
Reduced Expression of UPRmt Proteins HSP10, HSP60, HTRA2, OMA1, SPG7, and YME1L Is Associated with Accelerated Heart Failure in Humans

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Myocardial Samples
2.2. Protein Quantification by ELISA
2.3. Cell Culture
2.4. Pathological Analysis
2.5. Quantitative PCR
2.6. Statistical Analysis
3. Results
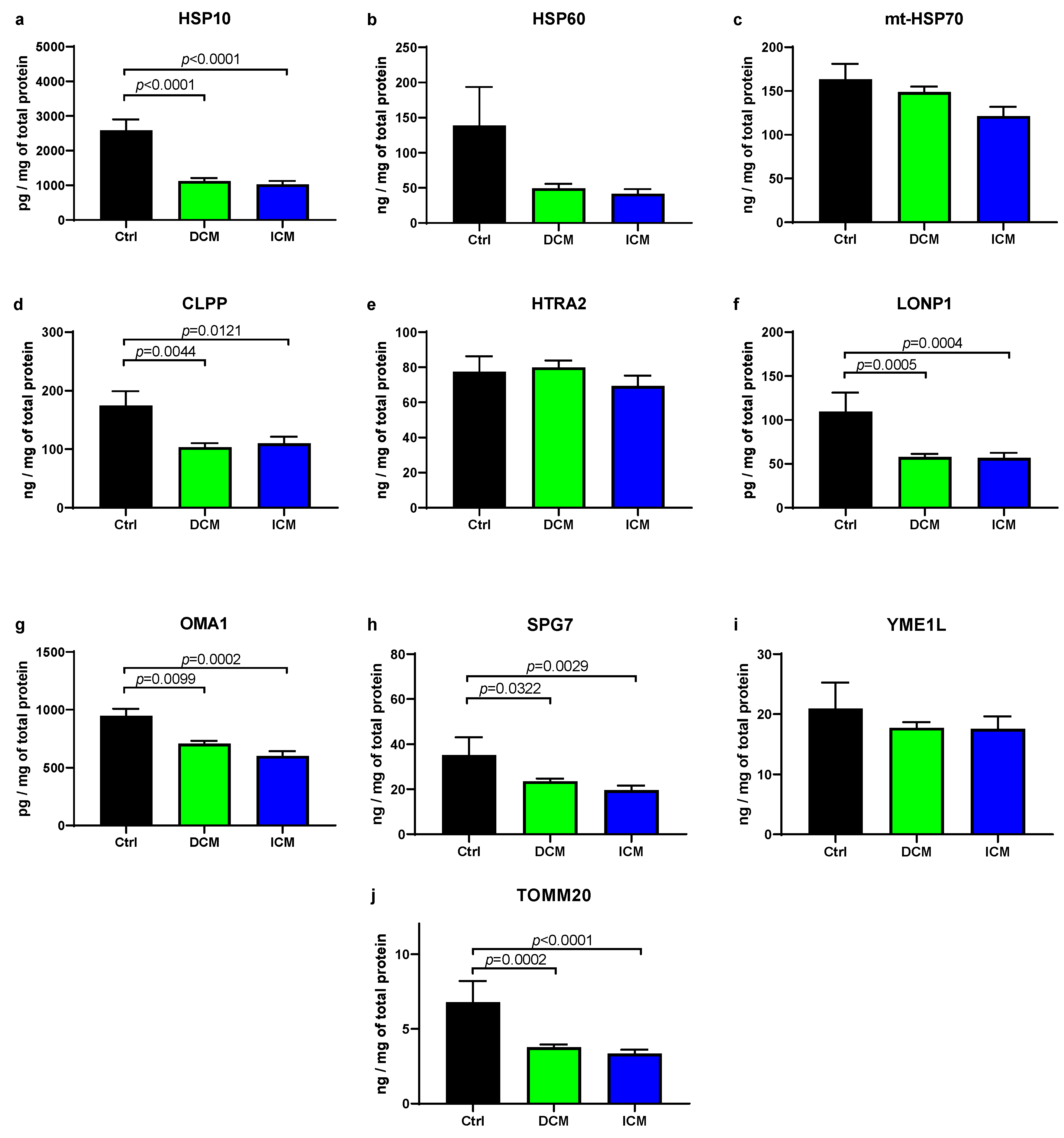
3.1. UPRmt Proteases and Chaperones Are Downregulated in Failing Human Myocardium
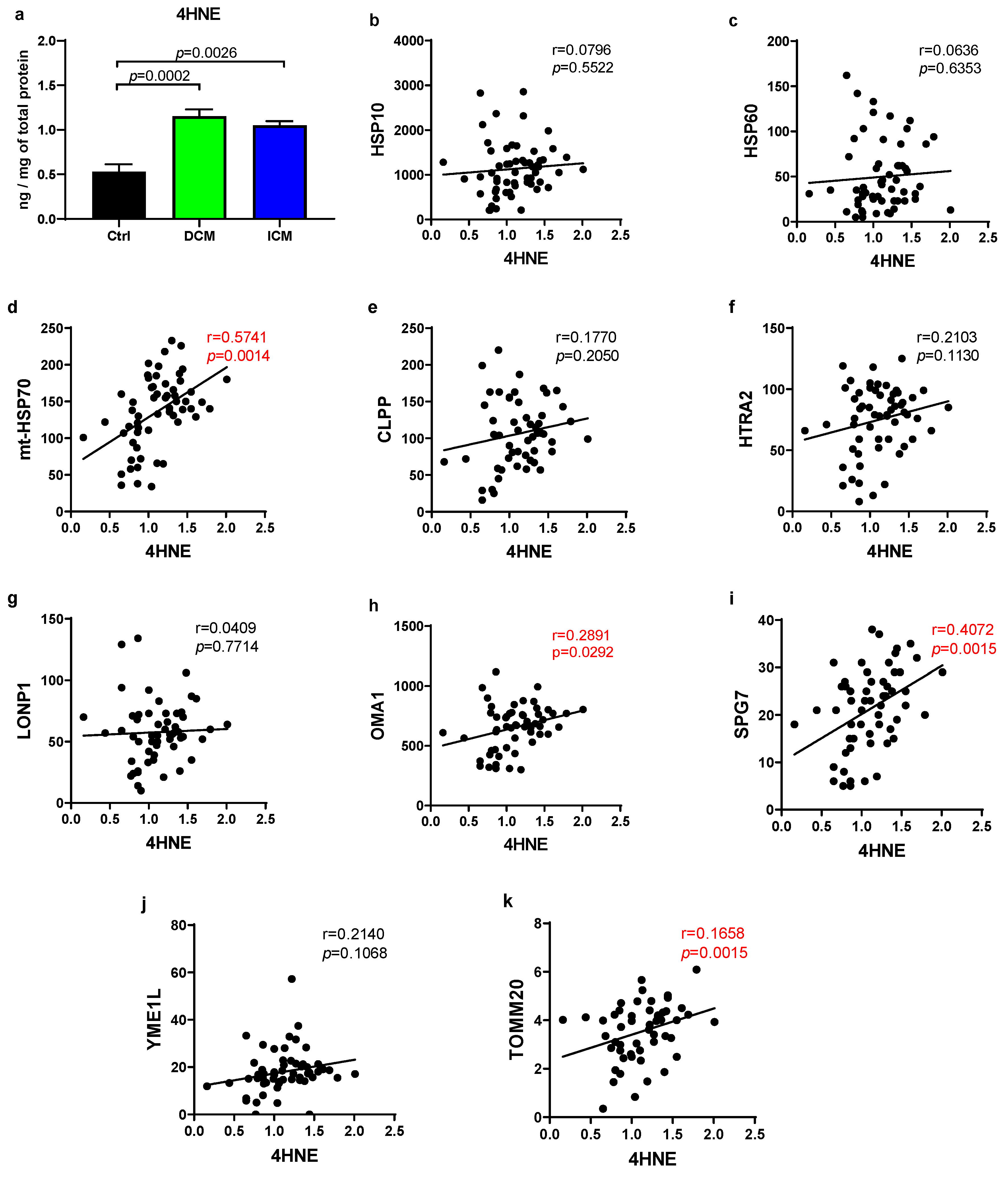
3.2. Human Failing Hearts Produce More ROS, Which Positively Correlate with Several UPRmt Proteins
3.3. UPRmt Genes Display Negative Correlation with the Degree of Cardiomyocyte Hypertrophy
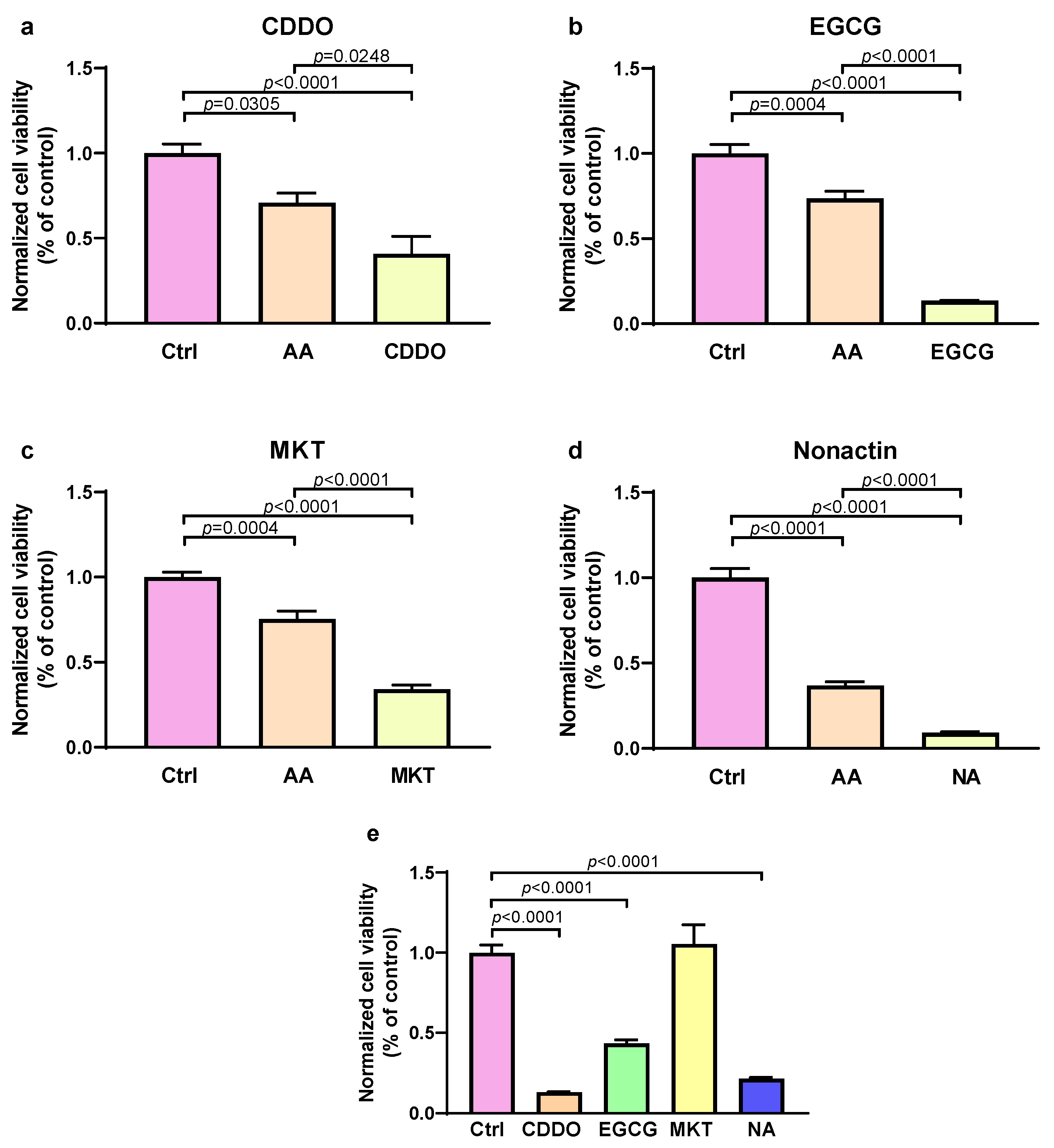
3.4. Inhibition of UPRmt Proteases and Chaperones Reduces Survival of Stressed Cells
3.5. Patients with Lower Expression of UPRmt Proteins Undergo Heart Transplantation or LVAD Implementation at Younger Age
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Briceno, N.; Schuster, A.; Lumley, M.; Perera, D. Ischaemic Cardiomyopathy: Pathophysiology, Assessment and the Role of Revascularisation. Heart 2016, 102, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Lakdawala, N.K.; Tschöpe, C.; Klingel, K. Dilated Cardiomyopathy: Causes, Mechanisms, and Current and Future Treatment Approaches. Lancet 2023, 402, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the Management of Cardiomyopathies: Developed by the Task Force on the Management of Cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Gallo, G.; Rubattu, S.; Volpe, M. Mitochondrial Dysfunction in Heart Failure: From Pathophysiological Mechanisms to Therapeutic Opportunities. Int. J. Mol. Sci. 2024, 25, 2667. [Google Scholar] [CrossRef]
- Liu, J.; He, X.; Zheng, S.; Zhu, A.; Wang, J. The Mitochondrial Unfolded Protein Response: A Novel Protective Pathway Targeting Cardiomyocytes. Oxid. Med. Cell. Longev. 2022, 2022, 6430342. [Google Scholar] [CrossRef]
- Svaguša, T.; Martinić, M.; Martinić, M.; Kovačević, L.; Šepac, A.; Miličić, D.; Bulum, J.; Starčević, B.; Sirotković-Skerlev, M.; Seiwerth, F.; et al. Mitochondrial Unfolded Protein Response, Mitophagy and Other Mitochondrial Quality Control Mechanisms in Heart Disease and Aged Heart. Croat. Med. J. 2020, 61, 126–138. [Google Scholar] [CrossRef]
