
Mucopolysaccharidosis Type I and α-Mannosidosis—Phenotypically Comparable but Genetically Different: Diagnostic and Therapeutic Considerations



Abstract
1. Introduction
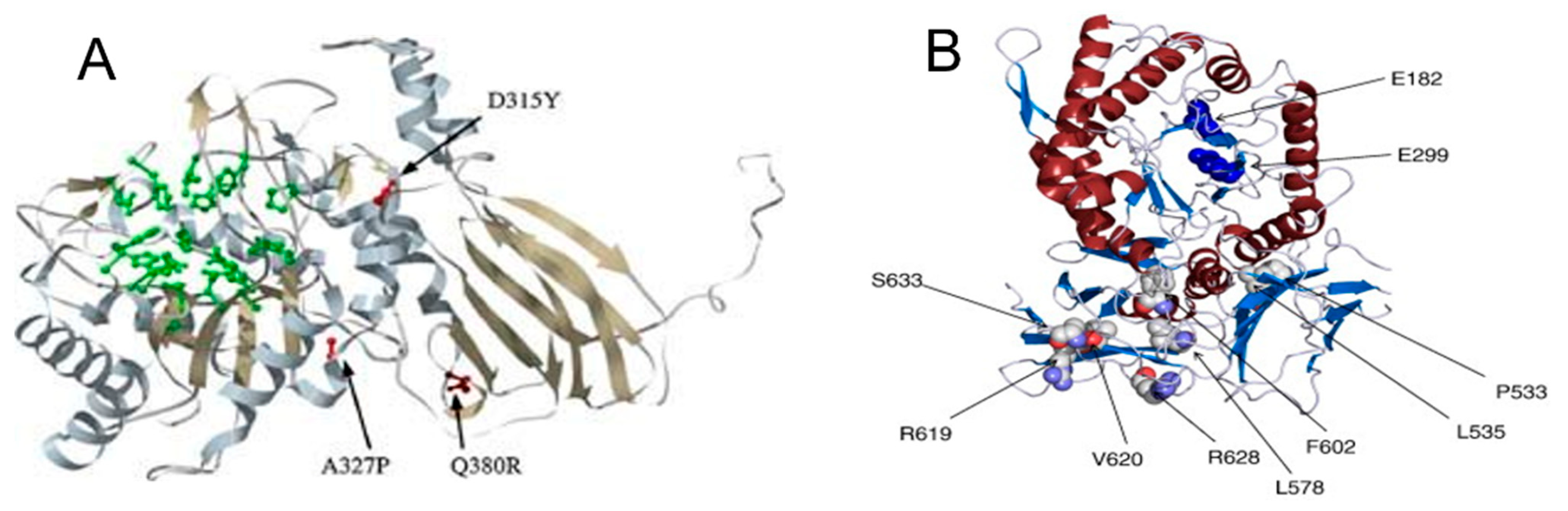
2. α-Mannosidosis Diagnosis—Genotype–Phenotype Correlation Analysis in Patients with Clinical Manifestations Like MPS
3. Misdiagnosis of α-Mannosidosis: A Case Study Comparing MPS and α-Mannosidosis from a Clinician’s Perspective
4. The Roles of Epigenetics in MPS-I and α-Mannosidosis
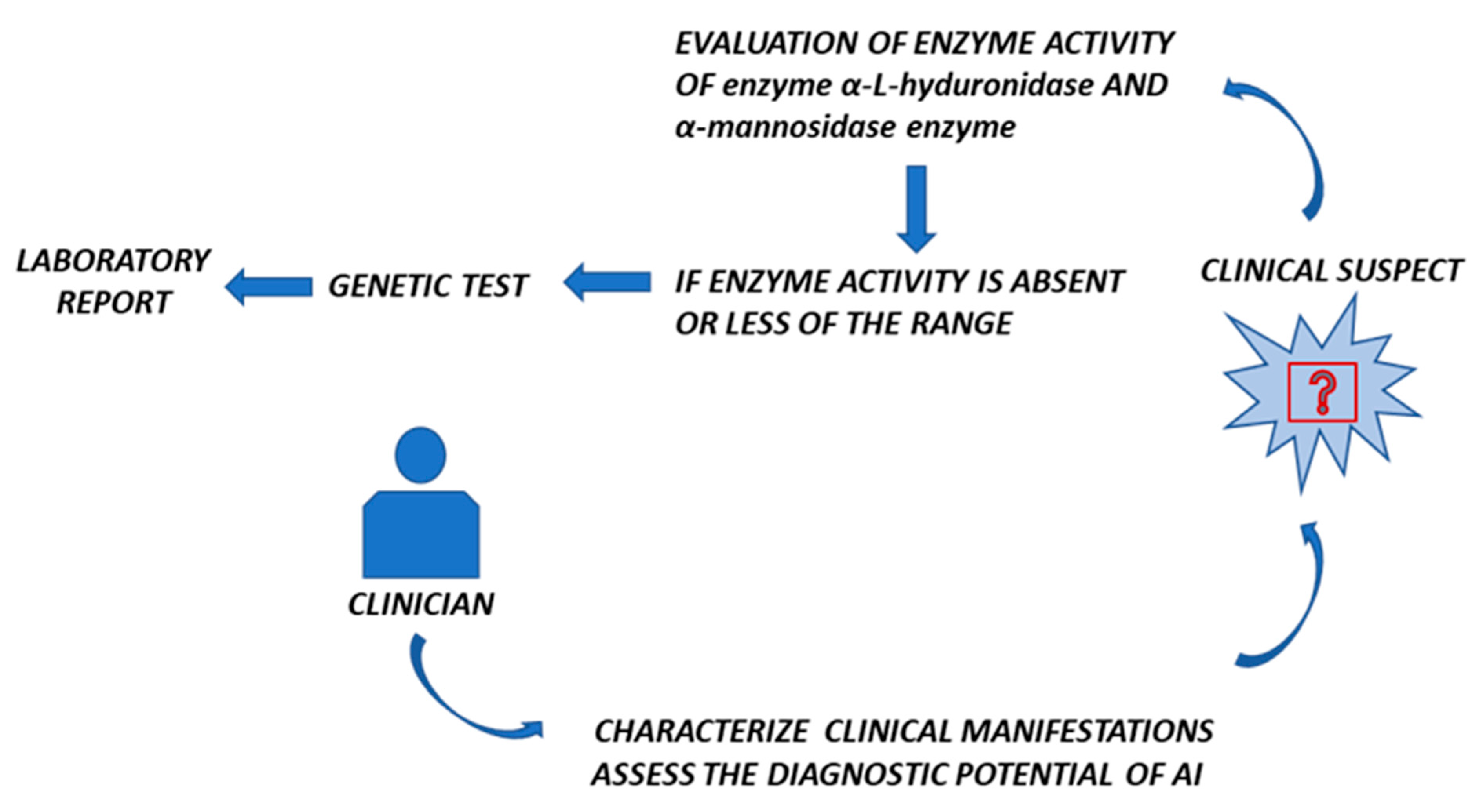
5. Could AI Be Useful to Distinguish MPS-I and α-Mannosidosis?
6. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wraith, J.E.; Jones, S. Mucopolysaccharidosis type I. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. 1), 102–106. [Google Scholar] [PubMed]
- Pérez-López, J.; Morales-Conejo, M.; López-Rodríguez, M.; Hermida-Ameijeiras, Á.; Moltó-Abad, M. Efficacy of laronidase therapy in patients with mucopolysaccharidosis type I who initiated enzyme replacement therapy in adult age. A systematic review and meta-analysis. Mol. Genet. Metab. 2017, 121, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Constantopoulos, G.; McComb, R.D.; Dekaban, A.S. Neurochemistry of the mucopolysaccharidoses: Brain glycosaminoglycans in normals and four types of mucopolysaccharidoses. J. Neurochem. 1976, 26, 901–908. [Google Scholar] [CrossRef]
- Hampe, C.S.; Eisengart, J.B.; Lund, T.C.; Orchard, P.J.; Swietlicka, M.; Wesley, J.; McIvor, R.S. Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology. Cells 2020, 9, 1838. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A. Mucopolysaccharidosis Type I. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2002. [Google Scholar]
- Segni e Sintomi Della MPS I in Bambini e Adulti. Available online: https://www.malattielisosomiali.it/it/mps1/segni-e-sintomi/bambini-e-adulti (accessed on 12 February 2025).
