
Redox Balance in Cancer in the Context of Tumor Prevention and Treatment


Abstract
1. Introduction
2. Methods
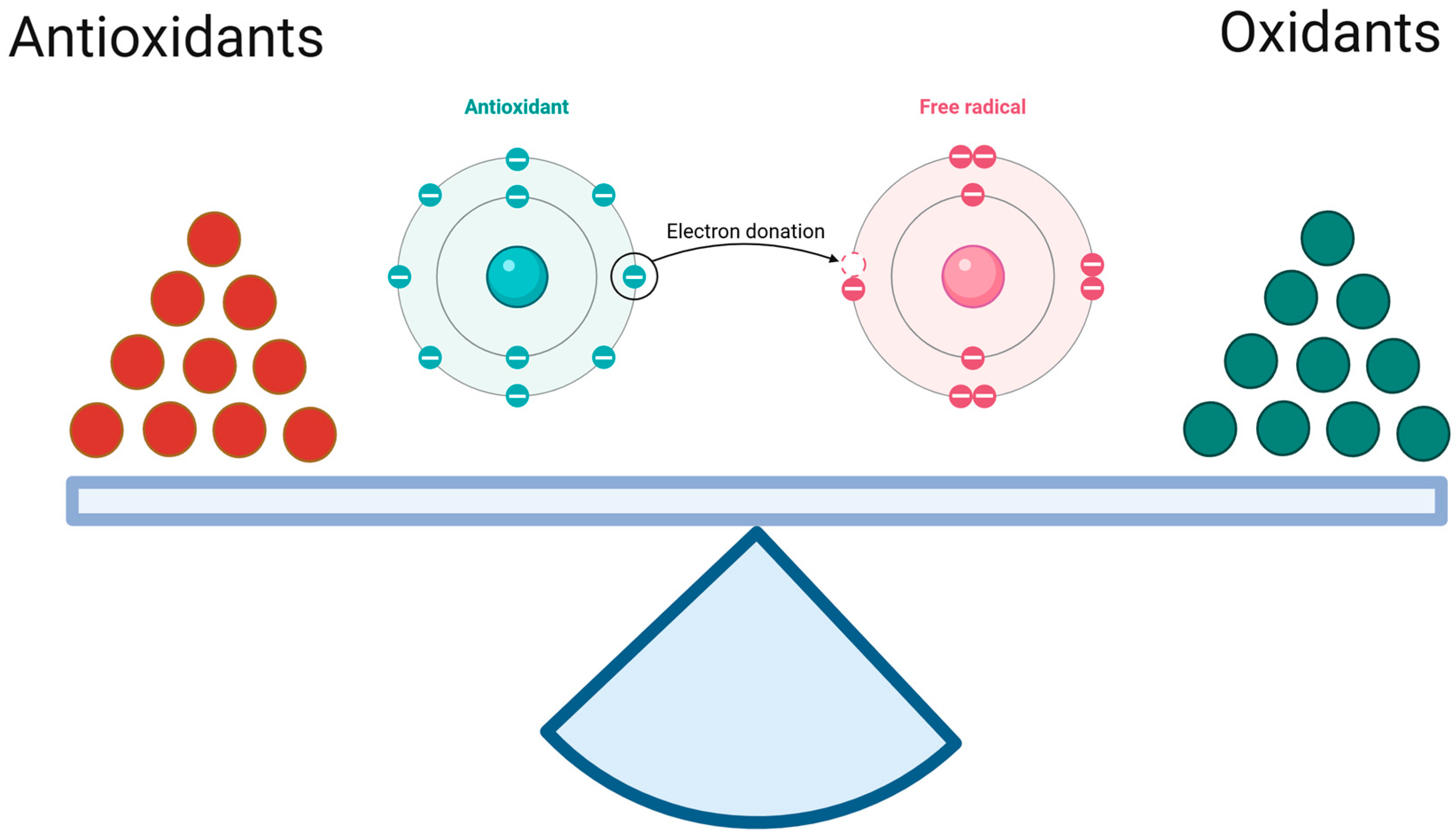
3. The Oxidant–Antioxidant Equilibrium
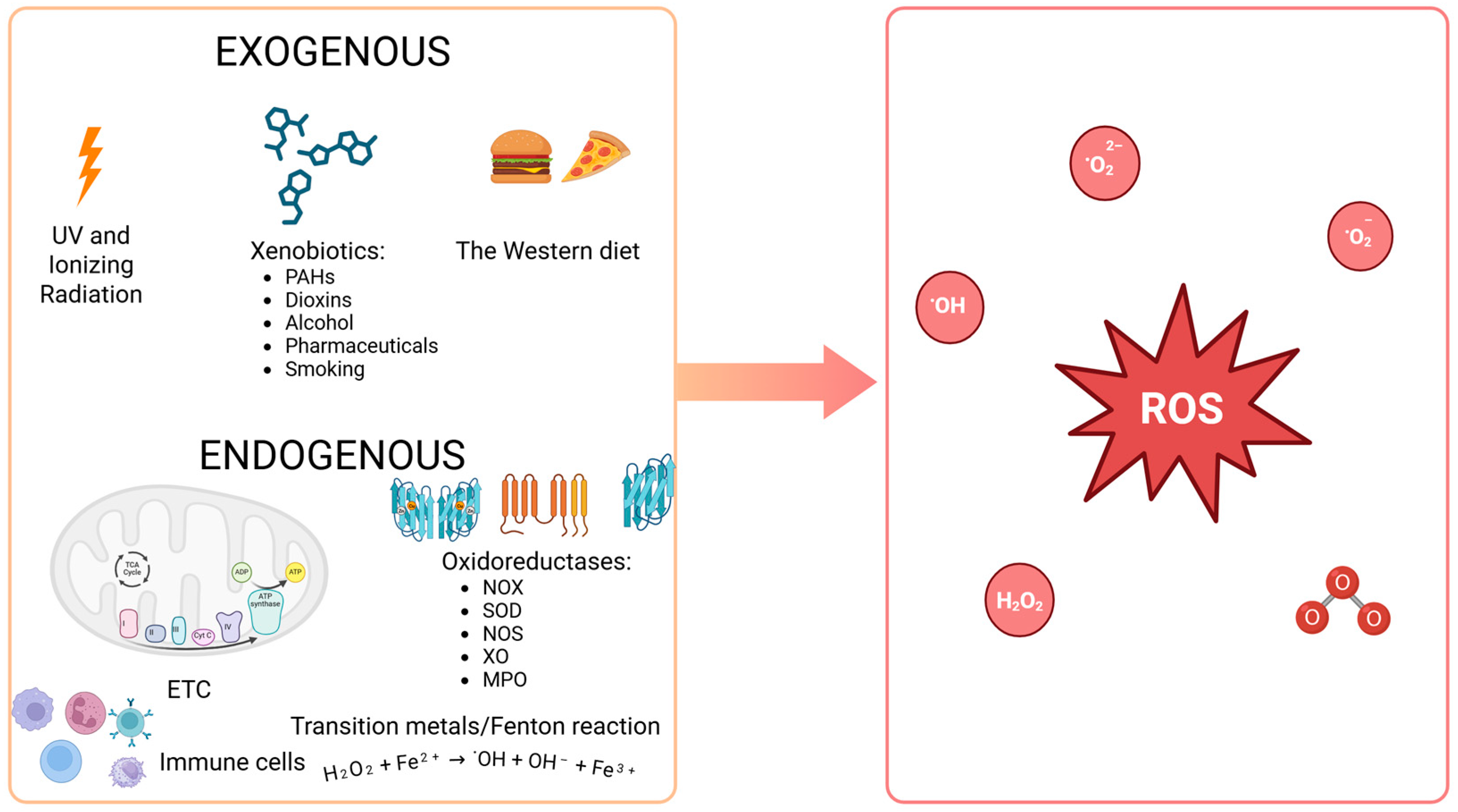
3.1. Reactive Oxygen Species
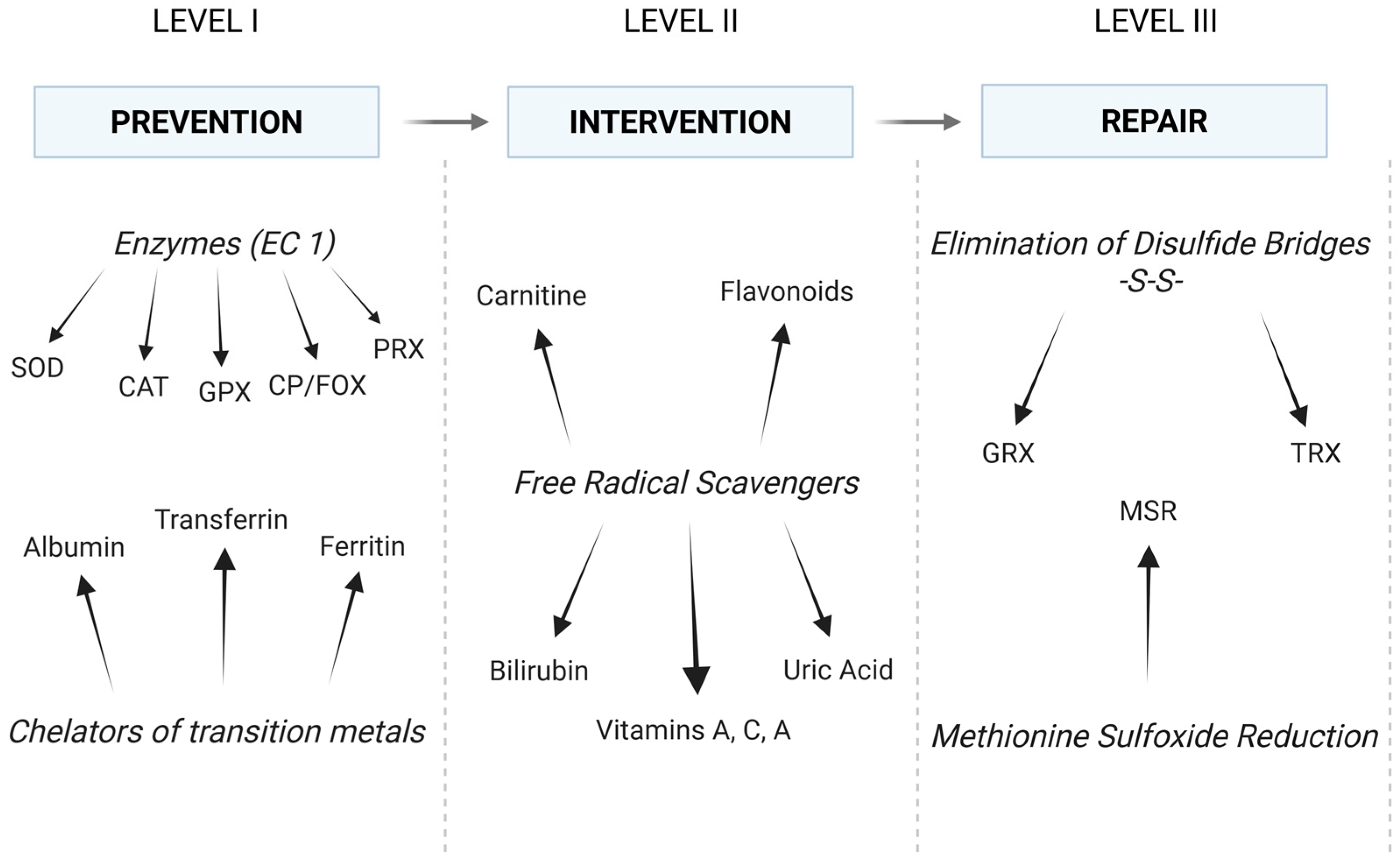
3.2. Antioxidant Capacity
4. The Redox Disturbances in Cancer
4.1. NRF2 and HIF Signaling Pathways
4.2. The Antioxidant System as an Anti-Cancer Strategy
5. Antioxidants—Cancer Prevention and Treatment
5.1. Benefits
5.2. Controversies
6. Conclusions and Future Prospects
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| NO˙ | nitric oxide |
| ˙OH | hydroxyl radical |
| 4-HNE | 4-hydroxynonenal |
| 8-iso-PGF2α | 8-iso-prastaglandin F2α |
| 8-OHdG | 8-hydroxy-2-deoxyguanosine |
| AGEs | advanced glycation end products |
| AMPK | 5′AMP-activated protein kinase |
| AOPPs | advanced protein oxidation products |
| AP-1 | activator protein-1 |
| CAT | catalase |
| CD | conjugated diens |
| GPX | glutathione peroxidase |
| GR | glutathione reductase |
| GRX | glutaredoxin |
| GSH | glutathione (reduced form) |
| GSSG | glutathione disulfide (the oxidized state of GSH) |
| H2O2 | hydrogen peroxide |
| HIF | hypoxia-inducible factor |
| HOCl | hypochlorous acid |
| IMA | ischemia-modified albumin |
| LOOH | lipid hydroperoxide |
| MDA | malondialdehyde |
| MSR | methionine sulfoxide reductase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor kappa B |
| NOS | nitric oxide synthase (iNOS: inducible NOS; eNOS: endothelial-specific NOS) |
| NOX | NADPH oxidase |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| O2˙− | superoxide anion radical |
| ONOO− | peroxynitrite |
| OSI | oxidative stress index |
| PPP | pentose phosphate pathway |
| PRX | peroxiredoxin |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SASP | senescence-associated secretory phenotype |
| SOD | superoxide dismutase |
| TABRS | thiobarbituric acid reactive substances |
| TAC | total antioxidant capacity |
| TRX | thioredoxin |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global Cancer Transitions According to the Human Development Index (2008–2030): A Population-based Study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2017, 19, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and Downsides of Reactive Oxygen Species for Cancer: The Roles of Reactive Oxygen Species in Tumorigenesis, Prevention, and Therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Fiaschi, T.; Chiarugi, P. Oxidative Stress, Tumor Microenvironment, and Metabolic Reprogramming: A Diabolic Liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef]
- Zahra, K.F.; Lefter, R.; Ali, A.; Abdellah, E.C.; Trus, C.; Ciobica, A.; Timofte, D. The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxidative Med. Cell. Longev. 2021, 2021, 9965916. [Google Scholar] [CrossRef]
- Li, D.; Yu, Q.; Wu, R.; Tuo, Z.; Wang, J.; Ye, L.; Shao, F.; Chaipanichkul, P.; Yoo, K.H.; Wei, W.; et al. Interactions Between Oxidative Stress and Senescence in Cancer: Mechanisms, Therapeutic Implications, and Future Perspectives. Redox Biol. 2024, 73, 103208. [Google Scholar] [CrossRef] [PubMed]
- Imbroisi Filho, R.; Ochioni, A.C.; Esteves, A.M.; Leandro, J.G.B.; Demaria, T.M.; Sola-Penna, M.; Zancan, P. Western Diet Leads to Aging-Related Tumorigenesis via Activation of the Inflammatory, UPR, and EMT Pathways. Cell Death Dis. 2021, 12, 643. [Google Scholar] [CrossRef] [PubMed]
- Nascimento Júnior, J.X.D.; Gomes, J.D.C.; Imbroisi Filho, R.; Valença, H.M.; Branco, J.R.; Araújo, A.B.; Moreira, A.O.E.; Crepaldi, L.D.; Paixão, L.P.; Ochioni, A.C.; et al. Dietary Caloric Input and Tumor Growth Accelerate Senescence and Modulate Liver and Adipose Tissue Crosstalk. Commun. Biol. 2025, 8, 18. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative Stress Inhibits Distant Metastasis by Human Melanoma Cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Guan, L.; Chen, Y.; Chen, P.; Sun, J.; Gonzalez, F.J.; Huang, M.; Bi, H. Lipidomics Reveals Carnitine Palmitoyltransferase 1C Protects Cancer Cells from Lipotoxicity and Senescence. J. Pharm. Anal. 2021, 11, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Kostić, A.; Jovanović Stojanov, S.; Podolski-Renić, A.; Nešović, M.; Dragoj, M.; Nikolić, I.; Tasić, G.; Schenone, S.; Pešić, M.; Dinić, J. Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells. Brain Sci. 2021, 11, 884. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Yun, Y.; Xue, J.; Jeon, B.; Kim, S. Doxorubicin-Induced Normal Breast Epithelial Cellular Aging and Its Related Breast Cancer Growth Through Mitochondrial Autophagy and Oxidative Stress Mitigated by Ginsenoside Rh2. Phytother. Res. 2020, 34, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Uruski, P.; Sepetowska, A.; Konieczna, C.; Pakuła, M.; Wyrwa, M.; Tussupkaliyev, A.; Tykarski, A.; Mikuła-Pietrasik, J.; Książek, K. Primary High-Grade Serous Ovarian Cancer Cells are Sensitive to Senescence Induced by Carboplatin and Paclitaxel In Vitro. Cell. Mol. Biol. Lett. 2021, 26, 44. [Google Scholar] [CrossRef]
- Ramesh, G.; Das, S.; Bola Sadashiva, S.R. Berberine, a Natural Alkaloid Sensitizes Human Hepatocarcinoma to Ionizing Radiation by Blocking Autophagy and Cell Cycle Arrest Resulting In Senescence. J. Pharm. Pharmacol. 2020, 72, 1893–1908. [Google Scholar] [CrossRef]
