
Endothelial–Mesenchymal Transition and Possible Role of Cytokines in Streptozotocin-Induced Diabetic Heart

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. STZ Injection
2.2. Determination of Cardiac Cytokines ROS, EndMT Markers (IL-18,6,33,10, lL-β1, ROS, Peroxynitrite (ONOO−), Nitric Oxide Synthases (NOS) Isoforms, and TNF-α Concentrations
2.3. Matrix Metalloproteinase 2 (MMP-2), MMP-2 Zymography
2.4. Western Blot Analyses of Cardiac Discoidin Domain Tyrosine Kinase Receptor 2 (DDR-2), Citrullinated Histone H3 (H3Cit), Vimentin
2.5. Determination of Cardiac Myeloperoxidase (MPO) Activity
2.6. Protein Content Measurement
2.7. Statistical Analysis
3. Results
3.1. The Expression of Various Cytokines in the Heart
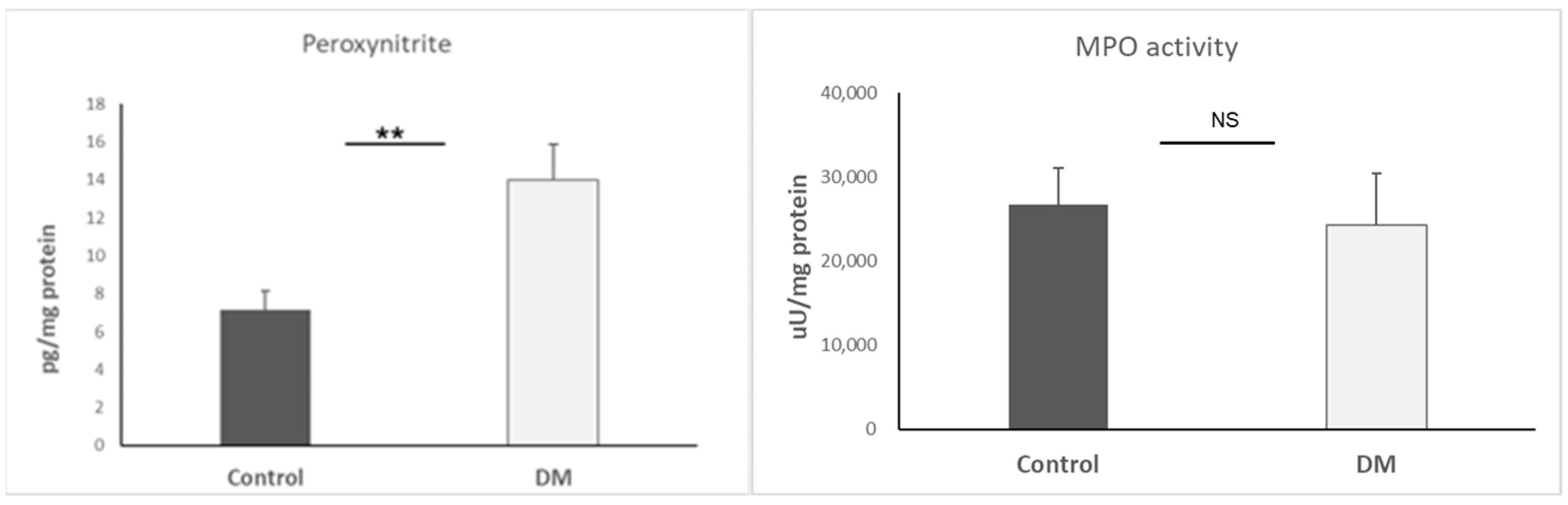
3.2. Basic ROS Examination
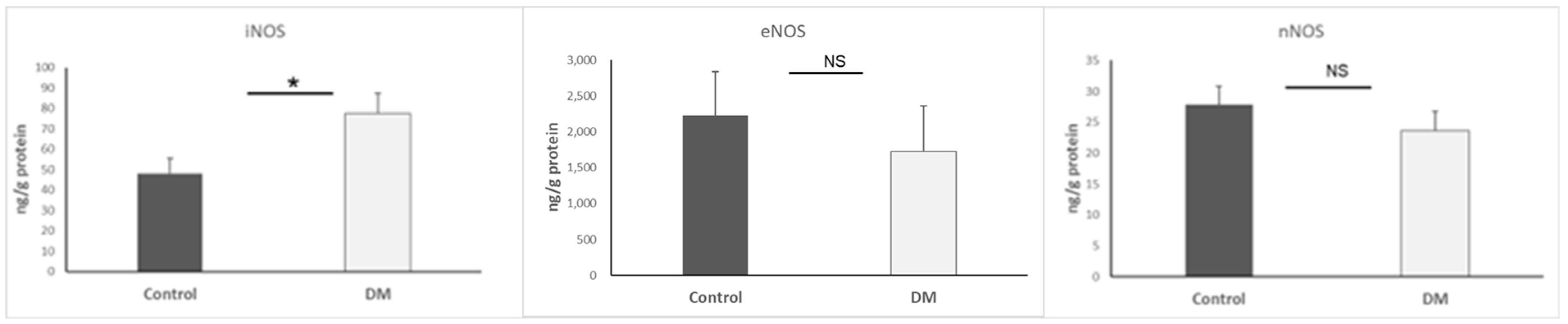
3.3. Nitric Oxide Synthases (NOS) Determination
3.4. The Assessment of EndMT by DDR-2, MMP-2, and TIMP-1
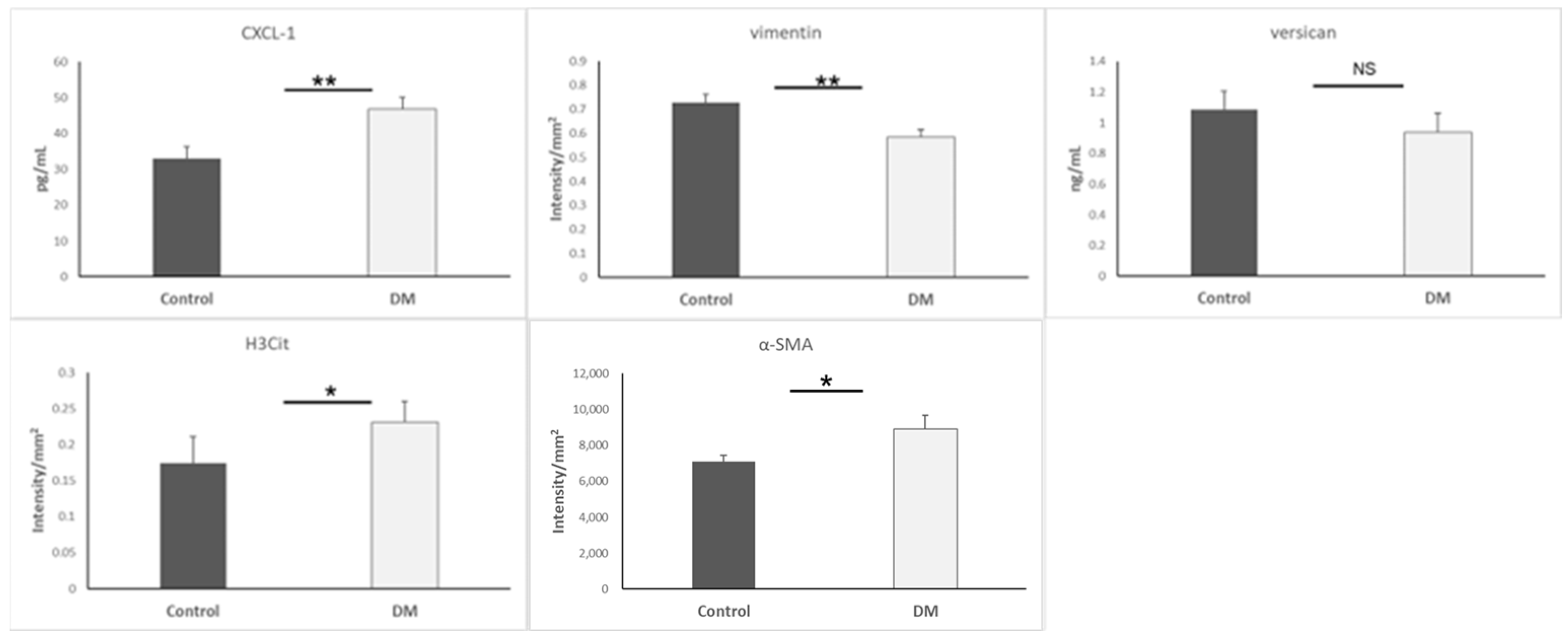
3.5. Biomarkers of Mesenchymal Cells and Neutrophils
3.6. TGF-β Level of Heart, Aorta, and Plasma
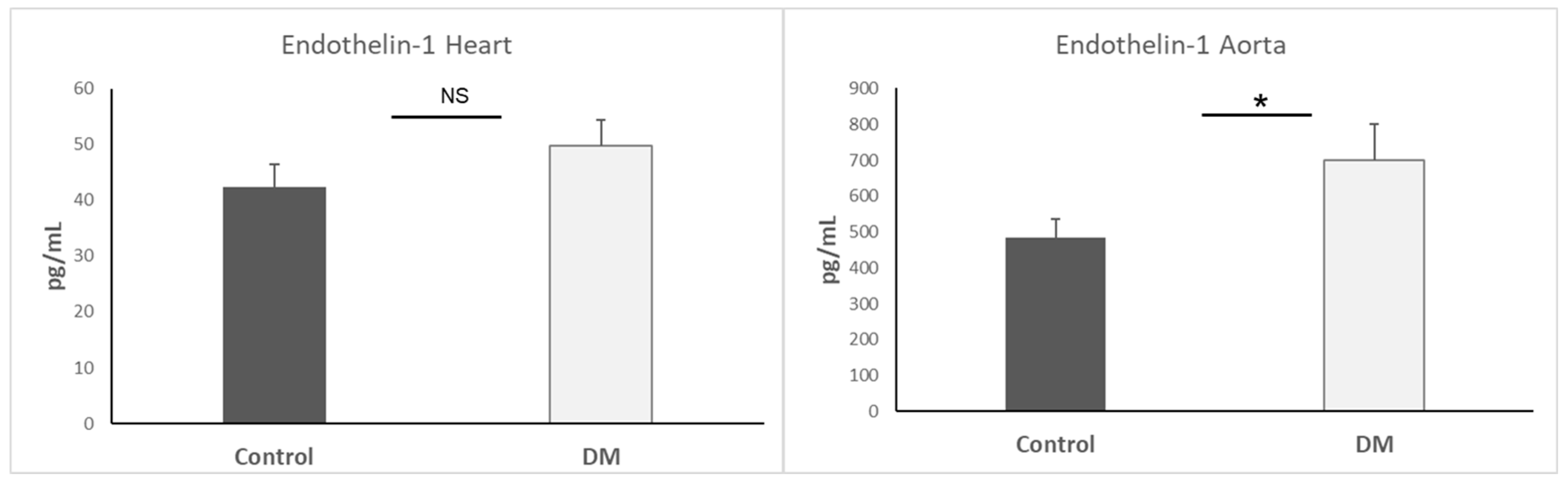
3.7. Endothelin-1 Expression in Heart and Aorta
4. Discussion
Table 1 and Table 2. Key Molecular Regulators and Therapeutic Targets Related to EndMT in Diabetic Cardiomyopathy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of Diabetes in Congestive Heart Failure: The Framingham Study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef] [PubMed]
- Rajbhandari, J.; Fernandez, C.J.; Agarwal, M.; Yeap, B.X.Y.; Pappachan, J.M. Diabetic Heart Disease: A Clinical Update. World J. Diabetes 2021, 12, 383–406. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Suriguga; Gong, M.; Liu, W.-J.; Cui, N.-X.; Wang, Y.; Du, X.; Yi, Z.-C. High Glucose Induced Endothelial to Mesenchymal Transition in Human Umbilical Vein Endothelial Cell. Exp. Mol. Pathol. 2017, 102, 377–383. [Google Scholar] [CrossRef]
- Delgado-Valero, B.; de la Fuente-Chávez, L.; Romero-Miranda, A.; Visitación Bartolomé, M.; Ramchandani, B.; Islas, F.; Luaces, M.; Cachofeiro, V.; Martínez-Martínez, E. Role of Endoplasmic Reticulum Stress in Renal Damage after Myocardial Infarction. Clin. Sci. 2021, 135, 143–159. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, Z.; Yuan, T.; Zhao, X.; Wang, M.; Liu, G.; Gan, L.; Qin, W. PD-1 Inhibition Disrupts Collagen Homeostasis and Aggravates Cardiac Dysfunction through Endothelial-Fibroblast Crosstalk and EndMT. Front. Pharmacol. 2025, 16, 1549487. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Chen, S.; Wang, H.; Chen, T.; Chakrabarti, S. Non-Coding RNA-Mediated Endothelial-to-Mesenchymal Transition in Human Diabetic Cardiomyopathy, Potential Regulation by DNA Methylation. Cardiovasc. Diabetol. 2023, 22, 303. [Google Scholar] [CrossRef]
