

Multi-Omics-Based Analysis of the Effect of Longevity Genes on the Immune Relevance of Colorectal Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Data Download
2.2. Comprehensive Analysis of Longevity-Related Genes in the TCGA Database
2.3. Consensus Clustering Classification
2.4. Construction of the Prognostic Model
2.5. Immune, Mismatch Repair and Stem Cell Index Analysis
2.6. Statistical Analysis
3. Results
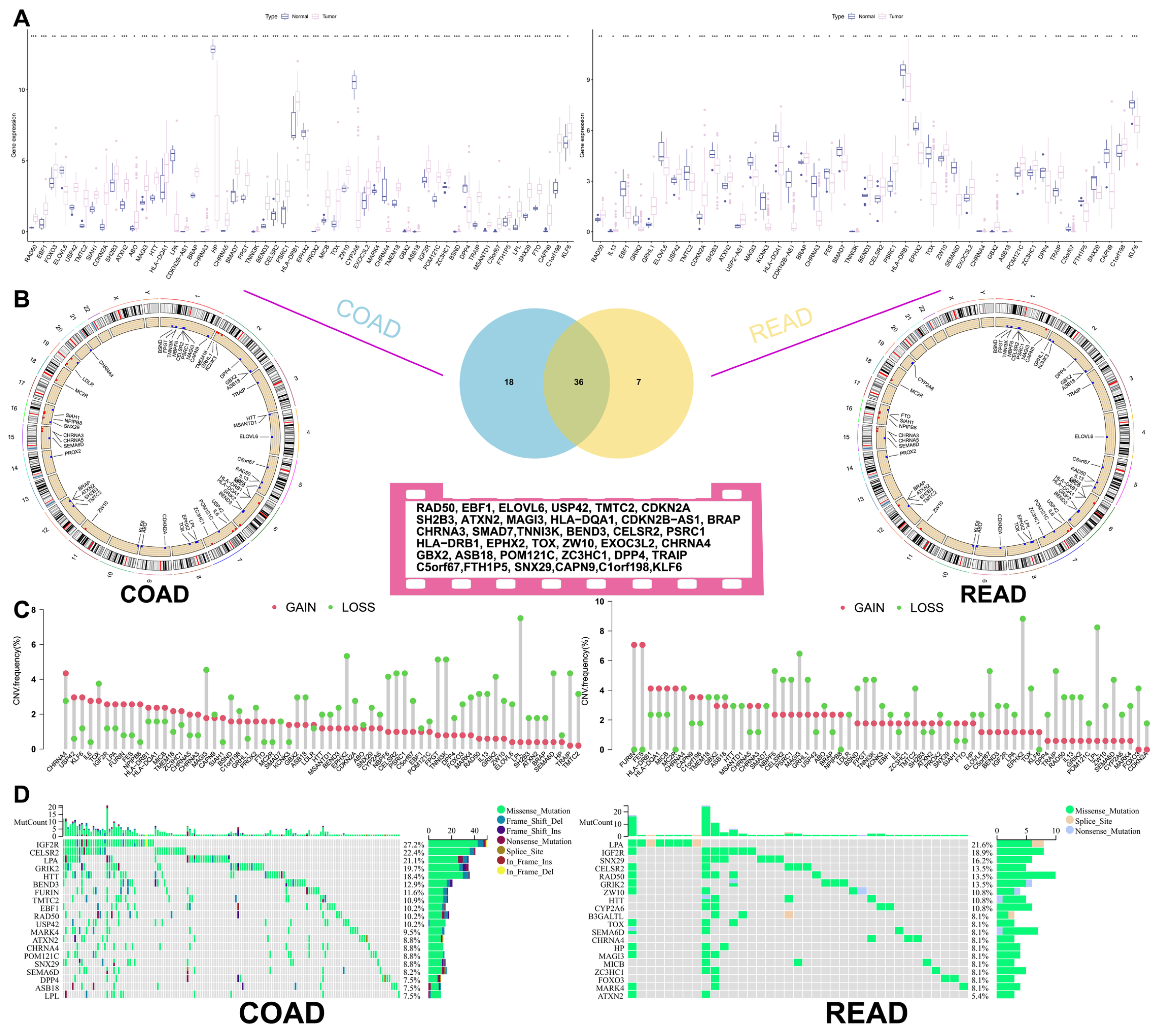
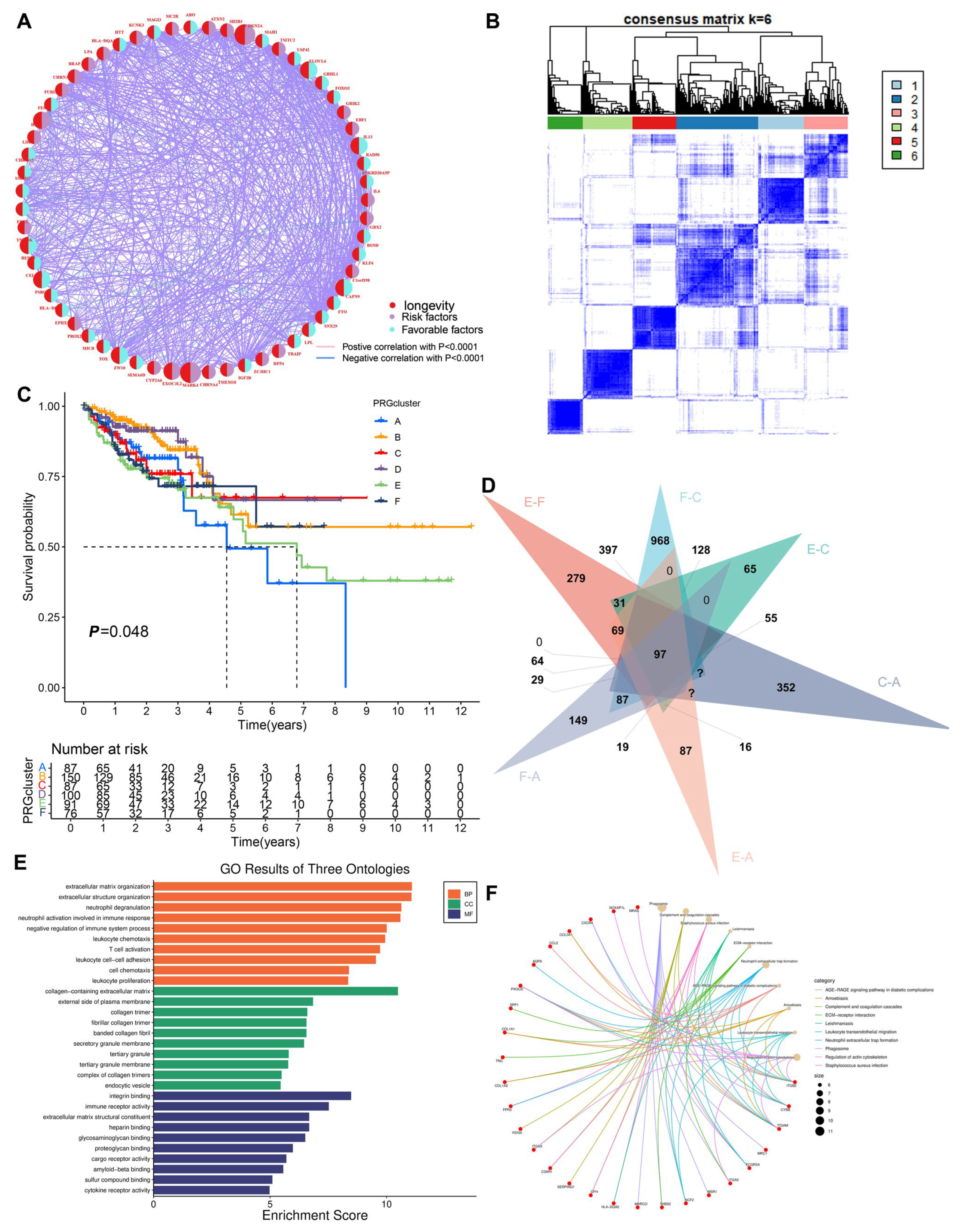
3.1. Correlation Analysis of Longevity-Related Genes in the TCGA Database
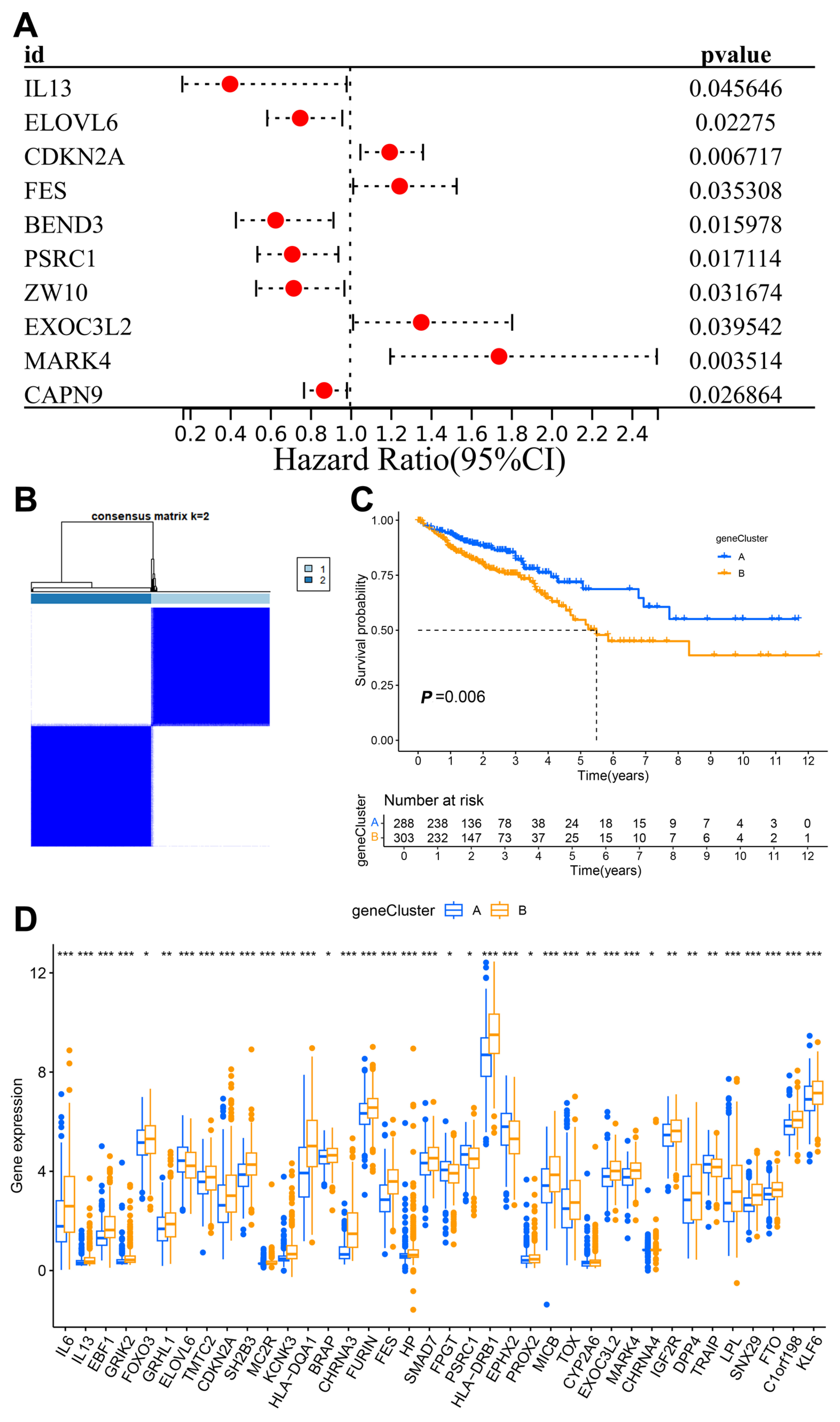
3.2. Construction of the Clustering Model
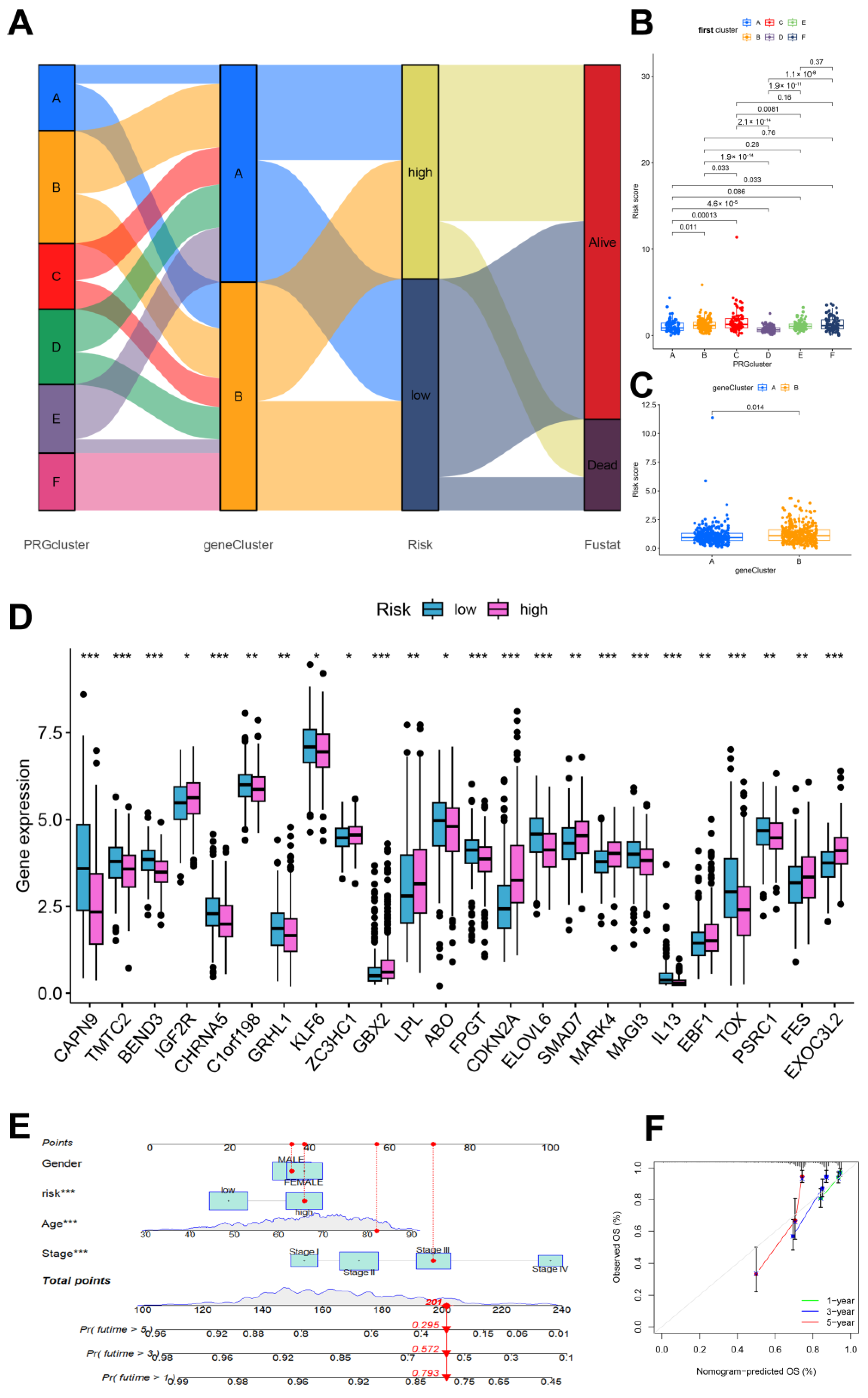
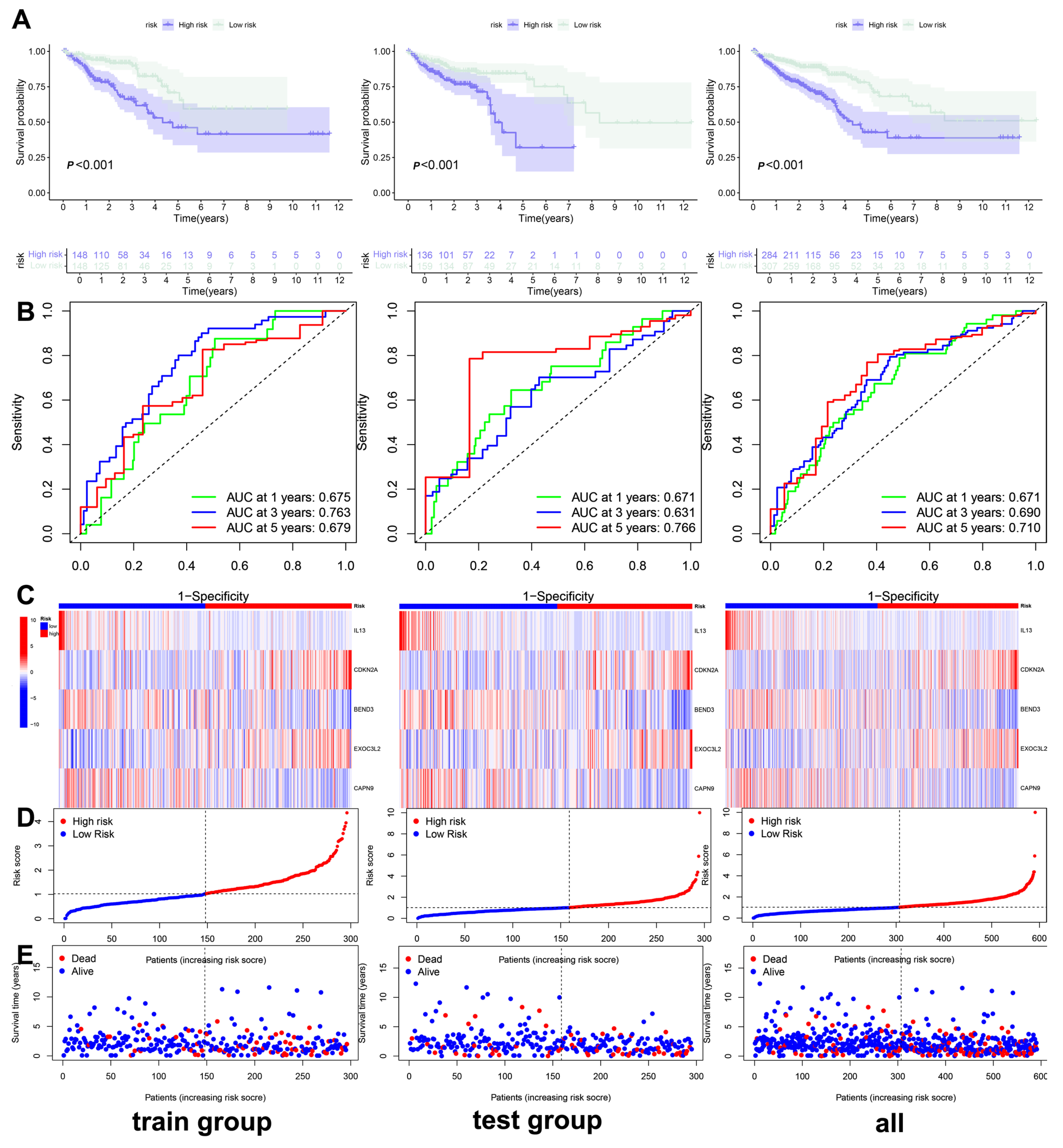
3.3. Construction and Validation of the Clinical Prediction Models
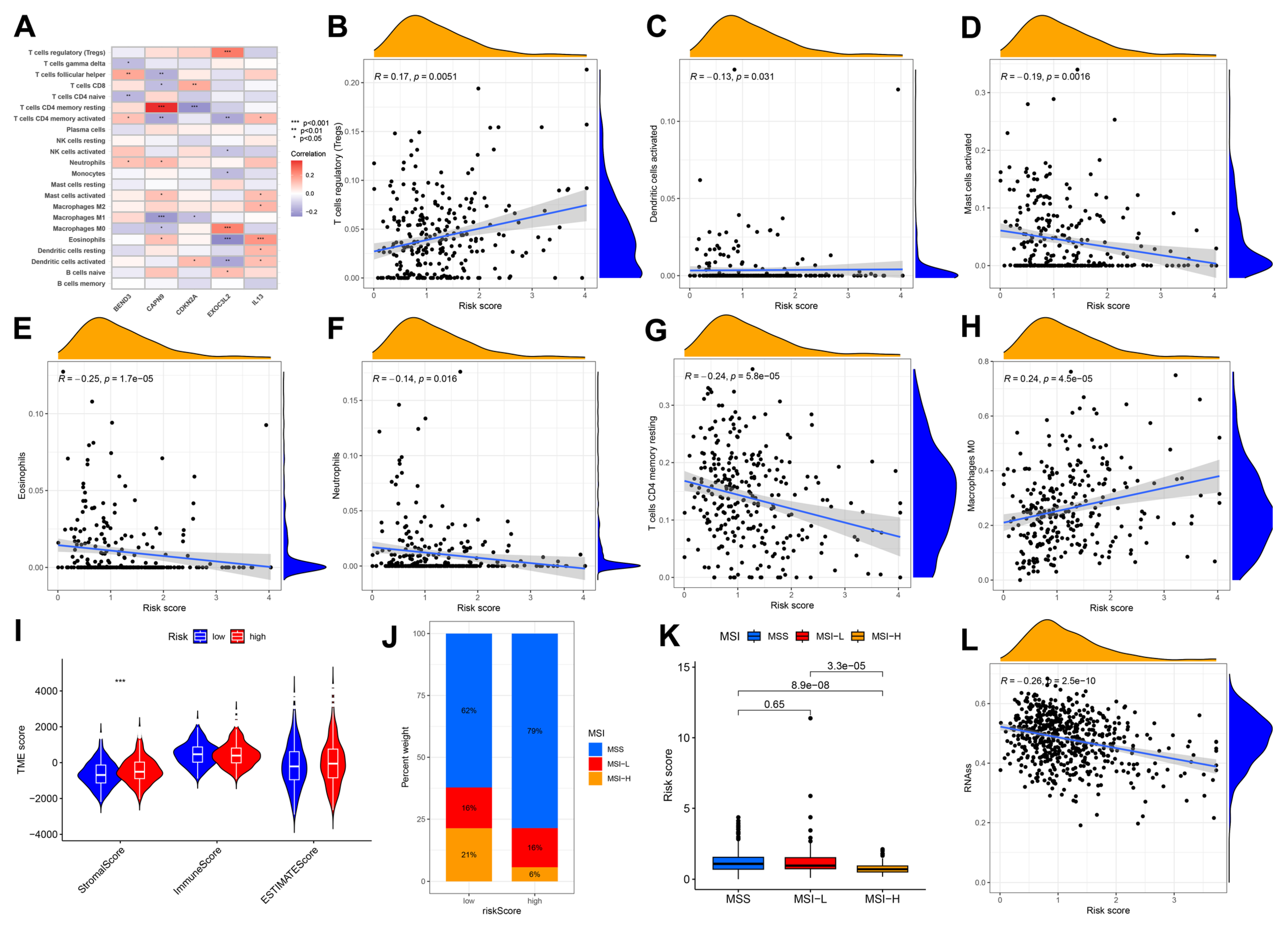
3.4. Immune Cell and Mismatch Repair Assays
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | colorectal cancer |
| LAGs | longevity-associated genes |
| TME | tumor microenvironment |
| CAR | chimeric antigen receptor |
| PCR | Polymerase Chain Reaction |
| GWAS | Genome-Wide Association Studies |
| PCA | principal component analysis |
| DEGs | differentially expressed genes |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LASSO | minimum absolute shrinkage and selection operator |
| AUC | area under the curve |
| OS | overall survival |