- Borkowska, A.; Olszewska, A.; Skarzynska, W.; Marciniak, M.; Skrzeszewski, M.; Kieda, C.; Was, H. High Hemin Concentration Induces Escape from Senescence of Normoxic and Hypoxic Colon Cancer Cells. Cancers 2022, 14, 4793. [Google Scholar] [CrossRef]
- Šima, J. Redox Reactions: Inconsistencies in Their Description. Found. Chem. 2013, 15, 57–64. [Google Scholar] [CrossRef]
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of Redox Regulation in Biology. Nat. Rev. Mol. Cell Biol. 2024, 25, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.C. Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. NADPH-The Forgotten Reducing Equivalent. Cold Spring Harb. Perspect. Biol. 2021, 13, a040550. [Google Scholar] [CrossRef]
- Koju, N.; Qin, Z.-H.; Sheng, R. Reduced Nicotinamide Adenine Dinucleotide Phosphate in Redox Balance and Diseases: A Friend or Foe? Acta Pharmacol. Sin. 2022, 43, 1889–1904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liew, C.W.; Handy, D.E.; Zhang, Y.; Leopold, J.A.; Hu, J.; Guo, L.; Kulkarni, R.N.; Loscalzo, J.; Stanton, R.C. High Glucose Inhibits Glucose-6-Phosphate Dehydrogenase, Leading to Increased Oxidative Stress and Beta-Cell Apoptosis. FASEB J. 2010, 24, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, M.; De Groot, H. NAD(P)H, a Directly Operating Antioxidant? FASEB J. 2001, 15, 1569–1574. [Google Scholar] [CrossRef]
- Del Campo, A.; Valenzuela, R.; Videla, L.A.; Zúñiga-Hernandez, J. Cellular Functional, Protective or Damaging Responses Associated with Different Redox Imbalance Intensities: A Comprehensive Review. Curr. Med. Chem. 2023, 30, 3927–3939. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Abe, C.; Miyazawa, T.; Miyazawa, T. Current Use of Fenton Reaction in Drugs and Food. Molecules 2022, 27, 5451. [Google Scholar] [CrossRef]
- Stanbury, D.M. The Principle of Detailed Balancing, the Iron-Catalyzed Disproportionation of Hydrogen Peroxide, and the Fenton Reaction. Dalton Trans. 2022, 51, 2135–2157. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Andrés Juan, C.; Plou, F.J.; Pérez-Lebeña, E. Superoxide Anion Chemistry—Its Role at the Core of the Innate Immunity. Int. J. Mol. Sci. 2023, 24, 1841. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Kopf, M. Redox Regulation of Immunometabolism. Nat. Rev. Immunol. 2021, 21, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Sutkowy, P.; Modrzejewska, M.; Porzych, M.; Woźniak, A. The Current State of Knowledge Regarding the Genetic Predisposition to Sports and Its Health Implications in the Context of the Redox Balance, Especially Antioxidant Capacity. Int. J. Mol. Sci. 2024, 25, 6915. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.C.; Winters-Stone, K.; Lee, A.; Schmitz, K.H. Cancer, Physical Activity, and Exercise. Compr. Physiol. 2012, 2, 2775–2809. [Google Scholar] [CrossRef]
- Ji, L.L.; Radak, Z.; Goto, S. Hormesis and Exercise: How the Cell Copes with Oxidative Stress. Am. J. Pharmacol. Toxicol. 2008, 3, 44–58. [Google Scholar] [CrossRef]
- Lu, Y.; Wiltshire, H.D.; Baker, J.S.; Wang, Q. Effects of High Intensity Exercise on Oxidative Stress and Antioxidant Status in Untrained Humans: A Systematic Review. Biology 2021, 10, 1272. [Google Scholar] [CrossRef]
- Le Gal, K.; Schmidt, E.E.; Sayin, V.I. Cellular Redox Homeostasis. Antioxidants 2021, 10, 1377. [Google Scholar] [CrossRef]
- Knoch, J.; Kamenisch, Y.; Kubisch, C.; Berneburg, M. Rare Hereditary Diseases with Defects in DNA-repair. Eur. J. Dermatol. 2012, 22, 443–455. [Google Scholar] [CrossRef]
- Möller, M.N.; Orrico, F.; Villar, S.F.; López, A.C.; Silva, N.; Donzé, M.; Thomson, L.; Denicola, A. Oxidants and Antioxidants in the Redox Biochemistry of Human Red Blood Cells. ACS Omega 2023, 8, 147–168. [Google Scholar] [CrossRef]
- Orrico, F.; Möller, M.N.; Cassina, A.; Denicola, A.; Thomson, L. Kinetic and Stoichiometric Constraints Determine the Pathway of H2O2 Consumption by Red Blood Cells. Free Radic. Biol. Med. 2018, 121, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, P.; Fita, I.; Loewen, P.C. Enzymology and Structure of Catalases. Adv. Inorg. Chem. 2000, 51, 51–106. [Google Scholar] [CrossRef]
- Kirkman, H.N.; Rolfo, M.; Ferraris, A.M.; Gaetani, G.F. Mechanisms of Protection of Catalase by NADPH: Kinetics and Stoichiometry. J. Biol. Chem. 1999, 274, 13908–13914. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research Progress of Glutathione Peroxidase Family (GPX) in redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians Against Oxidative Stress and Modulators of Peroxide Signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef]
- Hasan, A.A.; Kalinina, E.; Tatarskiy, V.; Shtil, A. The Thioredoxin System of Mammalian Cells and Its Modulators. Biomedicines 2022, 10, 1757. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and Thioredoxin Target Proteins: From Molecular Mechanisms to Functional Significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef]
