary finding genes: A reporting framework for clinical cytogeneticists. Genet. Med. Open 2024, 2, 101839. [Google Scholar] [CrossRef]
- Collins, R.L.; Glessner, J.T.; Porcu, E.; Lepamets, M.; Brandon, R.; Lauricella, C.; Han, L.; Morley, T.; Niestroj, L.M.; Ulirsch, J.; et al. A cross-disorder dosage sensitivity map of the human genome. Cell 2022, 185, 3041–3055.e25. [Google Scholar] [CrossRef]
- Nepomuceno, T.C.; Carvalho, M.A.; Rodrigue, A.; Simard, J.; Masson, J.Y.; Monteiro, A.N.A. PALB2 variants: Protein domains and cancer susceptibility. Trends Cancer 2021, 7, 188–197. [Google Scholar] [CrossRef]
- Kuzbari, Z.; Bandlamudi, C.; Loveday, C.; Garrett, A.; Mehine, M.; George, A.; Hanson, H.; Snape, K.; Kulkarni, A.; Allen, S.; et al. Germline-focused analysis of tumour-detected variants in 49,264 cancer patients: ESMO Precision Medicine Working Group recommendations. Ann. Oncol. 2023, 34, 215–227. [Google Scholar] [CrossRef]
- Fencer, M.G.; Krupa, K.A.; Bleich, G.C.; Grumet, S.; Eladoumikdachi, F.G.; Kumar, S.; Kowzun, M.J.; Potdevin, L.B. Diagnosis, management, and surveillance for patients with PALB2, CHEK2, and ATM gene mutations. Clin. Breast Cancer 2023, 23, e194–e199. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gianferante, M.; Karyadi, D.M.; Hartley, S.W.; Frone, M.N.; Luo, W.; Robison, L.L.; Armstrong, G.T.; Bhatia, S.; Dean, M.; et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in the childhood cancer survivor study. JNCI Cancer Spectr. 2021, 5, pkab007. [Google Scholar] [CrossRef]
- Denize, T.; Just, P.A.; Sibony, M.; Blons, H.; Timsit, M.O.; Drossart, T.; Jakubowicz, D.; Broudin, C.; Morini, A.; Molina, T.; et al. MET alterations in biphasic squamoid alveolar papillary renal cell carcinomas and clinicopathological features. Mod. Pathol. 2021, 34, 647–659. [Google Scholar] [CrossRef]
- Guo, R.; Offin, M.; Brannon, A.R.; Chang, J.; Chow, A.; Delasos, L.; Girshman, J.; Wilkins, O.; McCarthy, C.G.; Makhnin, A.; et al. MET exon 14-altered lung cancers and MET inhibitor resistance. Clin. Cancer Res. 2021, 27, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Guerin, C.; Vinchent, A.; Fernandes, M.; Damour, I.; Laratte, A.; Tellier, R.; Estevam, G.O.; Meneboo, J.P.; Villenet, C.; Descarpentries, C.; et al. MET variants with activating N-lobe mutations identified in hereditary papillary renal cell carcinomas still require ligand stimulation. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lyu, Y.; Li, L.; Wu, D.; Chen, J.; Lin, T.; Huang, Z.; Hu, J.; Wang, W.; Lin, H.; et al. Crizotinib inhibits activation of MET pathway caused by MET extracellular SEMA domain duplication. Lung Cancer 2020, 147, 64–70. [Google Scholar] [CrossRef]
- Lan, X.; Feng, M.; Lv, J.; Zhang, L.; Hu, P.; Wang, Y.; Zhang, Y.; Wang, S.; Liu, C.; Liu, C. A 23-year bibliometric analysis of the development of global research on hereditary renal carcinoma. Front. Oncol. 2024, 14, 1364997. [Google Scholar] [CrossRef]
- Sebai, M.; Tulasne, D.; Caputo, S.M.; Verkarre, V.; Fernandes, M.; Guérin, C.; Reinhart, F.; Adams, S.; Maugard, C.; Caron, O.; et al. Novel germline MET pathogenic variants in French patients with papillary renal cell carcinomas type I. Hum. Mutat. 2022, 43, 316–327. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Kidney Cancer v.2.2025. Available online: https://www.nccn.org/guidelines/category_1 (accessed on 12 December 2024).