| IL-13 | Interleukin 13 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.S.; Sun, K.; Zhang, Z.; Zeng, H.; Zou, S.; Chen, R.; Gu, X.; Wei, W.; He, J. Cancer incidence and mortality in China, 2022. Zhonghua Zhong Liu Za Zhi 2024, 46, 221–231. [Google Scholar] [CrossRef]
- Müller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Völkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-Cell Therapy in Autoimmune Disease—A Case Series with Follow-up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gou, R.; Chen, J.; Wang, Q.; Li, X.; Chang, J.; Chen, H.; Wang, X.; Wan, G. Catalase-positive Staphylococcus epidermidis based cryo-millineedle platform facilitates the photo-immunotherapy against colorectal cancer via hypoxia improvement. J. Colloid Interface Sci. 2024, 676, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yang, L.; Chen, G.; Xu, F.; Yang, F.; Yu, H.; Li, L.; Dong, X.; Han, J.; Cao, C.; et al. A Review on Drug Delivery System for Tumor Therapy. Front. Pharmacol. 2021, 12, 735446. [Google Scholar] [CrossRef]
- Beekman, M.; Nederstigt, C.; Suchiman, H.E.D.; Kremer, D.; van der Breggen, R.; Lakenberg, N.; Alemayehu, W.G.; de Craen, A.J.M.; Westendorp, R.G.J.; Boomsma, D.I.; et al. Genome-wide association study (GWAS)-identified disease risk alleles do not compromise human longevity. Proc. Natl. Acad. Sci. USA 2010, 107, 18046–18049. [Google Scholar] [CrossRef]