- De Luca, A.; Sanna, F.; Sallese, M.; Ruggiero, C.; Grossi, M.; Sacchetta, P.; Rossi, C.; De Laurenzi, V.; Di Ilio, C.; Favaloro, B. Methionine Sulfoxide Reductase a Down-Regulation in Human Breast Cancer Cells Results in a More Aggressive Phenotype. Proc. Natl. Acad. Sci. USA 2010, 107, 18628–18633. [Google Scholar] [CrossRef]
- He, D.; Feng, H.; Sundberg, B.; Yang, J.; Powers, J.; Christian, A.H.; Wilkinson, J.E.; Monnin, C.; Avizonis, D.; Thomas, C.J.; et al. Methionine Oxidation Activates Pyruvate Kinase M2 to Promote Pancreatic Cancer Metastasis. Mol. Cell 2022, 82, 3045–3060. [Google Scholar] [CrossRef]
- Zińczuk, J.; Maciejczyk, M.; Zaręba, K.; Pryczynicz, A.; Dymicka-Piekarska, V.; Kamińska, J.; Koper-Lenkiewicz, O.; Matowicka-Karna, J.; Kędra, B.; Zalewska, A.; et al. Pro-Oxidant Enzymes, Redox Balance and Oxidative Damage to Proteins, Lipids and DNA in Colorectal Cancer Tissue. Is Oxidative Stress Dependent on Tumour Budding and Inflammatory Infiltration? Cancers 2020, 12, 1636. [Google Scholar] [CrossRef]
- Salehi, S.S.; Mirmiranpour, H.; Rabizadeh, S.; Esteghamati, A.; Tomasello, G.; Alibakhshi, A.; Najafi, N.; Rajab, A.; Nakhjavani, M. Improvement in Redox Homeostasis after Cytoreductive Surgery in Colorectal Adenocarcinoma. Oxidative Med. Cell. Longev. 2021, 2021, 8864905. [Google Scholar] [CrossRef] [PubMed]
- Zińczuk, J.; Maciejczyk, M.; Zaręba, K.; Romaniuk, W.; Markowski, A.; Kędra, B.; Zalewska, A.; Pryczynicz, A.; Matowicka-Karna, J.; Guzińska-Ustymowicz, K. Antioxidant Barrier, Redox Status, and Oxidative Damage to Biomolecules in Patients with Colorectal Cancer. Can Malondialdehyde and Catalase Be Markers of Colorectal Cancer Advancement? Biomolecules 2019, 9, 637. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Wang, F.; Zhao, Y.S.; Pan, H.Z. Evaluation of Oxidative Stress in Colorectal Cancer Patients. Biomed. Environ. Sci. 2008, 21, 286–289. [Google Scholar] [CrossRef]
- Saygili, E.I.; Konukoglu, D.; Papila, C.; Akcay, T. Levels of Plasma Vitamin E, Vitamin C, TBARS, and Cholesterol in Male Patients with Colorectal Tumors. Biochemistry 2003, 68, 325–328. [Google Scholar] [CrossRef]
- Rašić, I.; Rašić, A.; Akšamija, G.; Radović, S. The Relationship Between Serum Level of Malondialdehyde and Progression of Colorectal Cancer. Acta Clin. Croat. 2018, 57, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Ellidag, H.Y.; Bulbuller, N.; Eren, E.; Abusoglu, S.; Akgol, E.; Cetiner, M.; Yılmaz, N. Ischemia-Modified Albumin: Could it Be a New Oxidative Stress Biomarker for Colorectal Carcinoma? Gut Liver 2013, 7, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Chandramathi, S.; Suresh, K.; Anita, Z.B.; Kuppusamy, U.R. Comparative Assessment of Urinary Oxidative Indices in Breast and Colorectal Cancer Patients. J. Cancer Res. Clin. Oncol. 2009, 135, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Dorf, J.; Zaręba, K.; Pryczynicz, A.; Matowicka-Karna, J.; Kędra, B.; Żukowski, P.; Zalewska, A.; Maciejczyk, M. Diagnostic Significance and Utility of Circulating Redox Biomarkers in Patients with Gastric Cancer—Preliminary Study. Ann. Med. 2023, 55, 2241472. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, L.; Rong, S.; Qu, H.; Zhang, Y.; Chang, D.; Pan, H.; Wang, W. Relation Between Gastric Cancer and Protein Oxidation, DNA Damage, and Lipid Peroxidation. Oxidative. Med. Cell. Longev. 2013, 2013, 543760. [Google Scholar] [CrossRef]
- Fidan, E.; Mentese, A.; Kavgaci, H.; Orem, A.; Fidan, S.; Uzun, A.; Ozdemir, F.; Aydin, F. Increased Ischemia-Modified Albumin Levels in Patients with Gastric Cancer. Neoplasma 2012, 59, 393–397. [Google Scholar] [CrossRef]
- Stachowicz-Stencel, T.; Synakiewicz, A.; Owczarzak, A.; Śliwińska, A.; Aleksandrowicz-Wrona, E.; Lysiak-Szydowska, W.; Balcerska, A. Ischemia-Modified Albumin as A Biochemical Marker in Children with Neuroblastoma and Soft Tissue Sarcomas. J. Clin. Lab. Anal. 2011, 25, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Kundaktepe, B.P.; Sozer, V.; Durmus, S.; Kocael, P.C.; Kundaktepe, F.O.; Papila, C.; Gelisgen, R.; Uzun, H. The Evaluation of Oxidative Stress Parameters in Breast and Colon Cancer. Medicine 2021, 100, e25104. [Google Scholar] [CrossRef] [PubMed]
- Sutkowy, P.; Czuczejko, J.; Małkowski, B.; Szewczyk-Golec, K.; Łopatto, R.; Maruszak, M.; Woźniak, A. Redox State and Lysosomal Activity in Women with Ovarian Cancer with Tumor Recurrence and Multiorgan Metastasis. Molecules 2021, 26, 4039. [Google Scholar] [CrossRef]
- Kilic, N.; Yavuz Taslipinar, M.; Guney, Y.; Tekin, E.; Onuk, E. An Investigation into the Serum Thioredoxin, Superoxide Dismutase, Malondialdehyde, and Advanced Oxidation Protein Products In Patients With Breast Cancer. Ann. Surg. Oncol. 2014, 21, 4139–4143. [Google Scholar] [CrossRef] [PubMed]
- Tesarová, P.; Kalousová, M.; Trnková, B.; Soukupová, J.; Argalásová, S.; Mestek, O.; Petruzelka, L.; Zima, T. Carbonyl and Oxidative Stress in Patients with Breast Cancer--Is There a Relation to the Stage of the Disease? Neoplasma 2007, 54, 219–224. [Google Scholar]
- Korkmaz, G.G.; Inal, B.B.; Ortakoylu, G.M.; Irmak, H.; Kara, A.A.; Gelisgen, R.; Ogurlu, O.; Uzun, H. Changes in Oxidative Stress Parameters and Antioxidant Status in Lung Cancer: Western Blot Analysis of Nitrotyrosine and Protein Carbonyls Content. Clin. Lab. 2014, 60, 599–607. [Google Scholar] [CrossRef]
