
Patient-Oriented In Vitro Studies in Duchenne Muscular Dystrophy: Validation of a 3D Skeletal Muscle Organoid Platform

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Immortalized Cell Lines Maintenance
2.2. 3D SkMO Formation
2.3. Contraction Assay on the Multi-Well Plate Format
2.4. Imaging of Calcium Transients
2.5. Immunofluorescence Staining
2.6. RNA Isolation, cDNA Synthesis, and Real Time-PCR
2.7. Microfluidic Setup
2.8. Statistical Analysis
3. Results
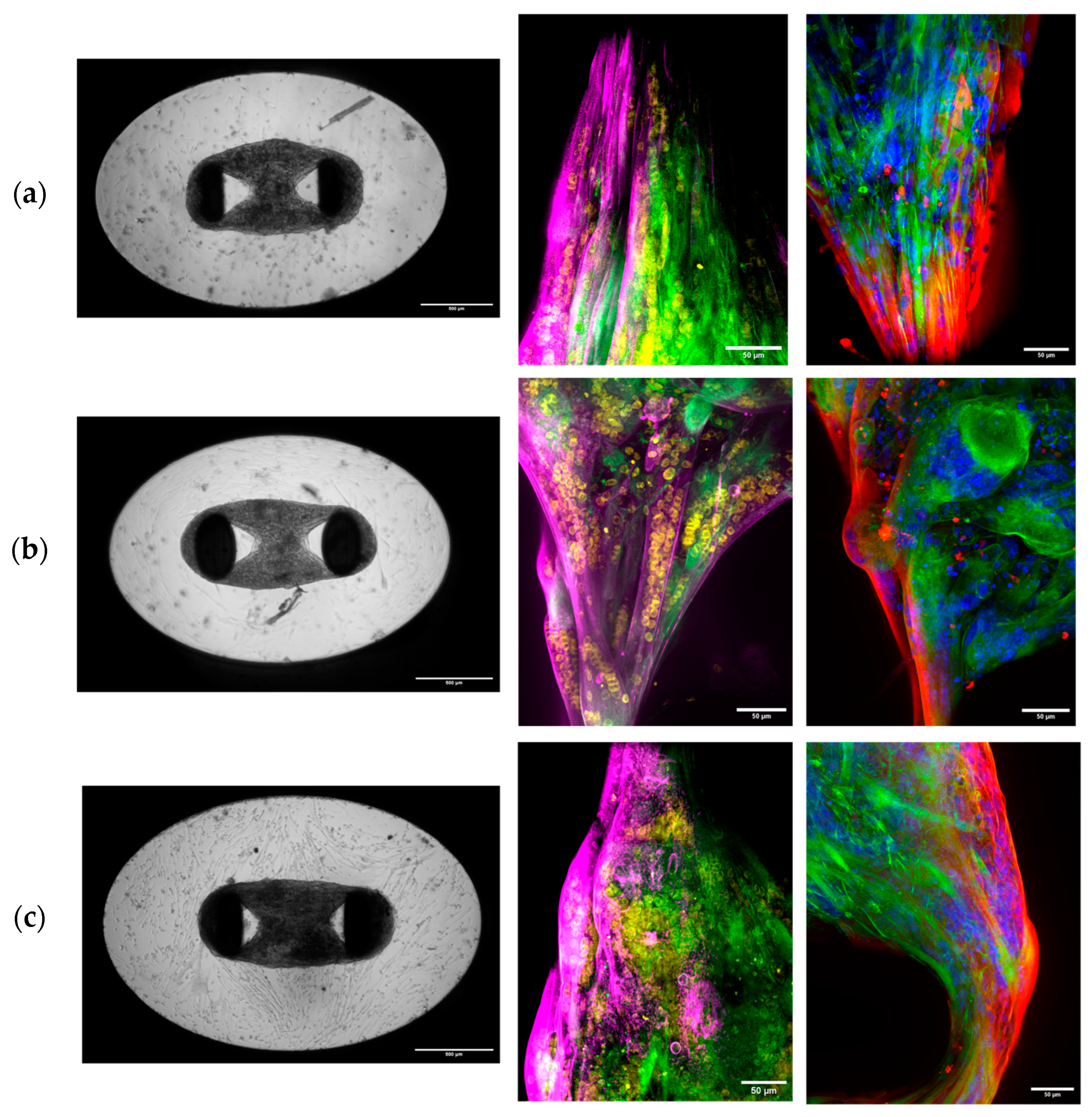
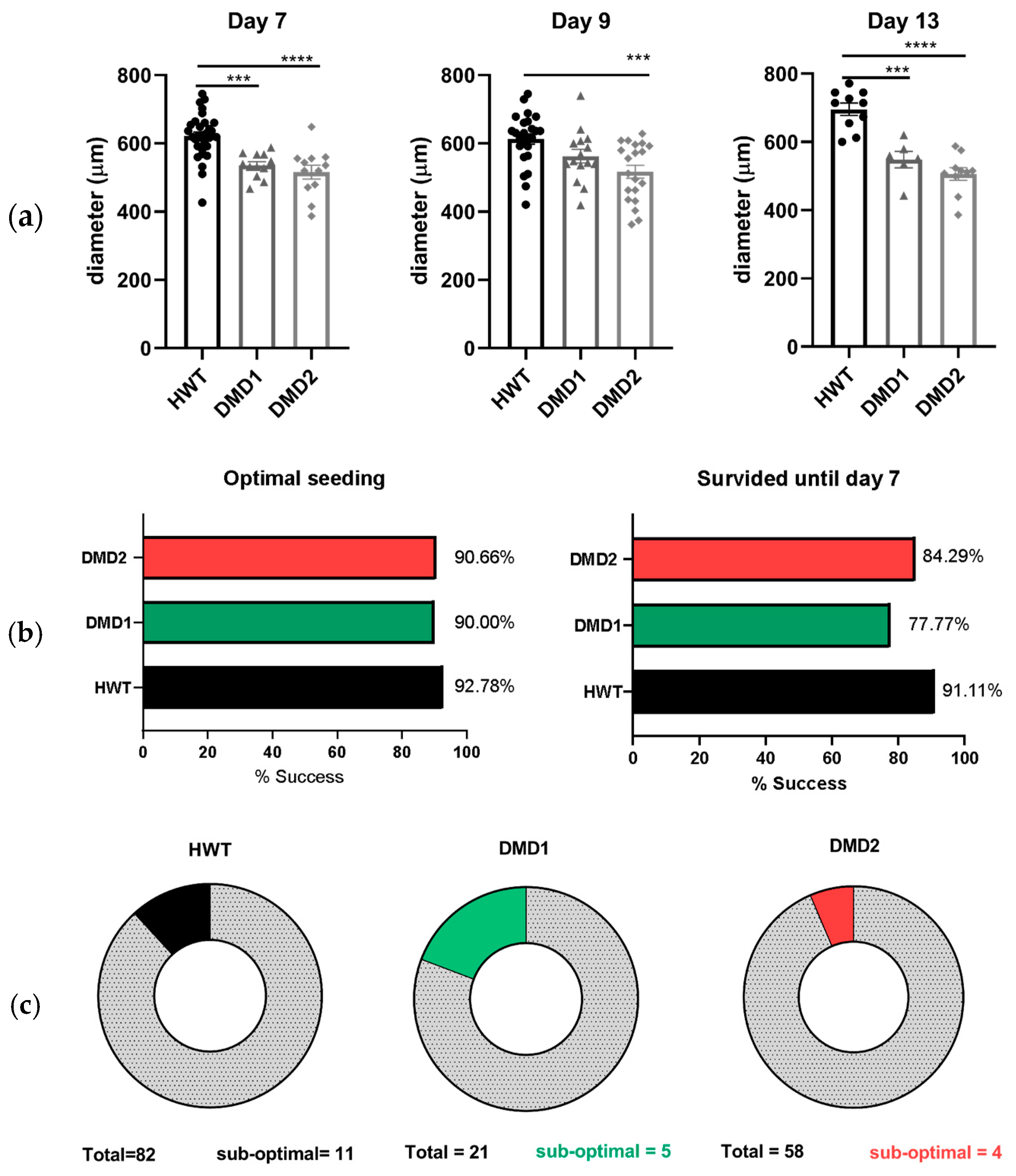
3.1. Monitoring 3D SkMO Formation and Assessment of Reproducibility
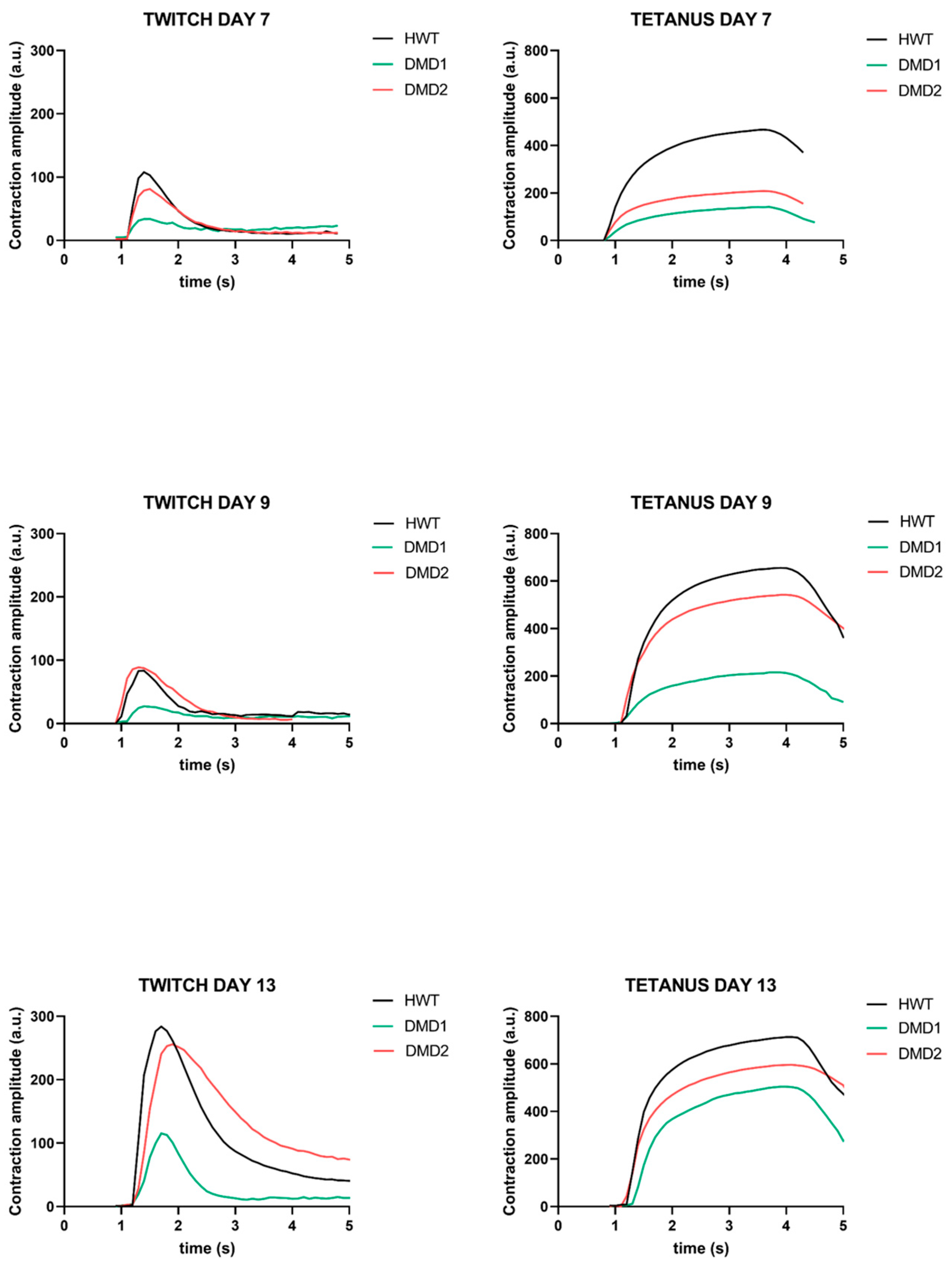
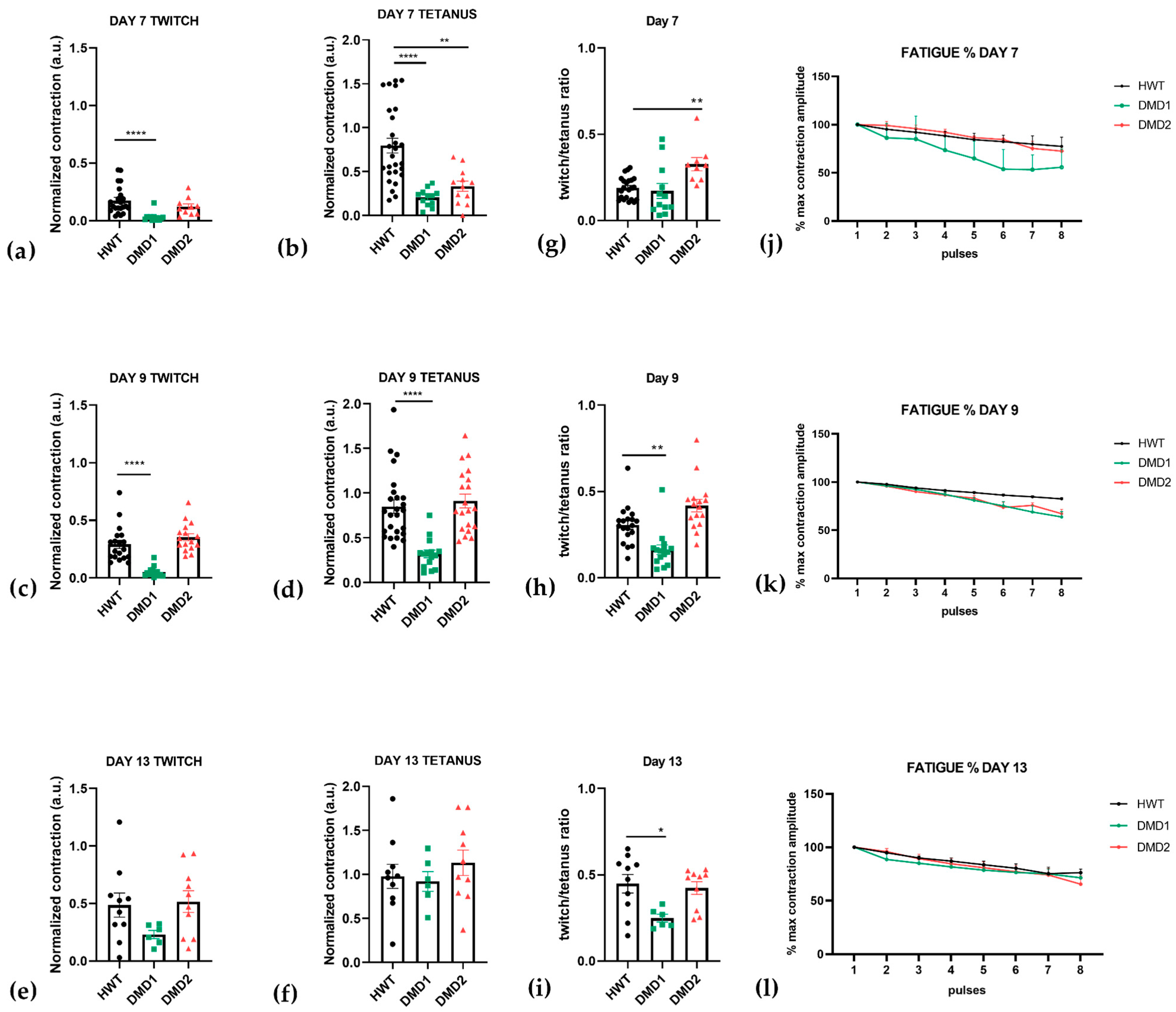
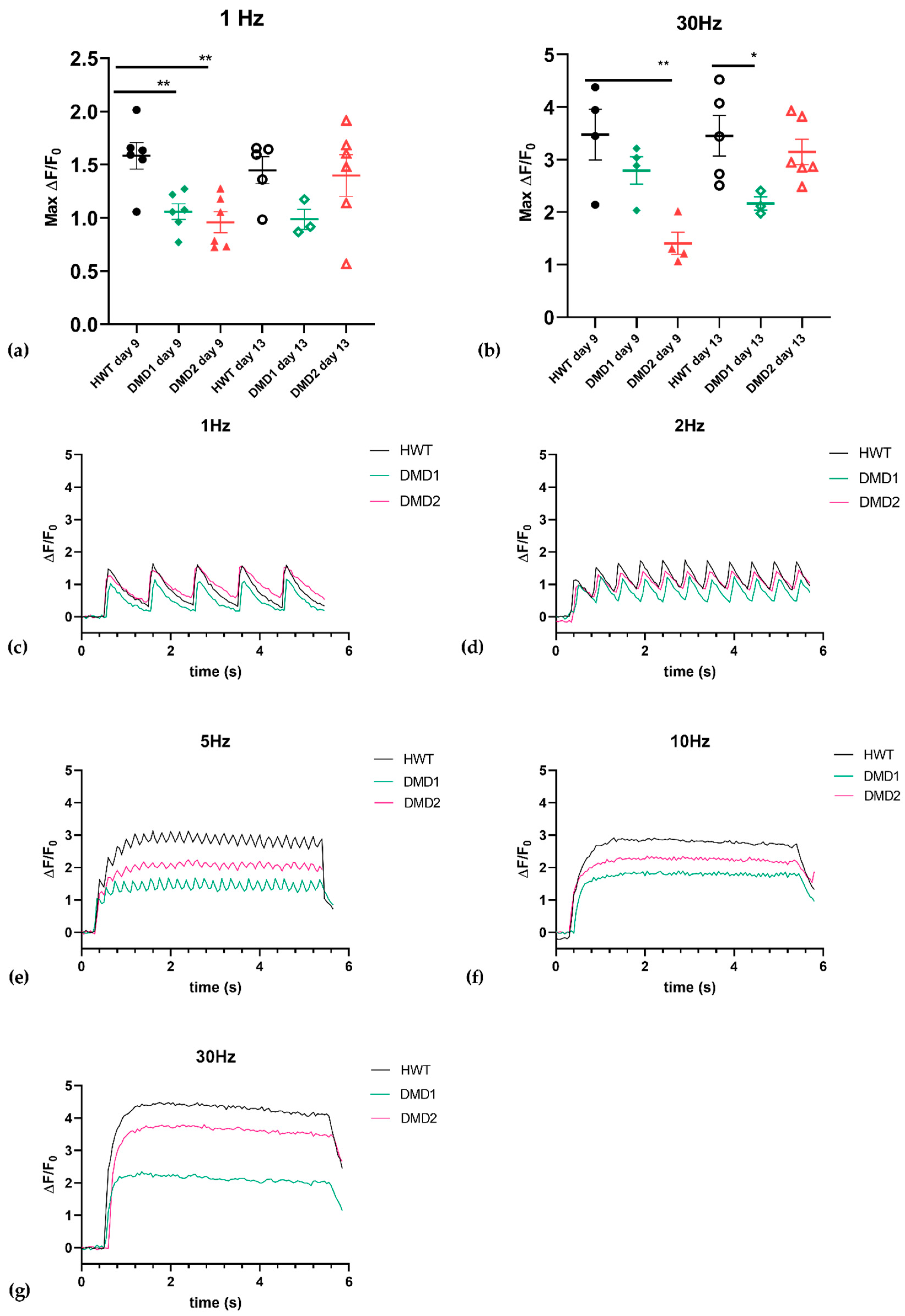
3.2. Functional Analysis Highlights Distinct Contraction Properties Among Cell Lines
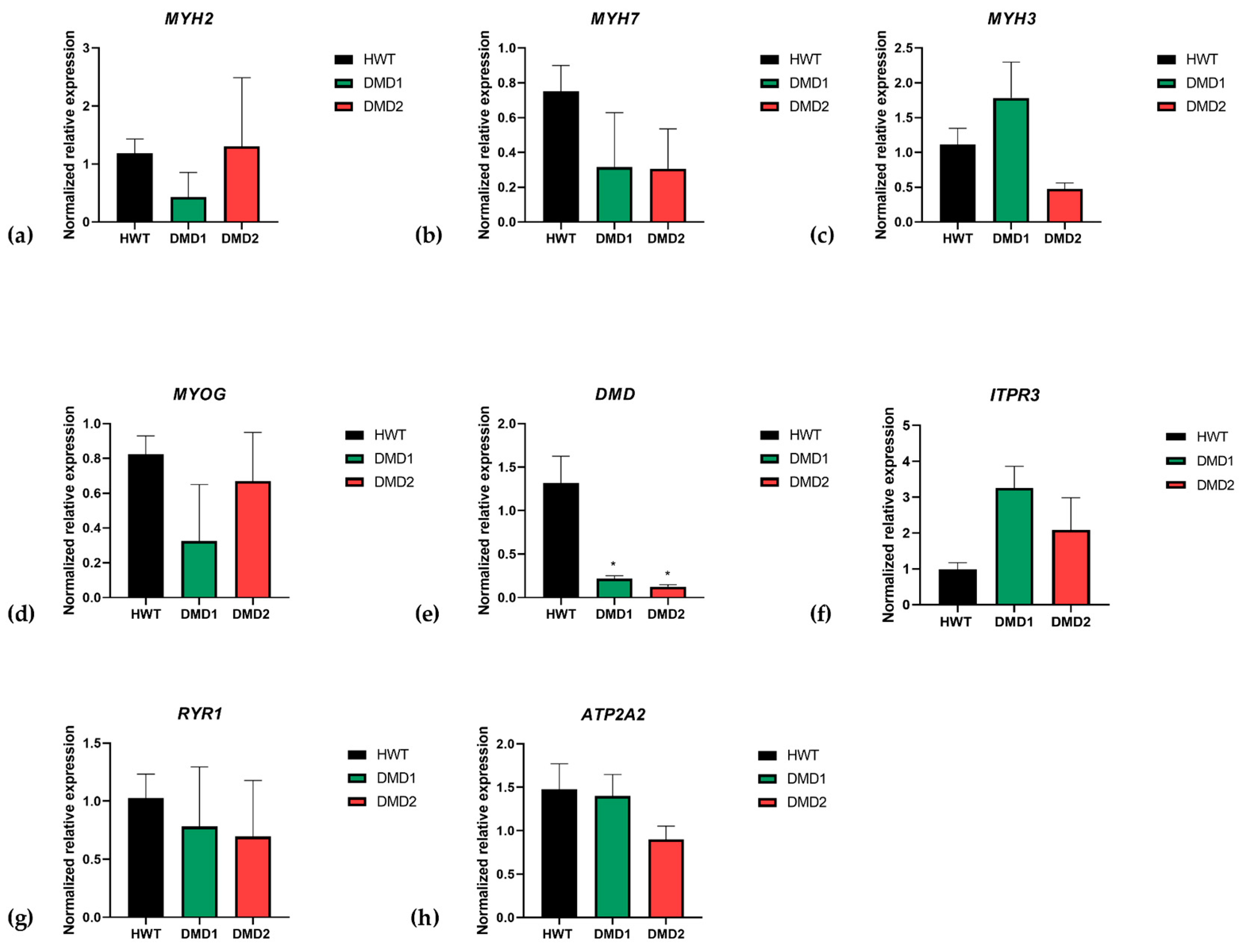
3.3. Gene Expression of Myogenesis Markers
3.4. Microfluidic Technology Recreates a Dynamic Environment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3D SkMO | three-dimensional skeletal muscle organoid |
References
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; Al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Jaber, A.; Palmieri, L.; Gicquel, E.; Richard, I.; Israeli, D. Advances and Challenges in Microdystrophin gene therapy for Duchenne Muscular Dystrophy: Progress and future directions. Med. Sci. 2024, 40, 46–51. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Dressman, D. Molecular pathophysiology and targeted therapeutics for muscular dystrophy. Trends Pharmacol. Sci. 2001, 22, 465–470. [Google Scholar] [CrossRef]