- Svagusa, T.; Sikiric, S.; Milavic, M.; Sepac, A.; Seiwerth, S.; Milicic, D.; Gasparovic, H.; Biocina, B.; Rudez, I.; Sutlic, Z.; et al. Heart Failure in Patients Is Associated with Downregulation of Mitochondrial Quality Control Genes. Eur. J. Clin. Investig. 2023, 53, e14054. [Google Scholar] [CrossRef]
- Wang, X.; Robbins, J. Proteasomal and Lysosomal Protein Degradation and Heart Disease. J. Mol. Cell. Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; González, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 Processing and Mitochondrial Fragmentation Cause Heart Failure in Mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Bárcena, C.; López-Otín, C.; Yehia, G.; et al. Mitochondrial LonP1 Protects Cardiomyocytes from Ischemia/Reperfusion Injury in Vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lei, J.; Wang, K.; Ma, L.; Liu, D.; Du, Y.; Wu, Y.; Zhang, S.; Wang, W.; Ma, X.; et al. Mitochondrial Omi/HtrA2 Promotes Caspase Activation Through Cleavage of HAX-1 in Aging Heart. Rejuvenation Res. 2017, 20, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-Y.; Chiu, Y.-C.; Lee, A.Y.-L.; Hwang, T.-L. Mitochondrial Lon Protease Controls ROS-Dependent Apoptosis in Cardiomyocyte under Hypoxia. Mitochondrion 2015, 23, 7–16. [Google Scholar] [CrossRef]
- Svaguša, T.; Sedlić, F.; Županić, S.; Manola, Š.; Bakoš, M.; Mirošević, V.; Livun, A. A Rare Form of LIM Domain-Binding Protein 3 (LDB3) Mutation Causes Hypertrophic Cardiomyopathy and Myofibrillar Myopathy Type 4. Clin. Genet. 2024, 106, 659–660. [Google Scholar] [CrossRef]
- Tayal, U.; Ware, J.S.; Lakdawala, N.K.; Heymans, S.; Prasad, S.K. Understanding the Genetics of Adult-Onset Dilated Cardiomyopathy: What a Clinician Needs to Know. Eur. Heart J. 2021, 42, 2384–2396. [Google Scholar] [CrossRef]
- Oh, S.-H.; Choi, Y.-B.; Kim, J.-H.; Weihl, C.C.; Ju, J.-S. Comparisons of ELISA and Western Blot Assays for Detection of Autophagy Flux. Data Brief 2017, 13, 696–699. [Google Scholar] [CrossRef]
- Crowe, A.R.; Yue, W. Semi-Quantitative Determination of Protein Expression Using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio-Protocol 2019, 9, e3465. [Google Scholar] [CrossRef]
- To, C.; Shilton, B.H.; Di Guglielmo, G.M. Synthetic Triterpenoids Target the Arp2/3 Complex and Inhibit Branched Actin Polymerization. J. Biol. Chem. 2010, 285, 27944–27957. [Google Scholar] [CrossRef]