- Yilmaz, I.A.; Akçay, T.; Cakatay, U.; Telci, A.; Ataus, S.; Yalçin, V. Relation Between Bladder Cancer and Protein Oxidation. Int. Urol. Nephrol. 2003, 35, 345–350. [Google Scholar] [CrossRef]
- Sawicka, E.; Kratz, E.M.; Szymańska, B.; Guzik, A.; Wesołowski, A.; Kowal, P.; Pawlik-Sobecka, L.; Piwowar, A. Preliminary Study on Selected Markers of Oxidative Stress, Inflammation and Angiogenesis in Patients with Bladder Cancer. Pathol. Oncol. Res. 2020, 26, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Heidari, F.; Rabizadeh, S.; Mansournia, M.A.; Mirmiranpoor, H.; Salehi, S.S.; Akhavan, S.; Esteghamati, A.; Nakhjavani, M. Inflammatory, Oxidative Stress and Anti-Oxidative Markers in Patients with Endometrial Carcinoma and Diabetes. Cytokine 2019, 120, 186–190. [Google Scholar] [CrossRef]
- Meyer, F.; Galan, P.; Douville, P.; Bairati, I.; Kegle, P.; Bertrais, S.; Estaquio, C.; Hercberg, S. Antioxidant Vitamin and Mineral Supplementation and Prostate Cancer Prevention in The SU.VI.MAX Trial. Int. J. Cancer 2005, 116, 182–186. [Google Scholar] [CrossRef]
- Li, H.; Kantoff, P.W.; Giovannucci, E.; Leitzmann, M.F.; Gaziano, J.M.; Stampfer, M.J.; Ma, J. Manganese Superoxide Dismutase Polymorphism, Prediagnostic Antioxidant Status, and Risk of Clinical Significant Prostate Cancer. Cancer Res. 2005, 65, 2498–24504. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Lin, H.Y.; Liao, P.L.; Huang, C.C.; Lin, L.L.; Hsu, W.M.; Chuang, J.H. Immunomodulator Polyinosinic-Polycytidylic Acid Enhances the Inhibitory Effect of 13-Cis-Retinoic Acid on Neuroblastoma Through a TLR3-Related Immunogenic-Apoptotic Response. Lab. Investig. 2020, 100, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.V.; Lee, J.; Choi, I.J.; Kim, Y.W.; Ryu, K.W.; Kim, J. Effect of Dietary Vitamin C on Gastric Cancer Risk in the Korean Population. World J. Gastroenterol. 2016, 22, 6257–6267. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C Selectively Kills KRAS and BRAF Mutant Colorectal Cancer Cells by Targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Takeshita, K.; Seeni, A.; Sugiura, S.; Tang, M.; Sato, S.Y.; Kuriyama, H.; Nakadate, M.; Abe, K.; Maeno, Y.; et al. Suppression of Prostate Cancer in a Transgenic Rat Model via Gamma-Tocopherol Activation of Caspase Signaling. Prostate 2009, 69, 644–651. [Google Scholar] [CrossRef]
- Quiles, J.L.; Farquharson, A.J.; Ramírez-Tortosa, M.C.; Grant, I.; Milne, L.; Huertas, J.R.; Battino, M.; Mataix, J.; Wahle, K.W. Coenzyme Q Differentially Modulates Phospholipid Hydroperoxide Glutathione Peroxidase Gene Expression and Free Radicals Production in Malignant and Non-Malignant Prostate Cells. Biofactors 2003, 18, 265–270. [Google Scholar] [CrossRef]
- Jeong, J.H.; An, J.Y.; Kwon, Y.T.; Rhee, J.G.; Lee, Y.J. Effects of Low Dose Quercetin: Cancer Cell-Specific Inhibition of Cell Cycle Progression. J. Cell Biochem. 2009, 106, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Na, M.H.; Kim, W.K. Alpha-Lipoic Acid Reduces Matrix Metalloproteinase Activity in MDA-MB-231 Human Breast Cancer Cells. Nutr. Res. 2010, 30, 403–409. [Google Scholar] [CrossRef]
- Keshavan, P.; Schwemberger, S.J.; Smith, D.L.; Babcock, G.F.; Zucker, S.D. Unconjugated Bilirubin Induces Apoptosis in Colon Cancer Cells by Triggering Mitochondrial Depolarization. Int. J. Cancer 2004, 112, 433–445. [Google Scholar] [CrossRef]
- Rao, P.; Suzuki, R.; Mizobuchi, S.; Yamaguchi, T.; Sasaguri, S. Bilirubin Exhibits a Novel Anti-Cancer Effect on Human Adenocarcinoma. Biochem. Biophys. Res. Commun. 2006, 342, 1279–1283. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, S.; Lee, J.H.; Lee, S.H. Melatonin Promotes Apoptosis of Colorectal Cancer Cells via Superoxide-mediated ER Stress by Inhibiting Cellular Prion Protein Expression. Anticancer Res. 2018, 38, 3951–3960. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Y.; Zhu, H.; Cai, R.; Wang, K.F.; Song, J.; Wang, R.X.; Zhou, R.X. Role of Transforming Growth Factor Β1 in the Inhibition of Gastric Cancer Cell Proliferation by Melatonin In Vitro and In Vivo. Oncol. Rep. 2019, 42, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef] [PubMed]
- Ambrosone, C.B.; Zirpoli, G.R.; Hutson, A.D.; McCann, W.E.; McCann, S.E.; Barlow, W.E.; Kelly, K.M.; Cannioto, R.; Sucheston-Campbell, L.E.; Hershman, D.L.; et al. Dietary Supplement Use During Chemotherapy and Survival Outcomes of Patients With Breast Cancer Enrolled in a Cooperative Group Clinical Trial (SWOG S0221). J. Clin. Oncol. 2020, 38, 804–814. [Google Scholar] [CrossRef]
- Hercberg, S.; Ezzedine, K.; Guinot, C.; Preziosi, P.; Galan, P.; Bertrais, S.; Estaquio, C.; Briançon, S.; Favier, A.; Latreille, J.; et al. Antioxidant Supplementation Increases The Risk of Skin Cancers in Women But Not in Men. J. Nutr. 2007, 137, 2098–2105. [Google Scholar] [CrossRef] [PubMed]
- Tanvetyanon, T.; Bepler, G. Beta-Carotene in Multivitamins and The Possible Risk of Lung Cancer Among Smokers Versus Former Smokers: A Meta-Analysis and Evaluation of National Brands. Cancer 2008, 113, 150–157. [Google Scholar] [CrossRef]
- Nayyar, A.S.; Khan, M.; Vijayalakshmi, K.R.; Suman, B.; Gayitri, H.C.; Anitha, M. Serum Total Protein, Albumin and Advanced Oxidation Protein Products (AOPP)--Implications in Oral Squamous Cell Carcinoma. Malays. J. Pathol. 2012, 34, 47–52. [Google Scholar]