- Mantuano, P.; Sanarica, F.; Conte, E.; Morgese, M.G.; Capogrosso, R.F.; Cozzoli, A.; Fonzino, A.; Quaranta, A.; Rolland, J.-F.; De Bellis, M.; et al. Effect of a long-term treatment with metformin in dystrophic mdx mice: A reconsideration of its potential clinical interest in Duchenne muscular dystrophy. Biochem. Pharmacol. 2018, 154, 89–103. [Google Scholar] [CrossRef]
- Nakamura, A.; Shiba, N.; Miyazaki, D.; Nishizawa, H.; Inaba, Y.; Fueki, N.; Maruyama, R.; Echigoya, Y.; Yokota, T. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J. Hum. Genet. 2017, 62, 459–463. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, J.; Shi, K.; Liu, Z. Drug development progress in duchenne muscular dystrophy. Front. Pharmacol. 2022, 13, 950651. [Google Scholar] [CrossRef]
- Pascual-Morena, C.; Lucerón-Lucas-Torres, M.; Martínez-García, I.; Rodríguez-Gutiérrez, E.; Patiño-Cardona, S.; Sequí-Domínguez, I. Efficacy and Safety of Vamorolone in Duchenne Muscular Dystrophy: A Systematic Review. Pediatr. Drugs 2024, 26, 695–707. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. Histone deacetylase inhibition with givinostat: A multi-targeted mode of action with the potential to halt the pathological cascade of Duchenne muscular dystrophy. Front. Cell Dev. Biol. 2025, 12, 1514898. [Google Scholar] [CrossRef]
- Wasala, N.B.; Chen, S.J.; Duan, D. Duchenne muscular dystrophy animal models for high-throughput drug discovery and precision medicine. Expert Opin. Drug Discov. 2020, 15, 443–456. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Models Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Boccanegra, B.; Mantuano, P.; Conte, E.; Cerchiara, A.G.; Tulimiero, L.; Quarta, R.; Caputo, E.; Sanarica, F.; Forino, M.; Spadotto, V.; et al. LKB1 signaling is altered in skeletal muscle of a Duchenne muscular dystrophy mouse model. Dis. Models Mech. 2023, 16, dmm049930. [Google Scholar] [CrossRef] [PubMed]