- Vincze, O.; Colchero, F.; Lemaître, J.-F.; Conde, D.A.; Pavard, S.; Bieuville, M.; Urrutia, A.O.; Ujvari, B.; Boddy, A.M.; Maley, C.C.; et al. Cancer risk across mammals. Nature 2022, 601, 263–267. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Zhang, Z.; Gladyshev, V.N.; Vijg, J. Comparative genetics of longevity and cancer: Insights from long-lived rodents. Nat. Rev. Genet. 2014, 15, 531–540. [Google Scholar] [CrossRef]
- Zhang, Y.; Sheng, C.; Fan, Z.; Liu, Y.; Liu, X.; Duan, H.; Dai, H.; Lyu, Z.; Yang, L.; Song, F.; et al. Risk-stratified screening and colorectal cancer incidence and mortality: A retrospective study from the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Prev. Med. 2024, 187, 108117. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A. The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia. Int. J. Mol. Sci. 2021, 22, 1565. [Google Scholar] [CrossRef]
- Hissong, E.; Crowe, E.P.; Yantiss, R.K.; Chen, Y.-T. Assessing colorectal cancer mismatch repair status in the modern era: A survey of current practices and re-evaluation of the role of microsatellite instability testing. Mod. Pathol. 2018, 31, 1756–1766. [Google Scholar] [CrossRef]
- Deelen, J.; Evans, D.S.; Arking, D.E.; Tesi, N.; Nygaard, M.; Liu, X.; Wojczynski, M.K.; Biggs, M.L.; van der Spek, A.; Atzmon, G.; et al. A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat. Commun. 2019, 10, 3669. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Ye, J.; Anderson, K.M.; Roychoudhury, S.; Rubin, E.H.; Halabi, S.; Chappell, R.J. Log-Rank Test vs MaxCombo and Difference in Restricted Mean Survival Time Tests for Comparing Survival Under Nonproportional Hazards in Immuno-oncology Trials: A Systematic Review and Meta-analysis. JAMA Oncol. 2022, 8, 1294–1300. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Choi, Y.J.; Kim, I.-K.; Lee, H.S.; Kim, H.; Baik, S.H.; Kim, N.K.; Lee, K.Y. LASSO-Based Machine Learning Algorithm for Prediction of Lymph Node Metastasis in T1 Colorectal Cancer. Cancer Res. Treat. 2021, 53, 773–783. [Google Scholar] [CrossRef]
- Pan, W.; Huang, W.; Zheng, J.; Meng, Z.; Pan, X. Construction of a prognosis model of head and neck squamous cell carcinoma pyroptosis and an analysis of immuno-phenotyping based on bioinformatics. Transl. Cancer Res. 2024, 13, 299–316. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Ruby, J.G.; Wright, K.M.; Rand, K.A.; Kermany, A.; Noto, K.; Curtis, D.; Varner, N.; Garrigan, D.; Slinkov, D.; Dorfman, I.; et al. Estimates of the Heritability of Human Longevity Are Substantially Inflated due to Assortative Mating. Genetics 2018, 210, 1109–1124. [Google Scholar] [CrossRef]
- Van den Berg, N.; Beekman, M.; Smith, K.R.; Janssens, A.; Slagboom, P.E. Historical demography and longevity genetics: Back to the future. Ageing Res. Rev. 2017, 38, 28–39. [Google Scholar] [CrossRef]