- Shikata, Y.; Kiga, M.; Futamura, Y.; Aono, H.; Inoue, H.; Kawada, M.; Osada, H.; Imoto, M. Mitochondrial Uncoupler Exerts a Synthetic Lethal Effect against β-Catenin Mutant Tumor Cells. Cancer Sci. 2017, 108, 772–784. [Google Scholar] [CrossRef]
- Du, G.J.; Zhang, Z.; Wen, X.D.; Yu, C.; Calway, T.; Yuan, C.S.; Wang, C.Z. Epigallocatechin Gallate (EGCG) Is the Most Effective Cancer Chemopreventive Polyphenol in Green Tea. Nutrients 2012, 4, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Rousaki, A.; Miyata, Y.; Jinwal, U.K.; Dickey, C.A.; Gestwicki, J.E.; Zuiderweg, E.R.P. Allosteric Drugs: The Interaction of Anti-Tumor Compound MKT-077 with Human Hsp70 Chaperones. J. Mol. Biol. 2011, 411, 614–632. [Google Scholar] [CrossRef] [PubMed]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell Death and Diseases Related to Oxidative Stress:4-Hydroxynonenal (HNE) in the Balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Schaur, R.J. Basic Aspects of the Biochemical Reactivity of 4-Hydroxynonenal. Mol. Asp. Med. 2003, 24, 149–159. [Google Scholar] [CrossRef]
- Inigo, J.R.; Chandra, D. The Mitochondrial Unfolded Protein Response (UPRmt): Shielding against Toxicity to Mitochondria in Cancer. J. Hematol. Oncol. 2022, 15, 98. [Google Scholar] [CrossRef]
- Heinzel, F.R.; Hohendanner, F.; Jin, G.; Sedej, S.; Edelmann, F. Myocardial Hypertrophy and Its Role in Heart Failure with Preserved Ejection Fraction. J. Appl. Physiol. 2015, 119, 1233–1242. [Google Scholar] [CrossRef]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear Protein Imbalance as a Conserved Longevity Mechanism. Nature 2013, 497, 451. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Suematsu, N.; Hayashidani, S.; Ichikawa, K.; Utsumi, H.; Machida, Y.; Egashira, K.; Takeshita, A. Direct Evidence for Increased Hydroxyl Radicals Originating from Superoxide in the Failing Myocardium. Circ. Res. 2000, 86, 152–157. [Google Scholar] [CrossRef]
- Maack, C.; Kartes, T.; Kilter, H.; Schäfers, H.-J.; Nickenig, G.; Böhm, M.; Laufs, U. Oxygen Free Radical Release in Human Failing Myocardium Is Associated with Increased Activity of Rac1-GTPase and Represents a Target for Statin Treatment. Circulation 2003, 108, 1567–1574. [Google Scholar] [CrossRef]
- Santin, Y.; Fazal, L.; Sainte-Marie, Y.; Sicard, P.; Maggiorani, D.; Tortosa, F.; Yücel, Y.Y.; Teyssedre, L.; Rouquette, J.; Marcellin, M.; et al. Mitochondrial 4-HNE Derived from MAO-A Promotes mitoCa2+ Overload in Chronic Postischemic Cardiac Remodeling. Cell Death Differ. 2020, 27, 1907–1923. [Google Scholar] [CrossRef]