- Camerino, G.M.; Cannone, M.; Giustino, A.; Massari, A.M.; Capogrosso, R.F.; Cozzoli, A.; De Luca, A. Gene expression in mdx mouse muscle in relation to age and exercise: Aberrant mechanical-metabolic coupling and implications for pre-clinical studies in Duchenne muscular dystrophy. Hum. Mol. Genet. 2014, 23, 5720–5732. [Google Scholar] [CrossRef]
- Capogrosso, R.F.; Mantuano, P.; Cozzoli, A.; Sanarica, F.; Massari, A.M.; Conte, E.; Fonzino, A.; Giustino, A.; Rolland, J.-F.; Quaranta, A.; et al. Contractile efficiency of dystrophic mdx mouse muscle: In vivo and ex vivo assessment of adaptation to exercise of functional end points. J. Appl. Physiol. 2017, 122, 828–843. [Google Scholar] [CrossRef]
- Jackson, E.L.; Lu, H. Three-dimensional models for studying development and disease: Moving on from organisms to organs-on-a-chip and organoids. Integr. Biol. 2016, 8, 672–683. [Google Scholar] [CrossRef]
- Khodabukus, A. Tissue-Engineered Skeletal Muscle Models to Study Muscle Function, Plasticity, and Disease. Front. Physiol. 2021, 12, 619710. [Google Scholar] [CrossRef]
- Young, J.; Margaron, Y.; Fernandes, M.; Duchemin-Pelletier, E.; Michaud, J.; Flaender, M.; Lorintiu, O.; Degot, S.; Poydenot, P. MyoScreen, a High-Throughput Phenotypic Screening Platform Enabling Muscle Drug Discovery. Slas Discov. 2018, 23, 790–806. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Leenaars, M.; Ritskes-Hoitinga, M. A gold standard publication checklist to improve the quality of animal studies, to fully integrate the Three Rs, and to make systematic reviews more feasible. Altern. Lab. Anim. 2010, 38, 167–182. [Google Scholar] [CrossRef]