- Wu, M.; Li, T.; Li, G.; Niu, B.; Wu, T.; Yan, L.; Wang, S.; He, S.; Huang, C.; Tong, W. LncRNA DANCR Deficiency Promotes High Glucose-Induced Endothelial to Mesenchymal Transition in Cardiac Microvascular Cells via the FoxO1/DDAH1/ADMA Signaling Pathway. Eur. J. Pharmacol. 2023, 950, 175732. [Google Scholar] [CrossRef]
- Karakasis, P.; Theofilis, P.; Patoulias, D.; Vlachakis, P.K.; Antoniadis, A.P.; Fragakis, N. Diabetes-Driven Atherosclerosis: Updated Mechanistic Insights and Novel Therapeutic Strategies. Int. J. Mol. Sci. 2025, 26, 2196. [Google Scholar] [CrossRef]
- Khan, A.W.; Jandeleit-Dahm, K.A. Atherosclerosis in Diabetes Mellitus: Novel Mechanisms and Mechanism-Based Therapeutic Approaches. Nat. Rev. Cardiol. 2025, 1–15. [Google Scholar] [CrossRef]
- Testai, L.; Brancaleone, V.; Flori, L.; Montanaro, R.; Calderone, V. Modulation of EndMT by Hydrogen Sulfide in the Prevention of Cardiovascular Fibrosis. Antioxidants 2021, 10, 910. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, A.; Jeddi, S. Streptozotocin as a Tool for Induction of Rat Models of Diabetes: A Practical Guide. EXCLI J. 2023, 22, 274. [Google Scholar] [CrossRef]
- Hsu, P.-C.; Huang, J.-C.; Tsai, W.-C.; Hung, W.-W.; Chang, W.-A.; Wu, L.-Y.; Chang, C.-Y.; Tsai, Y.-C.; Hsu, Y.-L. Tumor Necrosis Factor Receptor Superfamily Member 21 Induces Endothelial-Mesenchymal Transition in Coronary Artery Endothelium of Type 2 Diabetes Mellitus. Biomedicines 2022, 10, 1282. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Li, J.; Cai, L.; Chakrabarti, S.; Li, X. Cytokines and Diabetes Research. J. Diabetes Res. 2014, 2014, 1–2. [Google Scholar] [CrossRef]
- Bennici, G.; Almahasheer, H.; Alghrably, M.; Valensin, D.; Kola, A.; Kokotidou, C.; Lachowicz, J.; Jaremko, M. Mitigating Diabetes Associated with Reactive Oxygen Species (ROS) and Protein Aggregation through Pharmacological Interventions. RSC Adv. 2024, 14, 17448–17460. [Google Scholar] [CrossRef] [PubMed]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes Mellitus and Oxidative Stress—A Concise Review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef]
- Long, X.; Yuan, Q.; Tian, R.; Zhang, W.; Liu, L.; Yang, M.; Yuan, X.; Deng, Z.; Li, Q.; Sun, R. Efficient Healing of Diabetic Wounds by MSC-EV-7A Composite Hydrogel via Suppression of Inflammation and Enhancement of Angiogenesis. Biomater. Sci. 2024, 12, 1750–1760. [Google Scholar] [CrossRef]