- Tao, M.; Chen, J.; Cui, C.; Xu, Y.; Xu, J.; Shi, Z.; Yun, J.; Zhang, J.; Ou, G.-Z.; Liu, C.; et al. Identification of a longevity gene through evolutionary rate covariation of insect mito-nuclear genomes. Nat. Aging 2024, 4, 1076–1088. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Luo, J.; Yan, Y.; Zhang, Y.; Luo, C.; Li, N.; Zhou, Y.; Wu, D.; Dai, M.; Chen, H. Evaluation of long-term benefits and cost-effectiveness of nation-wide colorectal cancer screening strategies in China in 2020–2060: A modelling analysis. Lancet Reg. Health-West. Pac. 2024, 51, 101172. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Vale, N. Combining repurposed drugs to treat colorectal cancer. Drug Discov. Today 2022, 27, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, L.; Xie, Y.; Wang, F. Bregmannian consensus clustering for cancer subtypes analysis. Comput. Methods Programs Biomed. 2020, 189, 105337. [Google Scholar] [CrossRef]
- Manganaro, L.; Bianco, S.; Bironzo, P.; Cipollini, F.; Colombi, D.; Corà, D.; Corti, G.; Doronzo, G.; Errico, L.; Falco, P.; et al. Consensus clustering methodology to improve molecular stratification of non-small cell lung cancer. Sci. Rep. 2023, 13, 7759. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Wang, F.; Bai, G. Consensus Clustering Analysis Based on Enhanced-CT Radiomic Features: Esophageal Squamous Cell Carcinoma patients’ 3-Year Progression-Free Survival. Acad. Radiol. 2024, 31, 2807–2817. [Google Scholar] [CrossRef]
- Hershey, G.K. IL-13 receptors and signaling pathways: An evolving web. J. Allergy Clin. Immunol. 2003, 111, 677–690; quiz 691. [Google Scholar] [CrossRef]
