

Clinical Molecular Immunohistochemistry Mismatch Repair Mutations in Lynch Syndrome in Patients Under 50 Years: A Systematic Review

 ,
,
Abstract
1. Introduction
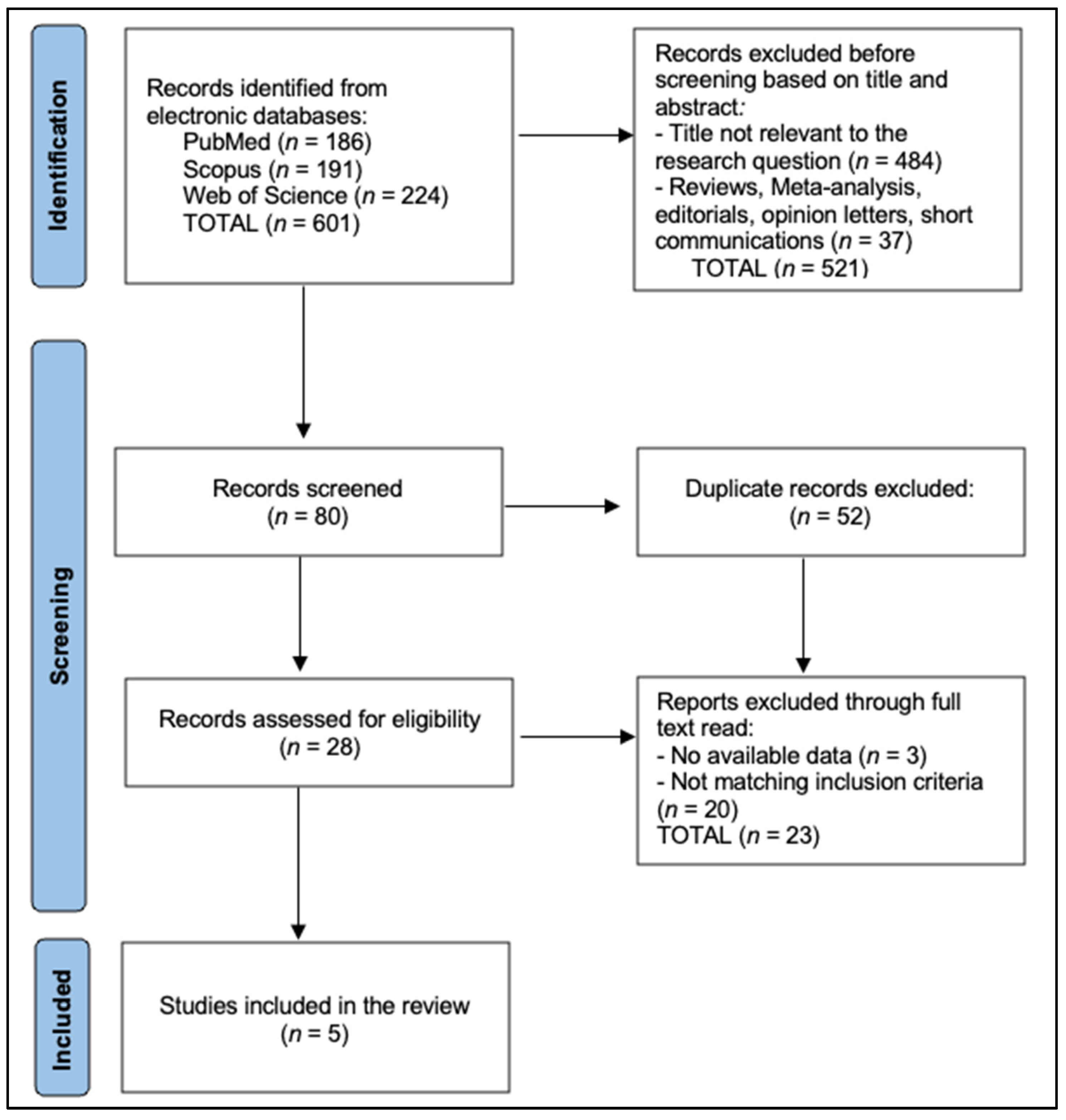
2. Materials and Methods
2.1. Eligibility Criteria and Research Question
2.2. Literature Search Strategy
2.3. Data Extraction and Outcomes of Interest
2.4. Quality Assessment and Risk of Bias
3. Results
4. Discussion
4.1. Summary of Evidence
4.2. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Shaukat, A.; Kahi, C.J.; Burke, C.A.; Rabeneck, L.; Sauer, B.G.; Rex, D.K. ACG Clinical Guidelines: Colorectal Cancer Screening 2021. Am. J. Gastroenterol. 2021, 116, 458–479. [Google Scholar] [CrossRef]
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New Clinical Criteria for Hereditary Nonpolyposis Colorectal Cancer (HNPCC, Lynch Syndrome) Proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on Genetic Evaluation and Management of Lynch Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef]
- Lipton, L.R.; Johnson, V.; Cummings, C.; Fisher, S.; Risby, P.; Eftekhar Sadat, A.T.; Cranston, T.; Izatt, L.; Sasieni, P.; Hodgson, S.V.; et al. Refining the Amsterdam Criteria and Bethesda Guidelines: Testing algorithms for the prediction of mismatch repair mutation status in the familial cancer clinic. J. Clin. Oncol. 2004, 22, 4934–4943. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; Chapelle, A.D.L.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Leslie, S.W.; McHugh, T.W. Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer). In StatPearls; StatPearls Publishing: Treasure Island, Finland, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK431096/ (accessed on 3 March 2025).