- Åstrand, H.; Rydén-Ahlgren, Å.; Sundkvist, G.; Sandgren, T.; Länne, T. Reduced Aortic Wall Stress in Diabetes Mellitus. Eur. J. Vasc. Endovasc. Surg. 2007, 33, 592–598. [Google Scholar] [CrossRef]
- Li, J.; Huynh, P.; Dai, A.; Wu, T.; Tu, Y.; Chow, B.; Kiriazis, H.; Du, X.-J.; Bach, L.A.; Wilkinson-Berka, J.L.; et al. Diabetes Reduces Severity of Aortic Aneurysms Depending on the Presence of Cell Division Autoantigen 1 (CDA1). Diabetes 2018, 67, 755–768. [Google Scholar] [CrossRef]
- Wang, E.; Wang, H.; Chakrabarti, S. Endothelial-to-Mesenchymal Transition: An Underappreciated Mediator of Diabetic Complications. Front. Endocrinol. 2023, 14, 1050540. [Google Scholar] [CrossRef]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. 2021, 1, e78. [Google Scholar] [CrossRef] [PubMed]
- Kupai, K.; Szucs, G.; Cseh, S.; Hajdu, I.; Csonka, C.; Csont, T.; Ferdinandy, P. Matrix Metalloproteinase Activity Assays: Importance of Zymography. J. Pharmacol. Toxicol. Methods 2010, 61, 205–209. [Google Scholar] [CrossRef]
- Mihai, C.; Iscru, D.F.; Druhan, L.J.; Elton, T.S.; Agarwal, G. Discoidin Domain Receptor 2 Inhibits Fibrillogenesis of Collagen Type 1. J. Mol. Biol. 2006, 361, 864–876. [Google Scholar] [CrossRef]
- Beghetti, M.; Black, S.M.; Fineman, J.R. Endothelin-1 in Congenital Heart Disease. Pediatr. Res. 2005, 57, 16R–20R. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of Interleukin (IL)-6-Type Cytokine Signalling and Its Regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Lo, H.; Lai, T.; Li, C.; Wu, W. TNF-α Induces CXCL1 Chemokine Expression and Release in Human Vascular Endothelial Cells in Vitro via Two Distinct Signaling Pathways. Acta Pharmacol. Sin. 2014, 35, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Kaminga, A.C.; Kinra, S.; Wen, S.W.; Liu, H.; Tan, X.; Liu, A. Chemokines in Type 1 Diabetes Mellitus. Front. Immunol. 2022, 12, 690082. [Google Scholar] [CrossRef]
- Monickaraj, F.; Acosta, G.; Cabrera, A.P.; Das, A. Transcriptomic Profiling Reveals Chemokine CXCL1 as a Mediator for Neutrophil Recruitment Associated with Blood-Retinal Barrier Alteration in Diabetic Retinopathy. Diabetes 2023, 72, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Maruszewska, A.; Bosiacki, M.; Chlubek, D.; Baranowska-Bosiacka, I. The Potential Importance of CXCL1 in the Physiological State and in Noncancer Diseases of the Cardiovascular System, Respiratory System and Skin. Int. J. Mol. Sci. 2022, 24, 205. [Google Scholar] [CrossRef]
- Takahashi, K.; Ohara, M.; Sasai, T.; Homma, H.; Nagasawa, K.; Takahashi, T.; Yamashina, M.; Ishii, M.; Fujiwara, F.; Kajiwara, T.; et al. Serum CXCL1 Concentrations Are Elevated in Type 1 Diabetes Mellitus, Possibly Reflecting Activity of Anti-Islet Autoimmune Activity. Diabetes Metab. Res. Rev. 2011, 27, 830–833. [Google Scholar] [CrossRef]