- Sedlic, F.; Muravyeva, M.Y.; Sepac, A.; Sedlic, M.; Williams, A.M.; Yang, M.; Bai, X.; Bosnjak, Z.J. Targeted Modification of Mitochondrial ROS Production Converts High Glucose-Induced Cytotoxicity to Cytoprotection: Effects on Anesthetic Preconditioning. J. Cell. Physiol. 2017, 232, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Sedlic, F.; Sepac, A.; Pravdic, D.; Camara, A.K.S.; Bienengraeber, M.; Brzezinska, A.K.; Wakatsuki, T.; Bosnjak, Z.J. Mitochondrial Depolarization Underlies Delay in Permeability Transition by Preconditioning with Isoflurane: Roles of ROS and Ca2+. Am. J. Physiol. Cell Physiol. 2010, 299, C506–C515. [Google Scholar] [CrossRef] [PubMed]
- Muravyeva, M.; Baotic, I.; Bienengraeber, M.; Lazar, J.; Bosnjak, Z.J.; Sedlic, F.; Warltier, D.C.; Kersten, J.R. Cardioprotection during Diabetes: The Role of Mitochondrial DNA. Anesthesiology 2014, 120, 870–879. [Google Scholar] [CrossRef]
- Sepac, A.; Sedlic, F.; Si-Tayeb, K.; Lough, J.; Duncan, S.A.; Bienengraeber, M.; Park, F.; Kim, J.; Bosnjak, Z.J. Isoflurane Preconditioning Elicits Competent Endogenous Mechanisms of Protection from Oxidative Stress in Cardiomyocytes Derived from Human Embryonic Stem Cells. Anesthesiology 2010, 113, 906–916. [Google Scholar] [CrossRef]
- Dai, D.-F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial Oxidative Stress Mediates Angiotensin II-Induced Cardiac Hypertrophy and Galphaq Overexpression-Induced Heart Failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury through Mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Zeng, C.; Duan, F.; Hu, J.; Luo, B.; Huang, B.; Lou, X.; Sun, X.; Li, H.; Zhang, X.; Yin, S.; et al. NLRP3 Inflammasome-Mediated Pyroptosis Contributes to the Pathogenesis of Non-Ischemic Dilated Cardiomyopathy. Redox Biol. 2020, 34, 101523. [Google Scholar] [CrossRef]
- Sedlic, F.; Pravdic, D.; Ljubkovic, M.; Marinovic, J.; Stadnicka, A.; Bosnjak, Z.J. Differences in Production of Reactive Oxygen Species and Mitochondrial Uncoupling as Events in the Preconditioning Signaling Cascade between Desflurane and Sevoflurane. Anesth. Analg. 2009, 109, 405–411. [Google Scholar] [CrossRef]
- Sedlic, F.; Pravdic, D.; Hirata, N.; Mio, Y.; Sepac, A.; Camara, A.K.; Wakatsuki, T.; Bosnjak, Z.J.; Bienengraeber, M. Monitoring Mitochondrial Electron Fluxes Using NAD(P)H-Flavoprotein Fluorometry Reveals Complex Action of Isoflurane on Cardiomyocytes. Biochim. Biophys. Acta 2010, 1797, 1749–1758. [Google Scholar] [CrossRef]