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic Predisposition to Colorectal Cancer: Syndromes, Genes, Classification of Genetic Variants and Implications for Precision Medicine. J. Pathol. 2019, 247, 574–588. [Google Scholar] [CrossRef]
- de Vos tot Nederveen Cappel, W.H.; Buskens, E.; van Duijvendijk, P.; Cats, A.; Menko, F.H.; Griffioen, G.; Slors, J.F.; Nagengast, F.M.; Kleibeuker, J.H.; Vasen, H.F. Decision analysis in the surgical treatment of colorectal cancer due to a mismatch repair gene defect. Gut 2003, 52, 1752–1755. [Google Scholar] [CrossRef]
- Engel, C.; Loeffler, M.; Steinke, V.; Rahner, N.; Holinski-Feder, E.; Dietmaier, W.; Schackert, H.K.; Goergens, H.; Doeberitz, M.v.K.; Goecke, T.O.; et al. Risks of Less Common Cancers in Proven Mutation Carriers with Lynch Syndrome. J. Clin. Oncol. 2012, 30, 4409–4415. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer Risks by Gene, Age, and Gender in 6350 Carriers of Pathogenic Mismatch Repair Variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef]
- Stoffel, E.M.; Murphy, C.C. Epidemiology and Mechanisms of the Increasing Incidence of Colon and Rectal Cancers in Young Adults. Gastroenterology 2020, 158, 341–353. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Barrow, E.; Hill, J.; Evans, D.G. Cancer risk in Lynch Syndrome. Fam. Cancer 2013, 12, 229–240. [Google Scholar] [CrossRef]
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J.; et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 404–412. [Google Scholar] [CrossRef]
- Vilar, E.; Tabernero, J. Molecular dissection of microsatellite instable colorectal cancer. Cancer Discov. 2013, 3, 502–511. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H. Screening for the Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, D.D.; Tan, Y.Y.; Walsh, M.D.; Clendenning, M.; Metcalf, A.M.; Ferguson, K.; Arnold, S.T.; Thompson, B.A.; Lose, F.A.; Parsons, M.T.; et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J. Clin. Oncol. 2014, 32, 90–100. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moreira, L.; Balaguer, F.; Lindor, N.; De La Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch Syndrome Among Patients with Colorectal Cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316, Erratum in Gut 2020, 69, e4. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- South, C.D.; Yearsley, M.; Martin, E.; Arnold, M.; Frankel, W.; Hampel, H. Immunohistochemistry staining for the mismatch repair proteins in the clinical care of patients with colorectal cancer. Genet. Med. 2009, 11, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Barzi, A.; Lenz, H.J.; Quinn, D.I.; Sadeghi, S. Comparative effectiveness of screening strategies for colorectal cancer. Cancer 2017, 123, 1516–1527. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vasen, H.F.A.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised Guidelines for the Clinical Management of Lynch Syndrome (HNPCC): Recommendations by a Group of European Experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef]
- Mensenkamp, A.R.; Vogelaar, I.P.; van Zelst–Stams, W.A.; Goossens, M.; Ouchene, H.; Hendriks–Cornelissen, S.J.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J. Somatic Mutations in MLH1 and MSH2 Are a Frequent Cause of Mismatch-Repair Deficiency in Lynch Syndrome–Like Tumors. Gastroenterology 2014, 146, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Niessen, R.C.; Berends, M.J.; Wu, Y.; Sijmons, R.H.; Hollema, H.; Ligtenberg, M.J.; de Walle, H.E.; de Vries, E.G.; Karrenbeld, A.; Buys, C.H.; et al. Identification of mismatch repair gene mutations in young patients with colorectal cancer and in patients with multiple tumours associated with hereditary non-polyposis colorectal cancer. Gut 2006, 55, 1781–1788. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Steinhagen, E.; Shia, J.; Markowitz, A.J.; Stadler, Z.K.; Salo-Mullen, E.E.; Zheng, J.; Lee-Kong, S.A.; Nash, G.M.; Offit, K.; Guillem, J.G. Systematic Immunohistochemistry Screening for Lynch Syndrome in Early Age-of-Onset Colorectal Cancer Patients Undergoing Surgical Resection. J. Am. Coll. Surg. 2012, 214, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, O.; Eguchi, H.; Chika, N.; Sakimoto, T.; Ishibashi, K.; Kumamoto, K.; Tamaru, J.-I.; Tachikawa, T.; Akagi, K.; Arai, T.; et al. Prevalence and Clinicopathologic/Molecular Characteristics of Mismatch Repair-Deficient Colorectal Cancer in the Under-50-Year-Old Japanese Population. Surg. Today 2017, 47, 1135–1143. [Google Scholar] [CrossRef]
- Wright, D.M.; Arnold, J.L.; Parry, B.; Hulme-Moir, M.; Winship, I.M.; Parry, S. Immunohistochemistry to Detect Hereditary Nonpolyposis Colorectal Cancer in Young Patients: The 7-Year Auckland Experience. Dis. Colon Rectum 2011, 54, 552–558. [Google Scholar] [CrossRef]
- Buchanan, D.D.; Clendenning, M.; Rosty, C.; Eriksen, S.V.; Walsh, M.D.; Walters, R.J.; Thibodeau, S.N.; Stewart, J.; Preston, S.; Win, A.K.; et al. Tumor testing to identify lynch syndrome in two Australian colorectal cancer cohorts. J. Gastroenterol. Hepatol. 2017, 32, 427–438. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Canard, G.; Lefevre, J.H.; Colas, C.; Coulet, F.; Svrcek, M.; Lascols, O.; Hamelin, R.; Shields, C.; Duval, A.; Fléjou, J.F.; et al. Screening for Lynch syndrome in colorectal cancer: Are we doing enough? Ann. Surg. Oncol. 2012, 19, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Castillejo, A.; Hernández-Illán, E.; Rodriguez-Soler, M.; Pérez-Carbonell, L.; Egoavil, C.; Barberá, V.M.; Castillejo, M.-I.; Guarinos, C.; Martínez-De-Dueñas, E.; Juan, M.-J.; et al. Prevalence of MLH1 constitutional epimutations as a cause of Lynch syndrome in unselected versus selected consecutive series of patients with colorectal cancer. J. Med. Genet. 2015, 52, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalli, A.; Carnevali, I.; Sahnane, N.; Leoni, E.; Furlan, D.; Berselli, M.; Sessa, F.; Tibiletti, M. Universal screening to identify Lynch syndrome: Two years of experience in a Northern Italian Center. Eur. J. Cancer Prev. 2020, 29, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Cavazza, A.; Radia, C.; Harlow, C.; Monahan, K.J. Experience of the implementation and outcomes of universal testing for Lynch syndrome in the United Kingdom. Colorectal Dis. 2019, 21, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Chika, N.; Eguchi, H.; Kumamoto, K.; Suzuki, O.; Ishibashi, K.; Tachikawa, T.; Akagi, K.; Tamaru, J.-I.; Okazaki, Y.; Ishida, H. Prevalence of Lynch syndrome and Lynch-like syndrome among patients with colorectal cancer in a Japanese hospital-based population. Jpn. J. Clin. Oncol. 2017, 47, 108–117, Erratum in Jpn. J. Clin. Oncol. 2017, 47, 191. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Study | First Author (Year) | Country | Study Period | Number of Patients (CRC < 50 Years) | MMR Screening Method | Additional Molecular Tests (BRAF/MLH1 Methylation) | Germline Testing Protocol |
|---|---|---|---|---|---|---|---|
| Study 1 [24] | Mensenkamp (2013) | Netherlands | 1997–2011 | 25 | IHC + MSI in MSI+ tumors | BRAF or MLH1 tests | Sanger + IonTorrent seq |
| Study 2 [25] | Niessen (2006) | Netherlands | 1996–2000 | 281 | IHC for MLH1, MSH2, MSH6, etc. | BRAF | Denaturing gradient + MLPA |
| Study 3 [26] | Steinhagen (2012) | USA | 2006–2010 | 198 | IHC for 4 MMR proteins | MLH1 methylation in MLH1/PMS2 loss | Standard germline panel |
| Study 4 [27] | Suzuki (2017) | Japan | 1996–2015 | 119 | IHC for 4 MMR proteins | BRAF V600E and MLH1 promoter if MLH1/PMS2 loss | Sanger + MLPA if needed |