- Wiesinger, T.; Schwarz, M.; Mechtler, T.P.; Liebmann-Reindl, S.; Streubel, B.; Kasper, D.C. α-Mannosidosis—An underdiagnosed lysosomal storage disease in individuals with an ‘MPS-like’ phenotype. Mol. Genet. Metab. 2020, 130, 149–152. [Google Scholar] [CrossRef]
- Govender, R.; Mubaiwa, L. Alpha-mannosidosis: A report of 2 siblings and review of the literature. J. Child Neurol. 2014, 29, 131–134. [Google Scholar] [CrossRef]
- Ficicioglu, C.; Stepien, K.M. Alpha-Mannosidosis. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2001. [Google Scholar]
- Ru, M.H.; Boelens, J.J.; Das, A.M.; Jones, S.A.; van der Lee, J.H.; Mahlaoui, N.; Mengel, E.; Offringa, M.; O’Meara, A.; Parini, R.; et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: Results of a European consensus procedure. Orphanet J. Rare Dis. 2011, 6, 55. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Santoro, L.; Monachesi, C.; Zampini, L.; Padella, L.; Galeazzi, T.; Santori, E.; Cordiali, R.; Dardis, A.; Catassi, C.; Boccieri, E.; et al. First experience of combined enzyme replacement therapy and hematopoietic stem cell transplantation in alpha-mannosidosis. Am. J. Med. Genet. A 2023, 191, 1948–1952. [Google Scholar] [CrossRef] [PubMed]
- Rempel, B.P.; Clarke, L.A.; Withers, S.G. A homology model for human alpha-l-iduronidase: Insights into human disease. Mol. Genet. Metab. 2005, 85, 28–37. [Google Scholar] [CrossRef]
- Beesley, C.E.; Meaney, C.A.; Greenland, G.; Adams, V.; Vellodi, A.; Young, E.P.; Winchester, B. Mutational analysis of 85 mucopolysaccharidosis type I families: Frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum. Genet. 2001, 109, 503–511. [Google Scholar]
- Lee-Chen, G.J.; Lin, S.P.; Tang, Y.F.; Chin, Y.W. Mucopolysaccharidosis type I: Characterization of novel mutations affecting alpha-L-iduronidase activity. Clin. Genet. 1999, 56, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Matte, U.; Yogalingam, G.; Brooks, D.; Leistner, S.; Schwartz, I.; Lima, L.; Norato, D.Y.; Brum, J.M.; Beesley, C.; Winchester, B.; et al. Identification and characterization of 13 new mutations in mucopolysaccharidosis type I patients. Mol. Genet. Metab. 2003, 78, 37–43. [Google Scholar] [CrossRef]
- Saito, S.; Ohno, K.; Maita, N.; Sakuraba, H. Structural and clinical implications of amino acid substitutions in α-L-iduronidase: Insight into the basis of mucopolysaccharidosis type I. Mol. Genet. Metab. 2014, 111, 107–112. [Google Scholar] [CrossRef]
- Amberger, J.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. McKusick’s Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res. 2009, 37, D793–D796. [Google Scholar] [CrossRef] [PubMed]
- Lenffer, J.; Nicholas, F.W.; Castle, K.; Rao, A.; Gregory, S.; Poidinger, M.; Mailman, M.D.; Ranganathan, S. OMIA (Online Mendelian Inheritance in Animals): An enhanced platform and integration into the Entrez search interface at NCBI. Nucleic Acids Res. 2006, 34, D599–D601. [Google Scholar] [CrossRef] [PubMed]
- Lyons, M.J.; Wood, T.; Espinoza, L.; Stensland, H.M.; Holden, K.R. Early onset alpha-mannosidosis with slow progression in three Hispanic male. Dev. Med. Child Neurol. 2007, 49, 854–857, Erratum in Dev. Med. Child Neurol. 2008, 50, 32. [Google Scholar] [CrossRef]
- Khan, J.M.; Ranganathan, S. A multi-species comparative structural bioinformatics analysis of inherited mutations in α-D-Mannosidase reveals strong genotype-phenotype correlation. BMC Genom. 2009, 10 (Suppl. 3), S33. [Google Scholar] [CrossRef]
- Öckerman, P.A. A generalised storage disorder resembling Hurler’s syndrome. Lancet 1967, 290, 239–241. [Google Scholar] [CrossRef]
- Loeb, H.; Vamos-Hurwitz, E. Mannosidosis. Arch. Neurol. 1977, 34, 650–651. [Google Scholar] [CrossRef]