- Pharaoh, G.; Pulliam, D.; Hill, S.; Sataranatarajan, K.; Van Remmen, H. Ablation of the Mitochondrial Complex IV Assembly Protein Surf1 Leads to Increased Expression of the UPRMT and Increased Resistance to Oxidative Stress in Primary Cultures of Fibroblasts. Redox Biol. 2016, 8, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Sutandy, F.X.R.; Gößner, I.; Tascher, G.; Münch, C. A Cytosolic Surveillance Mechanism Activates the Mitochondrial UPR. Nature 2023, 618, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ren, J.; Toan, S.; Mui, D. Role of Mitochondrial Quality Surveillance in Myocardial Infarction: From Bench to Bedside. Ageing Res. Rev. 2021, 66, 101250. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of Cardiac Hypertrophy and Heart Failure: Signaling Pathways and Novel Therapeutic Targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of Physiological and Pathological Cardiac Hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Raphael, C.; Briscoe, C.; Davies, J.; Ian Whinnett, Z.; Manisty, C.; Sutton, R.; Mayet, J.; Francis, D.P. Limitations of the New York Heart Association Functional Classification System and Self-Reported Walking Distances in Chronic Heart Failure. Heart 2007, 93, 476–482. [Google Scholar] [CrossRef]
- Shi, L.; Tan, Y.; Zheng, W.; Cao, G.; Zhou, H.; Li, P.; Cui, J.; Song, Y.; Feng, L.; Li, H.; et al. CTRP3 Alleviates Mitochondrial Dysfunction and Oxidative Stress Injury in Pathological Cardiac Hypertrophy by Activating UPRmt via the SIRT1/ATF5 Axis. Cell Death Discov. 2024, 10, 1–14. [Google Scholar] [CrossRef]
- Wang, Y.T.; Lim, Y.; McCall, M.N.; Huang, K.-T.; Haynes, C.M.; Nehrke, K.; Brookes, P.S. Cardioprotection by the Mitochondrial Unfolded Protein Response Requires ATF5. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H472–H478. [Google Scholar] [CrossRef]
- Wang, H.; Xu, X.; Fassett, J.; Kwak, D.; Liu, X.; Hu, X.; Falls, T.J.; Bell, J.C.; Li, H.; Bitterman, P.; et al. Double-Stranded RNA-Dependent Protein Kinase Deficiency Protects the Heart from Systolic Overload-Induced Congestive Heart Failure. Circulation 2014, 129, 1397–1406. [Google Scholar] [CrossRef]