- Robinson, N.B.; Krieger, K.; Khan, F.M.; Huffman, W.; Chang, M.; Naik, A.; Yongle, R.; Hameed, I.; Krieger, K.; Girardi, L.N.; et al. The current state of animal models in research: A review. Int. J. Surg. 2019, 72, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.; Estrada Chavez, B.; Sonam, S.; Nguyen, T.; Grenci, G.; Bigot, A.; Muchir, A.; Ladoux, B.; Cadot, B.; Le Grand, F.; et al. Bioengineering a miniaturized in vitro 3D myotube contraction monitoring chip to model muscular dystrophies. Biomaterials 2023, 293, 121935. [Google Scholar] [CrossRef]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Lad, H.; Fusto, A.; Tiper, Y.; Datye, A.; Nguyen, C.T.; Jacques, E.; Moyle, L.A.; Nguyen, T.; Musgrave, B.; et al. De novo revertant fiber formation and therapy testing in a 3D culture model of Duchenne muscular dystrophy skeletal muscle. Acta Biomater. 2021, 132, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Tejedera-Villafranca, A.; Montolio, M.; Ramón-Azcón, J.; Fernández-Costa, J.M. Mimicking sarcolemmal damagein vitro: A contractile 3D model of skeletal muscle for drug testing in Duchenne muscular dystrophy. Biofabrication 2023, 15, 045024. [Google Scholar] [CrossRef] [PubMed]
- Nesmith, A.P.; Wagner, M.A.; Pasqualini, F.S.; O’Connor, B.B.; Pincus, M.J.; August, P.R.; Parker, K.K. A human in vitro model of Duchenne muscular dystrophy muscle formation and contractility. J. Cell Biol. 2016, 215, 47–56. [Google Scholar] [CrossRef]
- In ‘t Groen, S.L.M.; Franken, M.; Bock, T.; Krüger, M.; de Greef, J.C.; Pijnappel, W.W.M.P. A knock down strategy for rapid, generic, and versatile modelling of muscular dystrophies in 3D-tissue-engineered-skeletal muscle. Skelet Muscle 2024, 14, 3. [Google Scholar] [CrossRef]