- Guo, L.-Y.; Yang, F.; Peng, L.-J.; Li, Y.-B.; Wang, A.-P. CXCL2, a New Critical Factor and Therapeutic Target for Cardiovascular Diseases. Clin. Exp. Hypertens. 2020, 42, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, Oxidative Stress and Therapeutic Strategies. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 2709–2729. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Szabó, C. Role of Peroxynitrite in the Pathogenesis of Cardiovascular Complications of Diabetes. Curr. Opin. Pharmacol. 2006, 6, 136–141. [Google Scholar] [CrossRef]
- Lin, K.-T.; Xue, J.-Y.; Sun, F.F.; Wong, P.Y.-K. Reactive Oxygen Species Participate in Peroxynitrite-Induced Apoptosis in HL-60 Cells. Biochem. Biophys. Res. Commun. 1997, 230, 115–119. [Google Scholar] [CrossRef]
- Prado, A.F.; Batista, R.I.M.; Tanus-Santos, J.E.; Gerlach, R.F. Matrix Metalloproteinases and Arterial Hypertension: Role of Oxidative Stress and Nitric Oxide in Vascular Functional and Structural Alterations. Biomolecules 2021, 11, 585. [Google Scholar] [CrossRef]
- Bassiouni, W.; Ali, M.A.M.; Schulz, R. Multifunctional Intracellular Matrix Metalloproteinases: Implications in Disease. FEBS J. 2021, 288, 7162–7182. [Google Scholar] [CrossRef]
- Hughes, B.G.; Schulz, R. Targeting MMP-2 to Treat Ischemic Heart Injury. Basic Res. Cardiol. 2014, 109, 424. [Google Scholar] [CrossRef] [PubMed]
- Viappiani, S.; Nicolescu, A.C.; Holt, A.; Sawicki, G.; Crawford, B.D.; León, H.; Van Mulligen, T.; Schulz, R. Activation and Modulation of 72kDa Matrix Metalloproteinase-2 by Peroxynitrite and Glutathione. Biochem. Pharmacol. 2009, 77, 826–834. [Google Scholar] [CrossRef]
- Egea, V.; Zahler, S.; Rieth, N.; Neth, P.; Popp, T.; Kehe, K.; Jochum, M.; Ries, C. Tissue Inhibitor of Metalloproteinase-1 (TIMP-1) Regulates Mesenchymal Stem Cells through Let-7f microRNA and Wnt/β-Catenin Signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E309–E316. [Google Scholar] [CrossRef]
- Bellafiore, M.; Battaglia, G.; Bianco, A.; Farina, F.; Palma, A.; Paoli, A. The Involvement of MMP-2 and MMP-9 in Heart Exercise-Related Angiogenesis. J. Transl. Med. 2013, 11, 283. [Google Scholar] [CrossRef]
- Feng, J.; Li, Y.; Li, Y.; Yin, Q.; Li, H.; Li, J.; Zhou, B.; Meng, J.; Lian, H.; Wu, M.; et al. Versican Promotes Cardiomyocyte Proliferation and Cardiac Repair. Circulation 2024, 149, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Angelov, S.N.; Hu, J.H.; Wei, H.; Airhart, N.; Shi, M.; Dichek, D.A. TGF-β (Transforming Growth Factor-β) Signaling Protects the Thoracic and Abdominal Aorta from Angiotensin II-Induced Pathology by Distinct Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2102–2113. [Google Scholar] [CrossRef]