- Almontashiri, N.A.M.; Chen, H.-H.; Mailloux, R.J.; Tatsuta, T.; Teng, A.C.T.; Mahmoud, A.B.; Ho, T.; Stewart, N.A.S.; Rippstein, P.; Harper, M.E.; et al. SPG7 Variant Escapes Phosphorylation-Regulated Processing by AFG3L2, Elevates Mitochondrial ROS, and Is Associated with Multiple Clinical Phenotypes. Cell Rep. 2014, 7, 834–847. [Google Scholar] [CrossRef]
- Lin, L.; Kim, S.C.; Wang, Y.; Gupta, S.; Davis, B.; Simon, S.I.; Torre-Amione, G.; Knowlton, A.A. HSP60 in Heart Failure: Abnormal Distribution and Role in Cardiac Myocyte Apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2238–H2247. [Google Scholar] [CrossRef] [PubMed]
- Sedlic, F.; Kovac, Z. Non-Linear Actions of Physiological Agents: Finite Disarrangements Elicit Fitness Benefits. Redox Biol. 2017, 13, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Duchen, M.R. Endothelial Mitochondria: Contributing to Vascular Function and Disease. Circ. Res. 2007, 100, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in Cardiomyocyte Isolation, Culture, and Gene Transfer. J. Mol. Cell. Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef]
- Alibrandi, L.; Lionetti, V. Interspecies Differences in Mitochondria: Implications for Cardiac and Vascular Translational Research. Vascul. Pharmacol. 2025, 159, 107476. [Google Scholar] [CrossRef]
- Sepac, A.; Si-Tayeb, K.; Sedlic, F.; Barrett, S.; Canfield, S.; Duncan, S.A.; Bosnjak, Z.J.; Lough, J.W. Comparison of Cardiomyogenic Potential among Human ESC and iPSC Lines. Cell Transplant. 2012, 21, 2523–2530. [Google Scholar] [CrossRef]
- Gwak, S.-J.; Bhang, S.H.; Kim, I.-K.; Kim, S.-S.; Cho, S.-W.; Jeon, O.; Yoo, K.J.; Putnam, A.J.; Kim, B.-S. The Effect of Cyclic Strain on Embryonic Stem Cell-Derived Cardiomyocytes. Biomaterials 2008, 29, 844–856. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-HF | HF | p-Value | |
|---|---|---|---|
| n | 6 | 59 | |
| Age (y), mean ± SEM | 56 ± 2 | 57 ± 1 | NS |
| Female gender | 2 (33%) | 8 (13%) | NS |
| Height (cm), mean ± SEM | 179 ± 5 | 176 ± 9 | NS |
| Body mass (kg), mean ± SEM | 104 ± 11 | 82 ± 14 | NS |
| BMI (kg/m2), mean ± SEM | 32.9 ± 4.5 | 26.3 ± 3.6 | NS |
| BSA (m2), mean ± SEM | 2.3 ± 0.1 | 2.0 ± 0.2 | NS |
| Smoking, n (%) | 2 (33%) | 18 (31%) | NS |
| AH, n (%) | 1 (17%) | 30 (50%) | NS |
| Dyslipidemia, n (%) | 1 (17%) | 30 (50%) | NS |
| ACEi, n (%) | 1 (17%) | 15 (25%) | NS |
| Statins, n (%) | 1 (17%) | 28 (47%) | NS |
| DCM | ICM | p-Value | |
|---|---|---|---|
| n | 30 | 29 | |
| Age (y), mean ± SEM | 58 ± 6 | 57 ± 7 | NS |