- Komar, Z.M.; van Gent, D.C.; Chakrabarty, S. Establishing a microfluidic tumor slice culture platform to study drug response. Curr. Protoc. 2023, 3, e693. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Quiros-Solano, W.F.; Kuijten, M.M.P.; Haspels, B.; Mallya, S.; Lo, C.S.Y.; Othman, A.; Silvestri, C.; van de Stolpe, A.; Gaio, N.; et al. A Microfluidic Cancer-on-Chip Platform Predicts Drug Response Using Organotypic Tumor Slice Culture. Cancer Res. 2022, 82, 510–520. [Google Scholar] [CrossRef]
- Bas-Cristóbal Menéndez, A.; Du, Z.; van den Bosch, T.P.P.; Othman, A.; Gaio, N.; Silvestri, C.; Quirós, W.; Lin, H.; Korevaar, S.; Merino, A.; et al. Creating a kidney organoid-vasculature interaction model using a novel organ-on-chip system. Sci. Rep. 2022, 12, 20699. [Google Scholar] [CrossRef]
- Sala, L.; van Meer, B.J.; Tertoolen, L.G.J.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.; Dieben, M.A.E.; Eschenhagen, T.; Giacomelli, E.; et al. MUSCLEMOTION: A Versatile Open Software Tool to Quantify Cardiomyocyte and Cardiac Muscle Contraction In Vitro and In Vivo. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef]
- Rivera-Arbeláez, J.M.; Keekstra, D.; Cofiño-Fabres, C.; Boonen, T.; Dostanic, M.; Ten Den, S.A.; Vermeul, K.; Mastrangeli, M.; van den Berg, A.; Segerink, L.I.; et al. Automated assessment of human engineered heart tissues using deep learning and template matching for segmentation and tracking. Bioeng. Transl. Med. 2023, 8, e10513. [Google Scholar] [CrossRef]
- Patel, L.; Roy, A.; Barlow, J.; O’Shea, C.; Nieves, D.; Azad, A.J.; Hall, C.; Davies, B.; Rath, P.; Pavlovic, D.; et al. Characterisation of infantile cardiomyopathy in Alström syndrome using ALMS1 knockout induced pluripotent stem cell derived cardiomyocyte model. Mol. Genet. Metab. 2024, 143, 108575. [Google Scholar] [CrossRef]