| Study 5 [28] | Wright (2011) | New Zealand | 2001–2007 | 243 | IHC for 2–4 proteins (evolved) | BRAF or Methylation | Sanger for relevant genes |
| Study | dMMR Prevalence (%, 95% CI) | Confirmed LS Among Total, n (%, 95% CI) | Classification of dMMR Cases |
|---|---|---|---|
| Mensenkamp et al. [24] | 52% | 0 (0%) | Primarily biallelic somatic hits |
| Niessen et al. [25] | 8.9% | 25 (8.9%) | All dMMR → LS confirmed |
| Steinhagen et al. [26] | 19.1% | 10 (5.1%) | 7 sporadic vs. 10 LS, rest VUS |
| Suzuki et al. [27] | 8.4% | 7 (5.9%) | 3 sporadic, 7 LS |
| Wright et al. [28] | 14% | 12 (5.0%) | Remainder sporadic or no final test |
| Study | Right-Sided Tumors in dMMR | Tumor-Infiltrating Lymphocytes (TILs) in dMMR | Mucinous/Signet Histology | Family History Mentioned |
|---|---|---|---|---|
| Mensenkamp et al. [24] | Not systematically reported | High TIL infiltration in many MSI + cases | Not systematically reported | N/A (unexplained MSI) |
| Niessen et al. [25] | 60% of dMMR were right-sided | 55% had TILs > moderate | 20% had mucinous components | 76% with first-degree hx |
| Steinhagen et al. [26] | 53% of dMMR were right-sided | 60% had TILs or Crohn-like reaction | 26% mucinous or signet | 55% with suspicious hx |
| Suzuki et al. [27] | 80% of dMMR were right-sided | 70% with moderate/high TILs | 50% mucinous or signet | 43% had first-degree hx |
| Wright et al. [28] | 57% of dMMR were right-sided | 39% had TIL mentioned in pathology | 30% mucinous or signet | 60% with suspicious hx |
| Study | Abnormal IHC Who Underwent Germline Testing (%) | Germline Mutation Confirmation (n/of Tested) | Somatic MLH1 Hypermethylation or BRAF V600E (%) | Follow-Up/Registry Referral Rate |
|---|---|---|---|---|
| Mensenkamp et al. [24] | Not all, but 25 “unexplained” MSI used | 0/25 (0%) LS found | Some tested, no universal data | High referral for further analysis |
| Niessen et al. [25] | 100% (25/25) with dMMR tested | 25/25 (100%) | Not explicitly stated | Many from known family registries |
| Steinhagen et al. [26] | 38 dMMR; 22 (57.8%) had germline testing | 10 (46% of tested); 5.1% of entire cohort | Some had MLH1 hypermethylation (3/4 MLH1 loss) | High counseling acceptance rate |
| Suzuki et al. [27] | 10 dMMR; 8 tested; 80% tested | 7/8 (88%); 5.9% entire cohort | 2/4 MLH1/PMS2 had hypermethylation, 1 had BRAF | Only 1 “possible LS” no final proof |
| Wright et al. [28] | 33 dMMR; 22 (67%) tested; 10 had LS (5%) | 10/22 (45%); 10 total new LS | Some tested for BRAF/Methylation, 7 sporadic | 7 not referred, 4 died soon |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manta, B.A.; Ilie, A.C.; Marc, F.; Nistor, D.; Mazilu, P.O.; Borza, C. Clinical Molecular Immunohistochemistry Mismatch Repair Mutations in Lynch Syndrome in Patients Under 50 Years: A Systematic Review. Biomedicines 2025, 13, 1062. https://doi.org/10.3390/biomedicines13051062
Manta BA, Ilie AC, Marc F, Nistor D, Mazilu PO, Borza C. Clinical Molecular Immunohistochemistry Mismatch Repair Mutations in Lynch Syndrome in Patients Under 50 Years: A Systematic Review. Biomedicines. 2025; 13(5):1062. https://doi.org/10.3390/biomedicines13051062
Chicago/Turabian StyleManta, Bogdan Adrian, Adrian Cosmin Ilie, Felicia Marc, Daciana Nistor, Patricia Octavia Mazilu, and Claudia Borza. 2025. "Clinical Molecular Immunohistochemistry Mismatch Repair Mutations in Lynch Syndrome in Patients Under 50 Years: A Systematic Review" Biomedicines 13, no. 5: 1062. https://doi.org/10.3390/biomedicines13051062
APA StyleManta, B. A., Ilie, A. C., Marc, F., Nistor, D., Mazilu, P. O., & Borza, C. (2025). Clinical Molecular Immunohistochemistry Mismatch Repair Mutations in Lynch Syndrome in Patients Under 50 Years: A Systematic Review. Biomedicines, 13(5), 1062. https://doi.org/10.3390/biomedicines13051062