| Female gender, n (%) | 3 (10%) | 5 (17%) | NS |
| Smoking, n (%) | 8 (26%) | 10 (34%) | NS |
| BMI (kg/m2), mean ± SEM | 26.7 ± 0.7 | 25.8 ± 0.6 | NS |
| SBP (mmHg), mean ± SEM | 108 ± 3 | 111 ± 2 | NS |
| DBP (mmHg), mean ± SEM | 73 ± 2 | 75 ± 2 | NS |
| MAP (mmHg), mean ± SEM | 85 ± 2 | 87 ± 9 | NS |
| ICD/CRT, n (%) | 18 (60%) | 15 (52%) | NS |
| AH, n (%) | 12 (40%) | 18 (62%) | NS |
| Dyslipidemia, n (%) | 8 (26%) | 22 (75%) | 0.0276 |
| AF, n (%) | 12 (40%) | 14 (48%) | NS |
| CKD, n (%) | 9 (30%) | 8 (27%) | NS |
| Hyperuricemia, n (%) | 8 (26%) | 7 (24%) | NS |
| VT/VF, n (%) | 18 (60%) | 13 (45%) | NS |
| LVEF (%), mean ± SEM | 22 ± 1 | 27 ± 2 | NS |
| ACI, n (%) | 7 (23%) | 8 (27%) | NS |
| ARB, n (%) | 13 (43%) | 13 (44%) | NS |
| Neprilysin inhibitors, n (%) | 12 (40%) | 10 (34%) | NS |
| BB, n (%) | 24 (80%) | 22 (75%) | NS |
| Statins, n (%) | 8 (26%) | 20 (68%) | 0.0017 |
| Diuretics, n (%) | 27 (90%) | 26 (89%) | NS |
| MRA, n (%) | 25 (83%) | 24 (83%) | NS |
| Amiodarone, n (%) | 18 (60%) | 9 (31%) | 0.0370 |
| Anticoagulants, n (%) | 21 (70%) | 20 (68%) | NS |
| GFR (mL/min/1.73 m2), mean ± SEM | 69 ± 4 | 72 ± 4 | NS |
| Regression | 0.0009 | 0.2 | 0.001 | 0.03 | ||||
| Dependent variable | HSPA9 | OMA1 | SPG7 | TOMM20 | ||||
| Independent variable | t | p | t | p | t | p | t | p |
| 4HNE | 3.76 | 0.0005 | 2.05 | 0.04 | 3.21 | 0.002 | 2.87 | 0.006 |
| Smoking | 1.80 | 0.08 | 0.58 | 0.57 | 1.64 | 0.11 | 0.85 | 0.40 |
| Sex | 1.46 | 0.15 | 0.2 | 0.84 | 2.38 | 0.02 | 0.59 | 0.56 |
| UPRmt Protein | DCM | ICM | all | |
|---|---|---|---|---|
| ageHTx/LVAD | p-value | 0.2246 | 0.2778 | 0.0635 |
| CLPP (Lo) | 56.5 ± 1.8 | 55.7 ± 2.6 | 55.8 ± 1.6 | |
| CLPP (Hi) | 59.3 ± 1.3 | 58.9 ± 1.00 | 59.2 ± 0.8 | |
| ageHTx/LVAD | p-value | 0.4278 | 0.0256 | 0.2639 |
| HSP10 (Lo) | 58.9 ± 1.8 | 54.6 ± 2.1 | 56.8 ± 1.4 | |
| HSP10 (Hi) | 57.1 ± 1.3 | 60.42 ± 1.0 | 59.0 ± 0.8 | |
| ageHTx/LVAD | p-value | 0.5000 | 0.0875 | 0.0032 |
| HSP60 (Lo) | 57.2 ± 1.8 | 55.2 ± 1.9 | 55.3 ± 1.3 | |
| HSP60 (Hi) | 58.8 ± 1.3 | 59.7 ± 1.5 | 60.2 ± 0.9 | |
| ageHTx/LVAD | p-value | 0.7936 | 0.1487 | 0.3871 |
| mt-HSP70 (Lo) | 58.3 ± 1.9 | 55.6 ± 1.7 | 57.0 ± 1.3 | |
| mt-HSP70 (Hi) | 57.7 ± 1.1 | 59.4 ± 1.9 | 58.5 ± 1.1 | |
| ageHTx/LVAD | p-value | 0.0181 | 0.1645 | 0.0055 |
| HTRA2 (Lo) | 55.4 ± 1.9 | 55.6 ± 2.0 | 55.5 ± 1.3 | |
| HTRA2 (Hi) | 60.6 ± 0.8 | 59.3 ± 1.6 | 60.1 ± 0.9 | |