- Vesga-Castro, C.; Aldazabal, J.; Vallejo-Illarramendi, A.; Paredes, J. Contractile force assessment methods for in vitro skeletal muscle tissues. eLife 2022, 11, e77204. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Costa, J.M.; Ortega, M.A.; Rodríguez-Comas, J.; Lopez-Muñoz, G.; Yeste, J.; Mangas-Florencio, L.; Fernández-González, M.; Martin-Lasierra, E.; Tejedera-Villafranca, A.; Ramon-Azcon, J. Training-on-a-Chip: A Multi-Organ Device to Study the Effect of Muscle Exercise on Insulin Secretion in Vitro. Adv. Mater. Technol. 2022, 8, 2200873. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fernández-Garibay, X.; Castaño, A.G.; De Chiara, F.; Hernández-Albors, A.; Balaguer-Trias, J.; Ramón-Azcón, J. Muscle-on-a-chip with an on-site multiplexed biosensing system for in situ monitoring of secreted IL-6 and TNF-α. Lab Chip 2019, 19, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Sitzia, C.; Cassinelli, L.; Colleoni, F.; Parolini, D.; Giovanella, U.; Maciotta, S.; Colombo, A.; Meregalli, M.; Torrente, Y. Inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ signaling mediates delayed myogenesis in Duchenne muscular dystrophy fetal muscle. Development 2016, 143, 658–669. [Google Scholar] [CrossRef]
- Bond, L.M.; Tumbarello, D.A.; Kendrick-Jones, J.; Buss, F. Small-molecule inhibitors of myosin proteins. Future Med. Chem. 2013, 5, 41–52. [Google Scholar] [CrossRef]
- Fernández-Costa, J.M.; Fernández-Garibay, X.; Velasco-Mallorquí, F.; Ramón-Azcón, J. Bioengineered in vitro skeletal muscles as new tools for muscular dystrophies preclinical studies. J. Tissue Eng. 2021, 12, 2041731420981339. [Google Scholar] [CrossRef]
- Kodippili, K.; Rudnicki, M.A. Satellite cell contribution to disease pathology in Duchenne muscular dystrophy. Front. Physiol. 2023, 14, 1180980. [Google Scholar] [CrossRef]