- Raffort, J.; Lareyre, F.; Clément, M.; Hassen-Khodja, R.; Chinetti, G.; Mallat, Z. Diabetes and Aortic Aneurysm: Current State of the Art. Cardiovasc. Res. 2018, 114, 1702–1713. [Google Scholar] [CrossRef]
- Rochmah, N.; Triastuti, I.W.; Deakandi, W.Y.; Irawan, R.; Endaryanto, A. Decreased Serum Transforming Growth Factor- Beta Levels in Indonesian Children with Type 1 Diabetes. J. Med. Pharm. Chem. Res. 2024, 6, 1478–1484. [Google Scholar] [CrossRef]
- Bai, P.; Lyu, L.; Yu, T.; Zuo, C.; Fu, J.; He, Y.; Wan, Q.; Wan, N.; Jia, D.; Lyu, A. Macrophage-Derived Legumain Promotes Pulmonary Hypertension by Activating the MMP (Matrix Metalloproteinase)-2/TGF (Transforming Growth Factor)-Β1 Signaling. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e130–e145. [Google Scholar] [CrossRef]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial Cell Death in Human Diabetes. Circ. Res. 2000, 87, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Håversen, L.; Sundelin, J.P.; Mardinoglu, A.; Rutberg, M.; Ståhlman, M.; Wilhelmsson, U.; Hultén, L.M.; Pekny, M.; Fogelstrand, P.; Bentzon, J.F.; et al. Vimentin Deficiency in Macrophages Induces Increased Oxidative Stress and Vascular Inflammation but Attenuates Atherosclerosis in Mice. Sci. Rep. 2018, 8, 16973. [Google Scholar] [CrossRef]
- Kondo, T.; Takahashi, M.; Yamasaki, G.; Sugimoto, M.; Kuse, A.; Morichika, M.; Nakagawa, K.; Sakurada, M.; Asano, M.; Ueno, Y. Immunohistochemical Analysis of Vimentin Expression in Myocardial Tissue from Autopsy Cases of Ischemic Heart Disease. Leg. Med. 2022, 54, 102003. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska-Podhorodecka, Z.; Ding, I.; Norouzi, M.; McCulloch, C.A. Impact of Vimentin on Regulation of Cell Signaling and Matrix Remodeling. Front. Cell Dev. Biol. 2022, 10, 869069. [Google Scholar] [CrossRef]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.-T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef]
- Abacar, K.; Macleod, T.; Direskeneli, H.; McGonagle, D. How Underappreciated Autoinflammatory (Innate Immunity) Mechanisms Dominate Disparate Autoimmune Disorders. Front. Immunol. 2024, 15, 1439371. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes Primes Neutrophils to Undergo NETosis, Which Impairs Wound Healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Yerra, V.G.; Advani, A. Histones and Heart Failure in Diabetes. Cell. Mol. Life Sci. 2018, 75, 3193–3213. [Google Scholar] [CrossRef]