- Bekhet, O.H.; Eid, M.E. The Interplay Between Reactive Oxygen Species and Antioxidants in Cancer Progression and Therapy: A Narrative Review. Transl. Cancer Res. 2021, 10, 4196–4206. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Gu, X.; Mu, C.; Zheng, R.; Zhang, Z.; Zhang, Q.; Liang, T. The Cancer Antioxidant Regulation System in Therapeutic Resistance. Antioxidants 2024, 13, 778. [Google Scholar] [CrossRef]
- Bae, T.; Hallis, S.P.; Kwak, M.K. Hypoxia, Oxidative Stress, and the Interplay of HIFs and NRF2 Signaling in Cancer. Exp. Mol. Med. 2024, 56, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 Regulatory Network Provides an Interface Between Redox and Intermediary Metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.E.; Levings, D.C.; Freeman, S.; Slattery, M. Identification of a Functional Antioxidant Response Element at the HIF1A Locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Gong, L.; Peng, Y.; Li, L.; Liu, G. Enhancer-Bound Nrf2 Licenses HIF-1α Transcription Under Hypoxia to Promote Cisplatin Resistance in Hepatocellular Carcinoma Cells. Aging 2020, 13, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Csiki, I.; Yanagisawa, K.; Haruki, N.; Nadaf, S.; Morrow, J.D.; Johnson, D.H.; Carbone, D.P. Thioredoxin-1 Modulates Transcription of Cyclooxygenase-2 via Hypoxia-Inducible Factor-1alpha in Non-Small Cell Lung Cancer. Cancer Res. 2006, 66, 143–150. [Google Scholar] [CrossRef]
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; et al. NQO1 Inhibits Proteasome-Mediated Degradation of HIF-1α. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef]
- Zheng, J.; Kim, S.J.; Saeidi, S.; Kim, S.H.; Fang, X.; Lee, Y.H.; Guillen-Quispe, Y.N.; Ngo, H.K.C.; Kim, D.H.; Kim, D.; et al. Overactivated NRF2 Induces Pseudohypoxia in Hepatocellular Carcinoma by Stabilizing HIF-1α. Free Radic. Biol. Med. 2023, 194, 347–356. [Google Scholar] [CrossRef]
- Malec, V.; Gottschald, O.R.; Li, S.; Rose, F.; Seeger, W.; Hänze, J. HIF-1 Alpha Signaling is Augmented During Intermittent Hypoxia by Induction of the Nrf2 Pathway in NOX1-Expressing Adenocarcinoma A549 Cells. Free Radic. Biol. Med. 2010, 48, 1626–1635. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, C.K.; Lee, H.; Jeoung, D.; Ha, K.S.; Kwon, Y.G.; Kim, K.W.; Kim, Y.M. Carbon Monoxide Promotes VEGF Expression by Increasing HIF-1alpha Protein Level via Two Distinct Mechanisms, Translational Activation and Stabilization of HIF-1alpha Protein. J. Biol. Chem. 2010, 285, 32116–32125. [Google Scholar] [CrossRef]
- Lee, S.; Hallis, S.P.; Jung, K.A.; Ryu, D.; Kwak, M.K. Impairment of HIF-1α-Mediated Metabolic Adaption by NRF2-Silencing in Breast Cancer Cells. Redox Biol. 2019, 24, 101210. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.A.; Lee, S.; Kwak, M.K. NFE2L2/NRF2 Activity is Linked to Mitochondria and AMP-Activated Protein Kinase Signaling in Cancers Through Mir-181c/Mitochondria-Encoded Cytochrome C Oxidase Regulation. Antioxid. Redox Signal. 2017, 27, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The Proto-Oncometabolite Fumarate Binds Glutathione to Amplify ROS-Dependent Signaling. Mol. Cell 2013, 51, 236–248. [Google Scholar] [CrossRef]
- Early, J.O.; Menon, D.; Wyse, C.A.; Cervantes-Silva, M.P.; Zaslona, Z.; Carroll, R.G.; Palsson-McDermott, E.M.; Angiari, S.; Ryan, D.G.; Corcoran, S.E.; et al. Circadian Clock Protein BMAL1 Regulates IL-1β in Macrophages via NRF2. Proc. Natl. Acad. Sci. USA 2018, 115, E8460–E8468. [Google Scholar] [CrossRef]
- Loboda, A.; Stachurska, A.; Florczyk, U.; Rudnicka, D.; Jazwa, A.; Wegrzyn, J.; Kozakowska, M.; Stalinska, K.; Poellinger, L.; Levonen, A.L.; et al. HIF-1 Induction Attenuates Nrf2-Dependent IL-8 Expression in Human Endothelial Cells. Antioxid. Redox Signal. 2009, 11, 1501–1517. [Google Scholar] [CrossRef]
- Triptolide. PubChem. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/107985 (accessed on 24 August 2024).
- Chen, F.; Liu, Y.; Wang, S.; Guo, X.; Shi, P.; Wang, W.; Xu, B. Triptolide, a Chinese Herbal Extract, Enhances Drug Sensitivity of Resistant Myeloid Leukemia Cell Lines Through Downregulation of HIF-1α and Nrf2. Pharmacogenomics 2013, 14, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, F.; Wang, S.; Guo, X.; Shi, P.; Wang, W.; Xu, B. Low-Dose Triptolide in Combination with Idarubicin Induces Apoptosis in AML Leukemic Stem-Like KG1a Cell Line by Modulation of the Intrinsic and Extrinsic Factors. Cell Death Dis. 2013, 4, e948. [Google Scholar] [CrossRef]
- Brusatol. PubChem. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/73432 (accessed on 24 August 2024).
- Lu, Y.; Wang, B.; Shi, Q.; Wang, X.; Wang, D.; Zhu, L. Brusatol Inhibits HIF-1 Signaling Pathway and Suppresses Glucose Uptake under Hypoxic Conditions in HCT116 Cells. Sci. Rep. 2016, 6, 39123. [Google Scholar] [CrossRef]
- Hallis, S.P.; Kim, S.K.; Lee, J.H.; Kwak, M.K. Association of NRF2 with HIF-2α-Induced Cancer Stem Cell Phenotypes in Chronic Hypoxic Condition. Redox Biol. 2023, 60, 102632. [Google Scholar] [CrossRef]
- Cardamonin. PubChem. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/641785 (accessed on 24 August 2024).