- Howard, Z.M.; Lowe, J.; Blatnik, A.J., 3rd; Roberts, D.; Burghes, A.H.; Bansal, S.S.; Rafael-Fortney, J.A. Early Inflammation in Muscular Dystrophy Differs between Limb and Respiratory Muscles and Increases with Dystrophic Severity. Am. J. Pathol. 2021, 191, 730–747. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Models Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef] [PubMed]
- Cerchiara, A.G.; Imbrici, P.; Quarta, R.; Cristiano, E.; Boccanegra, B.; Caputo, E.; Boccanegra, B.; Caputo, E.; Wells, D.J.; Cappellari, O.; et al. Ion channels as biomarkers of altered myogenesis in myofiber precursors of Duchenne muscular dystrophy. Ann. N. Y. Acad. Sci. 2024, 1534, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, A.; Haalstra, M.; Raghuraman, R.; Bielawski, K.; Bholasing, A.P.; van der Wal, E.; de Greef, J.C.; Pim Pijnappel, W.W.M. Real-time and Multichannel Measurement of Contractility of hiPSC-Derived 3D Skeletal Muscle using Fiber Optics-Based Sensing. Adv. Mater. Technol. 2023, 8, 2300845. [Google Scholar] [CrossRef]
- Agrawal, G.; Aung, A.; Varghese, S. Skeletal muscle-on-a-chip: An in vitro model to evaluate tissue formation and injury. Lab Chip 2017, 17, 3447–3461. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution | Final Concentration | |
|---|---|---|
| Mix A | X µL Cell suspension | |
| 0.10 µL Aprotinin (25 µg/mL in water) | 0.20 µg/mg fibrinogen | |
| 0.06 µL Thrombin (100 U/mL in 0.1% BSA) | 0.5 U/mg fibrinogen | |
| Mix B | 0.60 µL Geltrex | 20% v/v |
| 0.60 µL Fibrinogen (20 mg/mL in 0.9% NaCl) | 4 mg/mL |
| Gene Symbol | Gene Full Name | Unique Assay ID |
|---|---|---|
| MYH2 | Myosin, heavy chain 2 | qHsaCID0012805 |
| MYH7 | Myosin, heavy chain 7 | qHsaCID0011217 |
| MYH3 | Embryonic myosin heavy chain 3 | qHsaCID0015273 |
| MYOG | Myogenin (myogenic factor 4) | qHsaCED0043933 |
| DMD | Dystrophin | qHsaCID0010707 |