| ageHTx/LVAD | p-value | 0.2747 | 0.6744 | 0.7051 |
| LONP1 (Lo) | 59.2 ± 1.8 | 56.9 ± 2.1 | 58.0 ± 1.4 | |
| LONP1 (Hi) | 56.8 ± 1.3 | 57.6 ± 1.5 | 57.0 ± 0.9 | |
| ageHTx/LVAD | p-value | 0.5940 | 0.0094 | 0.0673 |
| OMA1 (Lo) | 57.5 ± 1.9 | 54.2 ± 2.0 | 56.2 ± 1.4 | |
| OMA1 (Hi) | 58.7 ± 1.3 | 60.8 ± 1.1 | 59.4 ± 0.8 | |
| ageHTx/LVAD | p-value | 0.0663 | 0.0159 | 0.0027 |
| 54.2 ± 2.0 | 56.0 ± 1.8 | 54.4 ± 2.0 | 55.3 ± 1.4 | |
| 60.8 ± 1.1 | 60.1 ± 1.0 | 60.6 ± 1.7 | 60.2 ± 0.7 | |
| ageHTx/LVAD | p-value | 0.7458 | 0.0850 | 0.0445 |
| YME1L (Lo) | 57.7 ± 1.8 | 54.9 ± 2.2 | 56.2 ± 1.4 | |
| YME1L (Hi) | 58.4 ± 1.3 | 59.7 ± 1.6 | 59.84 ± 0.9 | |
| ageHTx/LVAD | p-value | 0.2249 | 0.8490 | 0.3362 |
| TOMM20 (Lo) | 59.4 ± 1.2 | 57.2 ± 1.8 | 58.8 ± 1.1 | |
| TOMM20 (Hi) | 56.6 ± 1.8 | 57.7 ± 2.0 | 57.0 ± 1.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakovic, P.; Mirosevic, V.; Svagusa, T.; Sepac, A.; Kulic, A.; Milicic, D.; Gasparovic, H.; Rudez, I.; Urlic, M.; Sikiric, S.; et al. Reduced Expression of UPRmt Proteins HSP10, HSP60, HTRA2, OMA1, SPG7, and YME1L Is Associated with Accelerated Heart Failure in Humans. Biomedicines 2025, 13, 1142. https://doi.org/10.3390/biomedicines13051142
Bakovic P, Mirosevic V, Svagusa T, Sepac A, Kulic A, Milicic D, Gasparovic H, Rudez I, Urlic M, Sikiric S, et al. Reduced Expression of UPRmt Proteins HSP10, HSP60, HTRA2, OMA1, SPG7, and YME1L Is Associated with Accelerated Heart Failure in Humans. Biomedicines. 2025; 13(5):1142. https://doi.org/10.3390/biomedicines13051142
Chicago/Turabian StyleBakovic, Petra, Vid Mirosevic, Tomo Svagusa, Ana Sepac, Ana Kulic, Davor Milicic, Hrvoje Gasparovic, Igor Rudez, Marjan Urlic, Suncana Sikiric, and et al. 2025. "Reduced Expression of UPRmt Proteins HSP10, HSP60, HTRA2, OMA1, SPG7, and YME1L Is Associated with Accelerated Heart Failure in Humans" Biomedicines 13, no. 5: 1142. https://doi.org/10.3390/biomedicines13051142
APA StyleBakovic, P., Mirosevic, V., Svagusa, T., Sepac, A., Kulic, A., Milicic, D., Gasparovic, H., Rudez, I., Urlic, M., Sikiric, S., Seiwerth, S., Belina, D., Bakos, M., Vlahovic, M. K., Taradi, R., Zic, R., Ilic, I., Belev, B., Skoric, B., ... Sedlic, F. (2025). Reduced Expression of UPRmt Proteins HSP10, HSP60, HTRA2, OMA1, SPG7, and YME1L Is Associated with Accelerated Heart Failure in Humans. Biomedicines, 13(5), 1142. https://doi.org/10.3390/biomedicines13051142