- Kadakol, A.; Malek, V.; Goru, S.K.; Pandey, A.; Gaikwad, A.B. Telmisartan and Esculetin Combination Ameliorates Type 2 Diabetic Cardiomyopathy by Reversal of H3, H2A, and H2B Histone Modifications. Indian J. Pharmacol. 2017, 49, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Goldsmith, E.C.; Zhang, X.; Watson, J.; Hastings, J.; Potts, J.D. The Collagen Receptor DDR2 Is Expressed During Early Cardiac Development. Anat. Rec. 2010, 293, 762–769. [Google Scholar] [CrossRef]
- Kim, D.; You, E.; Jeong, J.; Ko, P.; Kim, J.-W.; Rhee, S. DDR2 Controls the Epithelial-Mesenchymal-Transition-Related Gene Expression via c-Myb Acetylation upon Matrix Stiffening. Sci. Rep. 2017, 7, 6847. [Google Scholar] [CrossRef]
- Mitchell, A.V.; Wu, J.; Meng, F.; Dong, L.; Block, C.J.; Song, W.; Zhang, B.; Li, J.; Wu, G. DDR2 Coordinates EMT and Metabolic Reprogramming as a Shared Effector of FOXQ1 and SNAI1. Cancer Res. Commun. 2022, 2, 1388–1403. [Google Scholar] [CrossRef]
- Du, J.-K.; Yu, Q.; Liu, Y.-J.; Du, S.-F.; Huang, L.-Y.; Xu, D.-H.; Ni, X.; Zhu, X.-Y. A Novel Role of Kallikrein-Related Peptidase 8 in the Pathogenesis of Diabetic Cardiac Fibrosis. Theranostics 2021, 11, 4207–4231. [Google Scholar] [CrossRef]
- Aukrust, S.G.; Holte, K.B.; Opstad, T.B.; Seljeflot, I.; Berg, T.J.; Helseth, R. NETosis in Long-Term Type 1 Diabetes Mellitus and Its Link to Coronary Artery Disease. Front. Immunol. 2022, 12, 799539. [Google Scholar] [CrossRef]
- Singh, A.; Bhatt, K.S.; Nguyen, H.C.; Frisbee, J.C.; Singh, K.K. Endothelial-to-Mesenchymal Transition in Cardiovascular Pathophysiology. Int. J. Mol. Sci. 2024, 25, 6180. [Google Scholar] [CrossRef] [PubMed]
- Giordo, R.; Ahmed, Y.M.; Allam, H.; Abusnana, S.; Pappalardo, L.; Nasrallah, G.K.; Mangoni, A.A.; Pintus, G. EndMT Regulation by Small RNAs in Diabetes-Associated Fibrotic Conditions: Potential Link with Oxidative Stress. Front. Cell Dev. Biol. 2021, 9, 683594. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-J.; Tsai, F.-C.; Chang, G.-J.; Chang, S.-H.; Huang, C.-C.; Chen, W.-J.; Yeh, Y.-H. miR-181b Targets Semaphorin 3A to Mediate TGF-β–Induced Endothelial-Mesenchymal Transition Related to Atrial Fibrillation. J. Clin. Investig. 2022, 132, e142548. [Google Scholar] [CrossRef]
- Mao, Y.; Jiang, L. MiR-200c-3p Promotes Ox-LDL-Induced Endothelial to Mesenchymal Transition in Human Umbilical Vein Endothelial Cells through SMAD7/YAP Pathway. J. Physiol. Sci. 2021, 71, 30. [Google Scholar] [CrossRef]
- Jordan, N.P.; Tingle, S.J.; Shuttleworth, V.G.; Cooke, K.; Redgrave, R.E.; Singh, E.; Glover, E.K.; Ahmad Tajuddin, H.B.; Kirby, J.A.; Arthur, H.M. MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis. Int. J. Mol. Sci. 2021, 22, 8629. [Google Scholar] [CrossRef]