- Jin, J.; Qiu, S.; Wang, P.; Liang, X.; Huang, F.; Wu, H.; Zhang, B.; Zhang, W.; Tian, X.; Xu, R.; et al. Cardamonin Inhibits Breast Cancer Growth by Repressing HIF-1α-Dependent Metabolic Reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 377. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 Enhances Resistance of Cancer Cells to Chemotherapeutic Drugs, The Dark Side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Cucci, M.A.; Grattarola, M.; Dianzani, C.; Muzio, G.; Pizzimenti, S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants 2021, 10, 510. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hong, X.; Zhao, F.; Ci, X.; Zhang, S. Targeting Nrf2 May Reverse the Drug Resistance in Ovarian Cancer. Cancer Cell Int. 2021, 21, 116. [Google Scholar] [CrossRef]
- Reinema, F.V.; Sweep, F.C.G.J.; Adema, G.J.; Peeters, W.J.M.; Martens, J.W.M.; Bussink, J.; Span, P.N. Tamoxifen Induces Radioresistance Through NRF2-Mediated Metabolic Reprogramming in Breast Cancer. Cancer Metab. 2023, 11, 3. [Google Scholar] [CrossRef]
- Marinello, P.C.; Panis, C.; Silva, T.N.X.; Binato, R.; Abdelhay, E.; Rodrigues, J.A.; Mencalha, A.L.; Lopes, N.M.D.; Luiz, R.C.; Cecchini, R.; et al. Metformin Prevention of Doxorubicin Resistance in MCF-7 and MDA-MB-231 Involves Oxidative Stress Generation and Modulation of Cell Adaptation Genes. Sci. Rep. 2019, 9, 5864. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 Inhibition Reverses the Resistance of Cisplatin-Resistant Head and Neck Cancer Cells to Artesunate-Induced Ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.; Shi, Y.; Wu, L.; Tan, Y.; Li, T.; Chen, Y.; Xia, J.; Hu, R. FAM117B Promotes Gastric Cancer Growth and Drug Resistance by Targeting the KEAP1/NRF2 Signaling Pathway. J. Clin. Investig. 2023, 133, e158705. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Li, Y.; Huang, H.; Huang, H.; Duan, Y.; Yuan, Z.; Zhu, W.; Mei, Z.; Luo, L.; Yan, P. Isoorientin Reverses Lung Cancer Drug Resistance by Promoting Ferroptosis via the SIRT6/Nrf2/GPX4 Signaling Pathway. Eur. J. Pharmacol. 2023, 954, 175853. [Google Scholar] [CrossRef]
- Levring, T.B.; Kongsbak, M.; Rode, A.K.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Human CD4+ T Cells Require Exogenous Cystine for Glutathione and DNA Synthesis. Oncotarget 2015, 6, 21853–21864. [Google Scholar] [CrossRef]
- Muri, J.; Heer, S.; Matsushita, M.; Pohlmeier, L.; Tortola, L.; Fuhrer, T.; Conrad, M.; Zamboni, N.; Kisielow, J.; Kopf, M. The Thioredoxin-1 System is Essential for Fueling DNA Synthesis During T-Cell Metabolic Reprogramming and Proliferation. Nat. Commun. 2018, 9, 1851. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Thut, H.; Feng, Q.; Kopf, M. Thioredoxin-1 Distinctly Promotes NF-Κb Target DNA Binding and NLRP3 Inflammasome Activation Independently of TXNIP. eLife 2020, 9, e53627. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Thut, H.; Heer, S.; Krueger, C.C.; Bornkamm, G.W.; Bachmann, M.F.; Kopf, M. The Thioredoxin-1 and Glutathione/Glutaredoxin-1 Systems Redundantly Fuel Murine B-Cell Development and Responses. Eur. J. Immunol. 2019, 49, 709–723. [Google Scholar] [CrossRef]
- Isakov, E.; Weisman-Shomer, P.; Benhar, M. Suppression of the Pro-Inflammatory NLRP3/Interleukin-1β Pathway in Macrophages by the Thioredoxin Reductase Inhibitor Auranofin. Biochim. Biophys. Acta. 2014, 1840, 3153–3161. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Thompson, M.A.; Tamayo, A.T.; Zuo, Z.; Lee, J.; Vega, F.; Ford, R.J.; Pham, L.V. Over-Expression of Thioredoxin-1 Mediates Growth, Survival, and Chemoresistance and Is a Druggable Target in Diffuse Large B-Cell Lymphoma. Oncotarget 2012, 3, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Gasdaska, P.Y.; Oblong, J.E.; Cotgreave, I.A.; Powis, G. The Predicted Amino Acid Sequence of Human Thioredoxin is Identical to that of the Autocrine Growth Factor Human Adult T-Cell Derived Factor (ADF): Thioredoxin mRNA is Elevated in Some Human Tumors. Biochim Biophys. Acta 1994, 1218, 292–296. [Google Scholar] [CrossRef]
- Raffel, J.; Bhattacharyya, A.K.; Gallegos, A.; Cui, H.; Einspahr, J.G.; Alberts, D.S.; Powis, G. Increased Expression of Thioredoxin-1 in Human Colorectal Cancer Is Associated with Decreased Patient Survival. J. Lab. Clin. Med. 2003, 142, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Bai, J.; Nishinaka, Y.; Ueda, S.; Sasada, T.; Ohshio, G.; Imamura, M.; Takabayashi, A.; Yamaoka, Y.; Yodoi, J. Expression of Thioredoxin and Glutaredoxin, Redox-Regulating Proteins, in Pancreatic Cancer. Cancer Detect. Prev. 2000, 24, 53–60. [Google Scholar]
- Lim, J.Y.; Yoon, S.O.; Hong, S.W.; Kim, J.W.; Choi, S.H.; Cho, J.Y. Thioredoxin and Thioredoxin-Interacting Protein as Prognostic Markers for Gastric Cancer Recurrence. World J. Gastroenterol. 2012, 18, 5581–5588. [Google Scholar] [CrossRef]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K.F. The Thioredoxin System in Breast Cancer Cell Invasion and Migration. Redox Biol. 2016, 8, 68–78. [Google Scholar] [CrossRef]
- Samaranayake, G.J.; Troccoli, C.I.; Huynh, M.; Lyles, R.D.Z.; Kage, K.; Win, A.; Lakshmanan, V.; Kwon, D.; Ban, Y.; Chen, S.X.; et al. Thioredoxin-1 Protects Against Androgen Receptor-Induced Redox Vulnerability in Castration-Resistant Prostate Cancer. Nat. Commun. 2017, 8, 1204. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Schneider, M.; Kölle, P.; Kuhlencordt, P.; Förster, H.; Beck, H.; Bornkamm, G.W.; Conrad, M. Loss of Thioredoxin Reductase 1 Renders Tumors Highly Susceptible to Pharmacologic Glutathione Deprivation. Cancer Res. 2010, 70, 9505–9514. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T.; et al. Impaired Mitochondrial Oxidative Phosphorylation Limits the Self-Renewal of T Cells Exposed to Persistent Antigen. Nat. Immunol. 2020, 21, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the Risk of Prostate Cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Tuveson, D.A. The Promise and Perils of Antioxidants for Cancer Patients. N. Engl. J. Med. 2014, 371, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, J.; Lian, S.; Zeng, Y.; He, S.; Xu, J.; Luo, L.; Yang, W.; Jiang, J. Targeting Metabolic–Redox Nexus to Regulate Drug Resistance: From Mechanism to Tumor Therapy. Antioxidants 2024, 13, 828. [Google Scholar] [CrossRef]
- Bouyahya, A.; El Menyiy, N.; Oumeslakht, L.; El Allam, A.; Balahbib, A.; Rauf, A.; Muhammad, N.; Kuznetsova, E.; Derkho, M.; Thiruvengadam, M.; et al. Preclinical and Clinical Antioxidant Effects of Natural Compounds against Oxidative Stress-Induced Epigenetic Instability in Tumor Cells. Antioxidants 2021, 10, 1553. [Google Scholar] [CrossRef]
- Luo, M.; Zhou, L.; Huang, Z.; Li, B.; Nice, E.C.; Xu, J.; Huang, C. Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants 2022, 11, 1128. [Google Scholar] [CrossRef]
- Guo, Q.; Li, L.; Hou, S.; Yuan, Z.; Li, C.; Zhang, W.; Zheng, L.; Li, X. The Role of Iron in Cancer Progression. Front Oncol. 2021, 11, 778492. [Google Scholar] [CrossRef]
- Suzuki, M.; Endo, M.; Shinohara, F.; Echigo, S.; Rikiishi, H. Differential Apoptotic Response of Human Cancer Cells to Organoselenium Compounds. Cancer Chemother. Pharmacol. 2010, 66, 475–484. [Google Scholar] [CrossRef]
- Wang, J.; Jiao, N.L.; Zheng, J. Effects of Se-Methylselenocysteine on Biological Behavior and Matrix Metalloproteinase-2 Expression in Human Breast Cancer MDA-MB-231 Cells. Ai Zheng = Aizheng = Chin. J. Cancer 2008, 27, 119–125. [Google Scholar]