| RYR1 | Ryanodine receptor 1 (skeletal) | qHsaCID0016836 |
| RPS18 | Ribosomal Protein S18 (reference gene) | qHsaCED0037454 |
| UBC | Ubiquitin C (reference gene) | qHsaCED0023867 |
| ITPR3 | Inositol 1,4,5-Trisphosphate Receptor Type 3 | qHsaCID0020985 |
| ATP2A2 | Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase 2 | qHsaCID0011088 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quarta, R.; Cristiano, E.; Han, M.K.L.; Boccanegra, B.; Marinelli, M.; Gaio, N.; Ohana, J.; Mouly, V.; Cappellari, O.; De Luca, A. Patient-Oriented In Vitro Studies in Duchenne Muscular Dystrophy: Validation of a 3D Skeletal Muscle Organoid Platform. Biomedicines 2025, 13, 1109. https://doi.org/10.3390/biomedicines13051109
Quarta R, Cristiano E, Han MKL, Boccanegra B, Marinelli M, Gaio N, Ohana J, Mouly V, Cappellari O, De Luca A. Patient-Oriented In Vitro Studies in Duchenne Muscular Dystrophy: Validation of a 3D Skeletal Muscle Organoid Platform. Biomedicines. 2025; 13(5):1109. https://doi.org/10.3390/biomedicines13051109
Chicago/Turabian StyleQuarta, Raffaella, Enrica Cristiano, Mitchell K. L. Han, Brigida Boccanegra, Manuel Marinelli, Nikolas Gaio, Jessica Ohana, Vincent Mouly, Ornella Cappellari, and Annamaria De Luca. 2025. "Patient-Oriented In Vitro Studies in Duchenne Muscular Dystrophy: Validation of a 3D Skeletal Muscle Organoid Platform" Biomedicines 13, no. 5: 1109. https://doi.org/10.3390/biomedicines13051109
APA StyleQuarta, R., Cristiano, E., Han, M. K. L., Boccanegra, B., Marinelli, M., Gaio, N., Ohana, J., Mouly, V., Cappellari, O., & De Luca, A. (2025). Patient-Oriented In Vitro Studies in Duchenne Muscular Dystrophy: Validation of a 3D Skeletal Muscle Organoid Platform. Biomedicines, 13(5), 1109. https://doi.org/10.3390/biomedicines13051109