- Wang, B.; Ge, Z.; Wu, Y.; Zha, Y.; Zhang, X.; Yan, Y.; Xie, Y. MFGE8 Is Down-regulated in Cardiac Fibrosis and Attenuates Endothelial-mesenchymal Transition through Smad2/3-Snail Signalling Pathway. J. Cell. Mol. Med. 2020, 24, 12799–12812. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmi, T.; Xu, X.; Tan, X.; Hulshoff, M.S.; Maamari, S.; Sossalla, S.; Zeisberg, M.; Zeisberg, E.M. Serelaxin Alleviates Cardiac Fibrosis through Inhibiting Endothelial-to-Mesenchymal Transition via RXFP1. Theranostics 2020, 10, 3905. [Google Scholar] [CrossRef]
- Gupta, S.; Mitra, A. Heal the Heart through Gut (Hormone) Ghrelin: A Potential Player to Combat Heart Failure. Heart Fail. Rev. 2021, 26, 417–435. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Moon, H.; Kim, R.; Kim, M.; Jeong, S.; Kim, H.; Kim, S.H.; Hwang, S.S.; Lee, M.Y. Epigallocatechin-3-Gallate Attenuates Myocardial Dysfunction via Inhibition of Endothelial-to-Mesenchymal Transition. Antioxidants 2023, 12, 1059. [Google Scholar] [CrossRef]
- Tsai, T.-H.; Lee, C.-H.; Cheng, C.-I.; Fang, Y.-N.; Chung, S.-Y.; Chen, S.-M.; Lin, C.-J.; Wu, C.-J.; Hang, C.-L.; Chen, W.-Y. Liraglutide Inhibits Endothelial-to-Mesenchymal Transition and Attenuates Neointima Formation after Endovascular Injury in Streptozotocin-Induced Diabetic Mice. Cells 2019, 8, 589. [Google Scholar] [CrossRef]
- Lin, S.; Du, L. The Therapeutic Potential of BRD4 in Cardiovascular Disease. Hypertens. Res. 2020, 43, 1006–1014. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor/Marker | Change in STZ-DM Model | Role in EndMT/Cardiovascular Pathophysiology | Key References |
|---|---|---|---|
| TGF-β | ↓ | Master EndMT inducer; low levels impair mesenchymal transition | [42,43,44] |
| CXCL-1 | ↑ | Chemokine attracting neutrophils; pro-inflammatory | [27,28,29,30,31] |
| TNF-α | ↑ | Inflammatory cytokine; upstream activator of CXCL-1 | [26] |
| IL-6 | ↑ | Pro-inflammatory; promotes endothelial dysfunction | [14,25] |
| IL-18 | ↑ | Inflammatory mediator; promotes cytokine storm | [14] |
| IL-33 | ↑ | Involved in innate immunity and inflammation | [14] |
| IFN-γ | ↑ | Stimulates Th1 responses and immune cell activation | [14] |
| Vimentin | ↓ | Mesenchymal marker; lower levels suggest impaired transition | [46,47,48,49,50,51] |
| α-SMA | ↑ | Mesenchymal marker: elevation indicates EndMT progression | [4,55] |
| Versican | ≈ | ECM proteoglycan; no significant change observed | [41] |
| DDR-2 | ↑ | Collagen receptor; increased in stiff/fibrotic matrix | [56,57,58] |
| MMP-2 | ↓ | Degrades ECM; decreased activity leads to fibrosis | [35,36,37,38] |