- Marins, M.; Curiati, M.A.; Gomes, C.P.; Martin, R.P.; Nicolicht-Amorim, P.; Yamamoto, J.U.D.S.; D’Almeida, V.; Martins, A.M.; Pesquero, J.B. α-mannosidosis diagnosis in Brazilian patients with MPS-like symptoms. Orphanet J. Rare Dis. 2024, 19, 439. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vargas-López, V.; Prada, L.F.; Alméciga-Díaz, C.J. Evidence of epigenetic landscape shifts in mucopolysaccharidosis IIIB and IVA. Sci. Rep. 2024, 14, 3961. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dai, W.; Qiao, X.; Fang, Y.; Guo, R.; Bai, P.; Liu, S.; Li, T.; Jiang, Y.; Wei, S.; Na, Z.; et al. Epigenetics-targeted drugs: Current paradigms and future challenges. Signal Transduct. Target. Ther. 2024, 9, 332. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moschetti, M.; Venezia, M.; Giacomarra, M.; Marsana, E.M.; Zizzo, C.; Duro, G.; D’Errico, A.; Colomba, P.; Duro, G. Highlights of Precision Medicine, Genetics, Epigenetics and Artificial Intelligence in Pompe Disease. Int. J. Mol. Sci. 2025, 26, 757. [Google Scholar] [CrossRef] [PubMed]
- Erol, Ç. Artificial Intelligence, Fabry Disease…. Anatol. J. Cardiol. 2023, 27, 178. [Google Scholar] [CrossRef]
- Hurvitz, N.; Dinur, T.; Revel-Vilk, S.; Agus, S.; Berg, M.; Zimran, A.; Ilan, Y. A Feasibility Open-Labeled Clinical Trial Using a Second-Generation Artificial-Intelligence-Based Therapeutic Regimen in Patients with Gaucher Disease Treated with Enzyme Replacement Therapy. J. Clin. Med. 2024, 13, 3325. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef]
- Liang, G.; Chan, M.F.; Tomigahara, Y.; Tsai, Y.C.; Gonzales, F.A.; Li, E.; Laird, P.W.; Jones, P.A. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell. Biol. 2002, 22, 480–491. [Google Scholar] [CrossRef]
- Merberg, D.; Moreland, R.; Su, Z.; Li, B.; Crooker, B.; Palmieri, K.; Moore, S.W.; Melber, A.; Boyanapalli, R.; Carey, G.; et al. Combined miRNA transcriptome and proteome analysis of extracellular vesicles in urine and blood from the Pompe mouse model. Ann. Med. 2024, 56, 2402503. [Google Scholar] [CrossRef]
- Hassan, S.; Sidransky, E.; Tayebi, N. The role of epigenetics in lysosomal storage disorders: Uncharted territory. Mol. Genet. Metab. 2017, 122, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Mironov, G.; Berezovski, M.V. Direct detection of endogenous MicroRNAs and their post-transcriptional modifications in cancer serum by capillary electrophoresis-mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 2891–2899. [Google Scholar] [CrossRef] [PubMed]
- Wojtara, M.; Rana, E.; Rahman, T.; Khanna, P.; Singh, H. Artificial intelligence in rare disease diagnosis and treatment. Clin. Transl. Sci. 2023, 16, 2106–2111. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Decherchi, S.; Pedrini, E.; Mordenti, M.; Cavalli, A.; Sangiorgi, L. Opportunities and Challenges for Machine Learning in Rare Diseases. Front. Med. 2021, 8, 747612. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A.; International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| MPS-I | α-Mannosidosis | ||
|---|---|---|---|
| MPS IH Hurler syndrome (severe form) | From 6 to 8 months
| Mild form | Within 10 years Slow progressin characterized by myopathy |
| Moderate | After 10 years Slow progression characterized by myopathy and skeletal abnormalities | ||
| Severe | Early life Rapid progression with central nervous system involvement leading to early death | ||
Main symptoms
| |||
| MPS IH/S Hurler–Scheie (mild form) | From 3 to 10 years
| ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venezia, M.; Vinci, M.; Colomba, P.; Zizzo, C.; Duro, G.; Moschetti, M. Mucopolysaccharidosis Type I and α-Mannosidosis—Phenotypically Comparable but Genetically Different: Diagnostic and Therapeutic Considerations. Biomedicines 2025, 13, 1199. https://doi.org/10.3390/biomedicines13051199
Venezia M, Vinci M, Colomba P, Zizzo C, Duro G, Moschetti M. Mucopolysaccharidosis Type I and α-Mannosidosis—Phenotypically Comparable but Genetically Different: Diagnostic and Therapeutic Considerations. Biomedicines. 2025; 13(5):1199. https://doi.org/10.3390/biomedicines13051199
Chicago/Turabian StyleVenezia, Marika, Martina Vinci, Paolo Colomba, Carmela Zizzo, Giovanni Duro, and Marta Moschetti. 2025. "Mucopolysaccharidosis Type I and α-Mannosidosis—Phenotypically Comparable but Genetically Different: Diagnostic and Therapeutic Considerations" Biomedicines 13, no. 5: 1199. https://doi.org/10.3390/biomedicines13051199
APA StyleVenezia, M., Vinci, M., Colomba, P., Zizzo, C., Duro, G., & Moschetti, M. (2025). Mucopolysaccharidosis Type I and α-Mannosidosis—Phenotypically Comparable but Genetically Different: Diagnostic and Therapeutic Considerations. Biomedicines, 13(5), 1199. https://doi.org/10.3390/biomedicines13051199