- Formentini, A.; Braun, P.; Fricke, H.; Link, K.-H.; Henne-Bruns, D.; Kornmann, M. Expression of interleukin-4 and interleukin-13 and their receptors in colorectal cancer. Int. J. Color. Dis. 2012, 27, 1369–1376. [Google Scholar] [CrossRef]
- Saigusa, S.; Tanaka, K.; Inoue, Y.; Toiyama, Y.; Okugawa, Y.; Iwata, T.; Mohri, Y.; Kusunoki, M. Low Serum Interleukin-13 Levels Correlate with Poorer Prognoses for Colorectal Cancer Patients. Int. Surg. 2014, 99, 223–229. [Google Scholar] [CrossRef]
- Bartolomé, R.A.; García-Palmero, I.; Torres, S.; López-Lucendo, M.; Balyasnikova, I.V.; Casal, J.I. IL13 Receptor α2 Signaling Requires a Scaffold Protein, FAM120A, to Activate the FAK and PI3K Pathways in Colon Cancer Metastasis. Cancer Res. 2015, 75, 2434–2444. [Google Scholar] [CrossRef]
- Matsui, S.; Okabayashi, K.; Tsuruta, M.; Shigegta, K.; Seishima, R.; Ishida, T.; Kondo, T.; Suzuki, Y.; Hasegawa, H.; Shimoda, M.; et al. Interleukin-13 and its signaling pathway is associated with obesity-related colorectal tumorigenesis. Cancer Sci. 2019, 110, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Liggett, W.H.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Patil, V.; Liu, J.; Dogan, H.; Tabatabai, G.; Yefet, L.S.; Behling, F.; Hoffman, E.; Bunda, S.; Yakubov, R.; et al. Increased mRNA expression of CDKN2A is a transcriptomic marker of clinically aggressive meningiomas. Acta Neuropathol. 2023, 146, 145–162. [Google Scholar] [CrossRef]