| TIMP-1 | ↓ | Inhibits MMPs; decreased expression disrupts ECM regulation | [39,40] |
| H3Cit | ↑ | Histone modification; indicates neutrophil activation and chromatin remodeling | [52,53,54] |
| Endothelin-1 | ↑ | Vasoconstrictor; elevated in diabetic aorta, promotes EndMT | [24] |
| Peroxynitrite | ↑ | ROS indicator; initiates oxidative damage and EndMT | [33] |
| iNOS | ↑ | Enzyme producing NO; high levels promote inflammation | [16,32,33,34] |
| Factor/Marker | Change in STZ-DM Model | Role in EndMT/Cardiovascular Pathophysiology | Key References |
|---|---|---|---|
| miR-200b | ↑ | Regulates EndMT via TGF-β/Smad pathway; inhibition reduces cardiac fibrosis | [62] |
| miR-181b | ↑ | Modulates TGF-β-induced EndMT via targeting Semaphorin 3A; implicated in atrial fibrillation | [63] |
| miR-200c-3p | ↑ | Promotes EndMT and intimal hyperplasia in graft vessels | [64] |
| miR-126-3p | ≈ | Dynamically regulated in EndMT; involved in fibrosis-related processes | [65] |
| MFGE8 | ↓ | Suppression promotes EndMT via Smad2/3-Snail signaling | [66] |
| Serelaxin | Therapeutic | Inhibits EndMT via RXFP1 receptor; reduces fibrosis | [67] |
| Ghrelin | Therapeutic | Inhibits EndMT; reduces cardiac fibrosis post-MI | [68] |
| EGCG | Therapeutic | Suppresses EndMT; improves cardiac function | [69] |
| Liraglutide | Therapeutic | Suppresses EndMT; reduces neointima formation in diabetic mice | [70] |
| BRD4 | ↑ | Its inhibition reduces EndMT and cardiac fibrosis | [71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.L.; Várkonyi, Á.; Csonka, Á.; Szász, A.; Várkonyi, T.; Pósa, A.; Kupai, K. Endothelial–Mesenchymal Transition and Possible Role of Cytokines in Streptozotocin-Induced Diabetic Heart. Biomedicines 2025, 13, 1148. https://doi.org/10.3390/biomedicines13051148
Kang HL, Várkonyi Á, Csonka Á, Szász A, Várkonyi T, Pósa A, Kupai K. Endothelial–Mesenchymal Transition and Possible Role of Cytokines in Streptozotocin-Induced Diabetic Heart. Biomedicines. 2025; 13(5):1148. https://doi.org/10.3390/biomedicines13051148
Chicago/Turabian StyleKang, Hsu Lin, Ákos Várkonyi, Ákos Csonka, András Szász, Tamás Várkonyi, Anikó Pósa, and Krisztina Kupai. 2025. "Endothelial–Mesenchymal Transition and Possible Role of Cytokines in Streptozotocin-Induced Diabetic Heart" Biomedicines 13, no. 5: 1148. https://doi.org/10.3390/biomedicines13051148
APA StyleKang, H. L., Várkonyi, Á., Csonka, Á., Szász, A., Várkonyi, T., Pósa, A., & Kupai, K. (2025). Endothelial–Mesenchymal Transition and Possible Role of Cytokines in Streptozotocin-Induced Diabetic Heart. Biomedicines, 13(5), 1148. https://doi.org/10.3390/biomedicines13051148