- Anjomshoa, M.; Amirheidari, B.; Janczak, J.; Sahihi, M.; Abolhassani, Y.; Farsinejad, A.; Forootanfar, H. In Vitro and In Silico Studies of a Zn(II) Complex as a Potential Therapeutic Agent for Breast Cancer. Sci. Rep. 2024, 14, 29138. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.G.; Gupta, P.; Harbach, R.H.; Guida, W.C.; Dou, Q.P. Organic Copper Complexes as a New Class of Proteasome Inhibitors and Apoptosis Inducers in Human Cancer Cells. Biochem. Pharmacol. 2004, 67, 1139–1151. [Google Scholar] [CrossRef]
- Nagai, M.; Vo, N.H.; Shin Ogawa, L.; Chimmanamada, D.; Inoue, T.; Chu, J.; Beaudette-Zlatanova, B.C.; Lu, R.; Blackman, R.K.; Barsoum, J. The Oncology Drug Elesclomol Selectively Transports Copper to the Mitochondria to Induce Oxidative Stress in Cancer Cells. Free Radic. Biol. Med. 2012, 52, 2142–2150. [Google Scholar] [CrossRef]
- Poller, J.M.; Zaloga, J.; Schreiber, E.; Unterweger, H.; Janko, C.; Radon, P.; Eberbeck, D.; Trahms, L.; Alexiou, C.; Friedrich, R.P. Selection of Potential Iron Oxide Nanoparticles for Breast Cancer Treatment Based on In Vitro Cytotoxicity and Cellular Uptake. Int. J. Nanomed. 2017, 12, 3207–3220. [Google Scholar] [CrossRef] [PubMed]
- Porte, S.; Audemard-Verger, A.; Wu, C.; Durand, A.; Level, T.; Giraud, L.; Lombès, A.; Germain, M.; Pierre, R.; Saintpierre, B.; et al. Iron Boosts Antitumor Type 1 T-cell Responses and Anti-PD1 Immunotherapy. Cancer Immunol. Res. 2024, 12, 1252–1267. [Google Scholar] [CrossRef]
- Yang, L.; Gao, Y.; Wei, J.; Cheng, Z.; Wu, S.; Zou, L.; Li, S.; Li, P. Selenium-Integrated Conjugated Oligomer Nanoparticles with High Photothermal Conversion Efficiency for NIR-II Imaging-Guided Cancer Phototheranostics In Vivo. J. Nanobiotechnol. 2023, 21, 314. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Anderson, K.E.; Harnack, L.J.; Folsom, A.R.; Jacobs, D.R., Jr. Heme Iron, Zinc, Alcohol Consumption, and Colon Cancer: Iowa Women’s Health Study. J. Natl. Cancer Inst. 2004, 96, 403–407. [Google Scholar] [CrossRef]
- Lin, L.C.; Que, J.; Lin, K.L.; Leung, H.W.; Lu, C.L.; Chang, C.H. Effects of Zinc Supplementation on Clinical Outcomes in Patients Receiving Radiotherapy for Head and Neck Cancers: A Double-Blinded Randomized Study. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 368–373. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, Y.; Cheng, Y.; Fu, D.; Chen, Z.; Wang, Y.; Zhang, L.; Yao, C.; Shi, L.; Li, M.; et al. Copper-Based Metal-Organic Framework Overcomes Cancer Chemoresistance through Systemically Disrupting Dynamically Balanced Cellular Redox Homeostasis. J. Am. Chem. Soc. 2022, 144, 4799–4809. [Google Scholar] [CrossRef] [PubMed]
- Beigi, F.H.; Jazi, S.S.; Shahbazi-Gahrouei, D.; Khaniabadi, P.M.; Hafezi, H.; Monajemi, R.; Amiri, G.R. Iron Oxide Nanoparticles Coated with Polydopamine as a Potential Nano-Photothermal Agent for Treatment of Melanoma Cancer: An In Vivo Study. Lasers Med. Sci. 2022, 37, 3413–3421. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Sun, W.; Zhang, C.; Dong, B.; Yang, J.; Hou, M.; Xiong, L.; Cai, B.; Liu, X.; Xue, W. Metabolic Control by Heat Stress Determining Cell Fate to Ferroptosis for Effective Cancer Therapy. ACS Nano 2021, 15, 7179–7194. [Google Scholar] [CrossRef] [PubMed]
- Sakallı Çetin, E.; Nazıroğlu, M.; Çiğ, B.; Övey, İ.S.; Aslan Koşar, P. Selenium Potentiates the Anticancer Effect of Cisplatin Against Oxidative Stress and Calcium Ion Signaling-Induced Intracellular Toxicity in MCF-7 Breast Cancer Cells: Involvement of the TRPV1 Channel. J. Recept. Signal Transduct. Res. 2017, 37, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Liu, T.; Li, Y.; Lau, J.; Yang, Z.; Fan, W.; Zhou, Z.; Shi, C.; Ke, C.; Bregadze, V.I.; et al. Fenton-Reaction-Acceleratable Magnetic Nanoparticles for Ferroptosis Therapy of Orthotopic Brain Tumors. ACS Nano 2018, 12, 11355–11365. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.; Reitz, L.K.; Vieira, F.G.K.; da Silva, E.L.; Di Pietro, P.F. Low to Moderate Adherence to 2018 Diet and Physical Exercise Recommendations of the World Cancer Research Fund/American Institute for Cancer Research is Associated with Prooxidant Biochemical Profile in Women Undergoing Adjuvant Breast Cancer Treatment. Nutr. Res. 2023, 109, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.; Milne, G.L.; Sandler, D.P.; Nichols, H.B. Oxidative Stress in Relation to Diet and Physical Activity Among Premenopausal Women. Br. J. Nutr. 2016, 116, 1416–1424. [Google Scholar] [CrossRef]
- Kasapović, J.; Pejić, S.; Todorović, A.; Stojiljković, V.; Pajović, S.B. Antioxidant Status and Lipid Peroxidation in The Blood of Breast Cancer Patients of Different Ages. Cell Biochem. Funct. 2008, 26, 723–730. [Google Scholar] [CrossRef]
- Monari, M.; Trinchero, A.; Calabrese, C.; Cattani, O.; Serrazanetti, G.P.; Foschi, J.; Fabbri, A.; Zahlane, D.; Di Febo, G.; Tonini, V.; et al. Superoxide Dismutase in Gastric Adenocarcinoma: Is It a Clinical Biomarker in The Development of Cancer? Biomarkers 2006, 11, 574–584. [Google Scholar] [CrossRef]
- Elchuri, S.; Oberley, T.; Qi, W.; Eisenstein, R.S.; Roberts, L.J.; van Remmen, H.; Epstein, C.J.; Huang, T.-T. CuZnSOD Deficiency Leads to Persistent and Widespread Oxidative Damage and Hepatocarcinogenesis Later in Life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef]
- Teoh-Fitzgerald, M.L.; Fitzgerald, M.P.; Jensen, T.J.; Futscher, B.W.; Domann, F.E. Genetic and Epigenetic Inactivation ff Extracellular Superoxide Dismutase Promotes an Invasive Phenotype in Human Lung Cancer by Disrupting ECM Homeostasis. Mol. Cancer Res. 2012, 10, 40–51. [Google Scholar] [CrossRef]
- Griess, B.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular Superoxide Dismutase and Its Role in Cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, J.G.; Alexander, M.S.; Cullen, J.J. Superoxide Dismutases in Pancreatic Cancer. Antioxidants 2017, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-M.; Hou, M.-F.; Wu, S.-H.; Hu, B.-W.; Yang, S.-F.; Chen, W.-T.; Chai, C.-Y.; Ma, H.; Tsai, L.-Y. Expression of Manganese Superoxide Dismutase in Patients with Breast Cancer. Kaohsiung J. Med. Sci. 2011, 27, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chaiswing, L.; Oberley, T.D.; Batinic-Haberle, I.; St. Clair, W.; Epstein, C.J.; St. Clair, D. A Mechanism-Based Antioxidant Approach for The Reduction of Skin Carcinogenesis. Cancer Res. 2005, 65, 1401–1405. [Google Scholar] [CrossRef] [PubMed]
- Epperly, M.W.; Wang, H.; Jones, J.A.; Dixon, T.; Montesinos, C.A.; Greenberger, J.S. Antioxidant-Chemoprevention Diet Ameliorates Late Effects of Total-Body Irradiation and Supplements Radioprotection by Mnsod-Plasmid Liposome Administration. Radiat. Res. 2011, 175, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Nicco, C.; Chéreau, C.; Laurent, A.; Weill, B.; Goldwasser, F.; Batteux, F. Improvement of The Therapeutic Index of Anticancer Drugs by The Superoxide Dismutase Mimic Mangafodipir. J. Natl. Cancer Inst. 2006, 98, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Gauter-Fleckenstein, B.; Fleckenstein, K.; Owzar, K.; Jiang, C.; Batinic-Haberle, I.; Vujaskovic, Z. Comparison of Two Mn Porphyrin-Based Mimics of Superoxide Dismutase in Pulmonary Radioprotection. Free Radic. Biol. Med. 2008, 44, 982–989. [Google Scholar] [CrossRef]
- Robbins, D.; Zhao, Y. The Role of Manganese Superoxide Dismutase in Skin Cancer. Enzym. Res. 2011, 2011, 409295. [Google Scholar] [CrossRef]
- Glorieux, C.; Dejeans, N.; Sid, B.; Beck, R.; Calderon, P.B.; Verrax, J. Catalase Overexpression in Mammary Cancer Cells Leads to A Less Aggressive Phenotype and An Altered Response to Chemotherapy. Biochem. Pharmacol. 2011, 82, 1384–1390. [Google Scholar] [CrossRef]