- Xing, X.; Cai, W.; Shi, H.; Wang, Y.; Li, M.; Jiao, J.; Chen, M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: A meta-analysis. Br. J. Cancer 2013, 108, 2542–2548. [Google Scholar] [CrossRef]
- Berg, M.; Nordgaard, O.; Kørner, H.; Oltedal, S.; Smaaland, R.; Søreide, J.A.; Søreide, K. Molecular Subtypes in Stage II-III Colon Cancer Defined by Genomic Instability: Early Recurrence-Risk Associated with a High Copy-Number Variation and Loss of RUNX3 and CDKN2A. PLoS ONE 2015, 10, e0122391. [Google Scholar] [CrossRef] [PubMed]
- Kurniawan, F.; Chetlangia, N.; Kamran, M.; Redon, C.E.; Pongor, L.; Sun, Q.; Lin, Y.-C.; Mohan, V.; Shaqildi, O.; Asoudegi, D.; et al. BEND3 safeguards pluripotency by repressing differentiation-associated genes. Proc. Natl. Acad. Sci. USA 2022, 119, e2107406119. [Google Scholar] [CrossRef]
- Zhao, Y.; Song, X.; Song, X.; Xie, L. Identification of Diagnostic Exosomal LncRNA-miRNA-mRNA Biomarkers in Colorectal Cancer Based on the ceRNA Network. Pathol. Oncol. Res. 2022, 28, 1610493. [Google Scholar] [CrossRef]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide Analysis of Genetic Loci Associated with Alzheimer Disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef]