- Jacoba, I.M.; Lu, Z. Hereditary papillary renal cell carcinoma. Semin. Diagn. Pathol. 2024, 41, 28–31. [Google Scholar] [CrossRef]
- Guerin, C.; Tulasne, D. Recording and classifying MET receptor mutations in cancers. eLife 2024, 13, e92762. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordinates, GRCh38 (chr7) | Duplication Size, bp | Duplicated MET Exon(s) | Phenotype | Sample Reference |
|---|---|---|---|---|
| 97419852–158923762 | 61,503,911 | All exons and flanking genes | Developmental disabilities, congenital anomalies | 147547 1 |
| 115459015–159325817 | 43,866,803 | All exons and flanking genes | Not reported | 152912 1 |
| 115059311–132281500 | 17,222,190 | All exons and flanking genes | Short stature, autism, gait imbalance, delayed gross motor development | 322280 2 |
| 114578028–125438062 | 10,860,035 | All exons and flanking genes | Autism, intellectual disability, bulbous nose, low-hanging columella, thin upper lip vermilion | 2362 2 |
| 114105646–116927595 | 2,821,950 | All exons and flanking genes | Hypotonia, abnormal pinna morphology, high and narrow palate, wide nasal bridge, feeding difficulties, adducted thumb, single transverse palmar crease, aplasia hypoplasia of the lungs, apnea | 439826 2 |
| 115015244–116710591 | 1,695,348 | 1–2 and 5′-flanking genes | Motor delay, mild intellectual disability | 509153 2 |
| 115042242–116687714 | 1,645,473 | Promoter and 5′-flanking genes | Developmental disabilities, congenital anomalies | 59715 1 |
| 116699075–117504373 * | 805,299 | 2–21 and 3′-flanking genes | Papillary RC | 583430 1 |
| 116756194–117318372 | 562,179 | 6–21 and 3′-flanking genes | Hypotonia, seizure disorder, autism with high cognitive abilities, mild global developmental delay, hypertelorism, prominent forehead | 281389 2 |
| 116740252–116841718 * | 101,467 | 5–21 | Papillary RC | Cases #1 and #2 |
| 116740619–116839316 | 98,698 | 5–21 | Not reported | 505381 2 |
| 116740852–116798386 * | 57,535 | 5–21 | RC (type not defined) | 252900 1 |
| 116699075–116700294 * | 1220 | 2 | Papillary RC | 583929 1 |
| 116699084–116700284 * | 1201 | 2 | RC (type not defined) | 220130 1 |
| 116755345–116755525 * | 181 | 6 | Papillary RC, RC (type not defined) | 643144 1 (2 samples) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhaylenko, D.S.; Kuryakova, N.B.; Bostanova, F.M.; Zabnenkova, V.V.; Ryzhkova, O.P.; Volodin, I.V.; Zaletaev, D.V.; Pustoshilov, D.V.; Kutsev, S.I.; Strelnikov, V.V. Patients with Papillary Renal Cancer and Germline Duplication of MET Exons 5-21. Biomedicines 2025, 13, 1329. https://doi.org/10.3390/biomedicines13061329
Mikhaylenko DS, Kuryakova NB, Bostanova FM, Zabnenkova VV, Ryzhkova OP, Volodin IV, Zaletaev DV, Pustoshilov DV, Kutsev SI, Strelnikov VV. Patients with Papillary Renal Cancer and Germline Duplication of MET Exons 5-21. Biomedicines. 2025; 13(6):1329. https://doi.org/10.3390/biomedicines13061329
Chicago/Turabian StyleMikhaylenko, Dmitry S., Natalya B. Kuryakova, Fatima M. Bostanova, Viktoria V. Zabnenkova, Oksana P. Ryzhkova, Ilya V. Volodin, Dmitry V. Zaletaev, Dmitry V. Pustoshilov, Sergey I. Kutsev, and Vladimir V. Strelnikov. 2025. "Patients with Papillary Renal Cancer and Germline Duplication of MET Exons 5-21" Biomedicines 13, no. 6: 1329. https://doi.org/10.3390/biomedicines13061329
APA StyleMikhaylenko, D. S., Kuryakova, N. B., Bostanova, F. M., Zabnenkova, V. V., Ryzhkova, O. P., Volodin, I. V., Zaletaev, D. V., Pustoshilov, D. V., Kutsev, S. I., & Strelnikov, V. V. (2025). Patients with Papillary Renal Cancer and Germline Duplication of MET Exons 5-21. Biomedicines, 13(6), 1329. https://doi.org/10.3390/biomedicines13061329