- De Oliveira, V.A.; da Motta, L.L.; De Bastiani, M.A.; Lopes, F.M.; Müller, C.B.; Gabiatti, B.P.; França, F.S.; Castro, M.A.; Klamt, F. In Vitro Evaluation of Antitumoral Efficacy of Catalase in Combination with Traditional Chemotherapeutic Drugs Against Human Lung Adenocarcinoma Cells. Tumour Biol. 2016, 37, 10775–10784. [Google Scholar] [CrossRef] [PubMed]
- Bracalente, C.; Ibañez, I.L.; Berenstein, A.; Notcovich, C.; Cerda, M.B.; Klamt, F.; Chernomoretz, A.; Durán, H. Reprogramming Human A375 Amelanotic Melanoma Cells by Catalase Overexpression: Upregulation of Antioxidant Genes Correlates with Regression of Melanoma Malignancy and with Malignant Progression when Downregulated. Oncotarget 2016, 7, 41154–41171. [Google Scholar] [CrossRef]
- Deshpande, K.C.; Kulkarni, M.M.; Rajput, D.V. Evaluation of Glutathione Peroxidase in The Blood and Tumor Tissue of Oral Squamous Cell Carcinoma Patients. J. Oral Maxillofac. Pathol. 2018, 22, 447. [Google Scholar] [CrossRef]
- Cook, J.A.; Pass, H.I.; Iype, S.N.; Friedman, N.; DeGraff, W.; Russo, A.; Mitchell, J.B. Cellular Glutathione and Thiol Measurements From Surgically Resected Human Lung Tumor and Normal Lung Tissue. Cancer Res. 1991, 51, 4287–4294. [Google Scholar] [PubMed]
- Lee, F.Y.; Vessey, A.; Rofstad, E.; Siemann, D.W.; Sutherland, R.M. Heterogeneity of Glutathione Content in Human Ovarian Cancer. Cancer Res. 1989, 49, 5244–5248. [Google Scholar] [PubMed]
- Perry, R.R.; Mazetta, J.A.; Levin, M.; Barranco, S.C. Glutathione Levels and Variability in Breast Tumors and Normal Tissue. Cancer 1993, 72, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Kudo, H.; Mio, T.; Kokunai, T.; Tamaki, N.; Sumino, K.; Matsumoto, S. Quantitative Analysis of Glutathione in Human Brain Tumors. J. Neurosurg. 1990, 72, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hinkhouse, M.M.; Sun, W.; Weydert, C.J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Redox Regulation of Pancreatic Cancer Cell Growth: Role of Glutathione Peroxidase in The Suppression of The Malignant Phenotype. Hum. Gene Ther. 2004, 15, 239–250. [Google Scholar] [CrossRef]
- Liu, J.; Du, J.; Zhang, Y.; Sun, W.; Smith, B.J.; Oberley, L.W.; Cullen, J.J. Suppression of the Malignant Phenotype in Pancreatic Cancer by Overexpression of Phospholipid Hydroperoxide Glutathione Peroxidase. Hum. Gene Ther. 2006, 17, 105–116. [Google Scholar] [CrossRef]
- George, S.; Abrahamse, H. Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants 2020, 9, 1156. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Lu, W.; Ghergurovich, J.M.; Guo, L.; Blair, I.A.; Rabinowitz, J.D.; Yang, X. Upregulation of Antioxidant Capacity and Nucleotide Precursor Availability Suffices for Oncogenic Transformation. Cell Metab. 2021, 33, 94–109.e8. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 Interaction in Non-Small-Cell Lung Cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 Regulates Serine Biosynthesis in Non–Small Cell Lung Cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Gaya-Bover, A.; Hernández-López, R.; Alorda-Clara, M.; Ibarra de la Rosa, J.M.; Falcó, E.; Fernández, T.; Company, M.M.; Torrens-Mas, M.; Roca, P.; Oliver, J.; et al. Antioxidant Enzymes Change in Different Non-Metastatic Stages in Tumoral and Peritumoral Tissues of Colorectal Cancer. Int. J. Biochem. Cell Biol. 2020, 120, 105698. [Google Scholar] [CrossRef] [PubMed]
- Kocot, J.; Kiełczykowska, M.; Dąbrowski, W.; Piłat, J.; Rudzki, S.; Musik, I. Total Antioxidant Status Value and Superoxide Dismutase Activity in Human Colorectal Cancer Tissue Depending on the Stage of the Disease: A Pilot Study. Adv. Clin. Exp. Med. 2013, 22, 431–437. [Google Scholar] [PubMed]
- Qiu, W.; Jiang, J.; Zhan, Z.; Huang, L.; Deng, J.; Ye, J.; Li, G.; Liao, K.; Zhang, H.; Ding, Y.; et al. Prognostic Impact of Pretreatment Serum Superoxide Dismutase Activity in Patients with Locoregionally Advanced Nasopharyngeal Carcinoma. Int. J. Biol. Markers 2022, 37, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; Tangpong, J.; Chaiswing, L.; Oberley, T.D.; St. Clair, D.K. Manganese Superoxide Dismutase Is A P53-Regulated Gene That Switches Cancers Between Early and Advanced Stages. Cancer Res. 2011, 71, 6684–6695. [Google Scholar] [CrossRef]
- Kim, A. Modulation of MnSOD in Cancer: Epidemiological and Experimental Evidence. Toxicol. Res. 2010, 26, 83–93. [Google Scholar] [CrossRef]
- Palma, F.R.; He, C.; Danes, J.M.; Paviani, V.; Coelho, D.R.; Gantner, B.N.; Bonini, M.G. Mitochondrial Superoxide Dismutase: What the Established, the Intriguing, and the Novel Reveal About a Key Cellular Redox Switch. Antioxid. Redox Signal. 2020, 32, 701–714. [Google Scholar] [CrossRef]
- Gallicchio, L.; Boyd, K.; Matanoski, G.; Tao, X.G.; Chen, L.; Lam, T.K.; Shiels, M.; Hammond, E.; Robinson, K.A.; Caulfield, L.E.; et al. Carotenoids and The Risk of Developing Lung Cancer: A Systematic Review. Am. J. Clin. Nutr. 2008, 88, 372–383. [Google Scholar] [CrossRef]
- Mfouo Tynga, I.; Abrahamse, H. Nano-Mediated Photodynamic Therapy for Cancer: Enhancement of Cancer Specificity and Therapeutic Effects. Nanomaterials 2018, 8, 923. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type/Study Material | Effect | References |
|---|---|---|
| Colon/colonic mucosa, peripheral blood | ↑AC and ↑OS | [50,51] |
| Colon/peripheral blood | ↑OS | [52,53,54,55,56,62] |
| Colon/urine | ↑OS | [57] |
| Gastric/peripheral blood | ↑OS | [58,59,60] |
| Ovarian/peripheral blood | Mild redox anomalies (tendencies toward ↑AC and ↑OS) | [63] |
| Breast/peripheral blood, urine | ↑OS | [57,62,64,65] |
| Lung, bladder, endometrial, oral/peripheral blood | ↑OS | [66,67,68,69] |
| Caner type/antioxidant | Positive | |
| Prostate/β-carotene, α-tocopherol, vitamin C, lycopene + minerals (Se, Zn) | Reducing cancer incidence | [70,71] |
| Neuroblastoma/vitamin A | Anticancer properties | [72] |
| Gastric, colon/vitamin C | Cancer prevention and anticancer properties | [73,74] |
| Prostate/vitamin E, coenzyme Q10 | Anticancer properties | [75,76] |
| Breast/quercetin, α-lipoic acid | Anticancer properties | [77,78] |
| Colon/bilirubin, melatonin | Anticancer properties | [79,80,81] |
| Gastric/melatonin | Anticancer properties | [82] |
| Negative | ||
| Lung/N-acetylcysteine and vitamin E | Tumor progression | [83] |
| Breast/vitamins A, C and E, carotenoids, coenzyme Q10 | Increased risk of tumor recurrence | [84] |
| Skin/vitamins C, and E, β-carotene + minerals (Se, Zn) | Increased incidence of cancer in women | [85] |
| Lung/β-carotene + vitamins (multivitamin supplement) | Increased risk of cancer in tobacco smokers | [86] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutkowy, P.; Czeleń, P. Redox Balance in Cancer in the Context of Tumor Prevention and Treatment. Biomedicines 2025, 13, 1149. https://doi.org/10.3390/biomedicines13051149
Sutkowy P, Czeleń P. Redox Balance in Cancer in the Context of Tumor Prevention and Treatment. Biomedicines. 2025; 13(5):1149. https://doi.org/10.3390/biomedicines13051149
Chicago/Turabian StyleSutkowy, Paweł, and Przemysław Czeleń. 2025. "Redox Balance in Cancer in the Context of Tumor Prevention and Treatment" Biomedicines 13, no. 5: 1149. https://doi.org/10.3390/biomedicines13051149
APA StyleSutkowy, P., & Czeleń, P. (2025). Redox Balance in Cancer in the Context of Tumor Prevention and Treatment. Biomedicines, 13(5), 1149. https://doi.org/10.3390/biomedicines13051149