- Logan, J.G.; Yun, S.; Bao, Y.; Farber, E.; Farber, C.R. RNA-sequencing analysis of differential gene expression associated with arterial stiffness. Vascular 2020, 28, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Wang, S.; Guo, L.; Liu, L.; Liu, R.; Li, Q.; Zheng, M.; Zhao, G.; Wen, J. Expression and effect of Calpain9 gene genetic polymorphism on slaughter indicators and intramuscular fat content in chickens. Poult. Sci. 2018, 97, 3414–3420. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, G.; Liu, S.; Wang, H.; Chen, B. MiR-585-3p suppresses tumor proliferation and migration by directly targeting CAPN9 in high grade serous ovarian cancer. J. Ovarian Res. 2021, 14, 90. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Min, G.; Wang, H.; Jiang, L. Multi-Omics-Based Analysis of the Effect of Longevity Genes on the Immune Relevance of Colorectal Cancer. Biomedicines 2025, 13, 1085. https://doi.org/10.3390/biomedicines13051085
Huang Y, Min G, Wang H, Jiang L. Multi-Omics-Based Analysis of the Effect of Longevity Genes on the Immune Relevance of Colorectal Cancer. Biomedicines. 2025; 13(5):1085. https://doi.org/10.3390/biomedicines13051085
Chicago/Turabian StyleHuang, Yichu, Guangtao Min, Hongpeng Wang, and Lei Jiang. 2025. "Multi-Omics-Based Analysis of the Effect of Longevity Genes on the Immune Relevance of Colorectal Cancer" Biomedicines 13, no. 5: 1085. https://doi.org/10.3390/biomedicines13051085
APA StyleHuang, Y., Min, G., Wang, H., & Jiang, L. (2025). Multi-Omics-Based Analysis of the Effect of Longevity Genes on the Immune Relevance of Colorectal Cancer. Biomedicines, 13(5), 1085. https://doi.org/10.3390/biomedicines13051085