
Construction of Diagnostic Model for Regulatory T Cell-Related Genes in Sepsis Based on Machine Learning

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
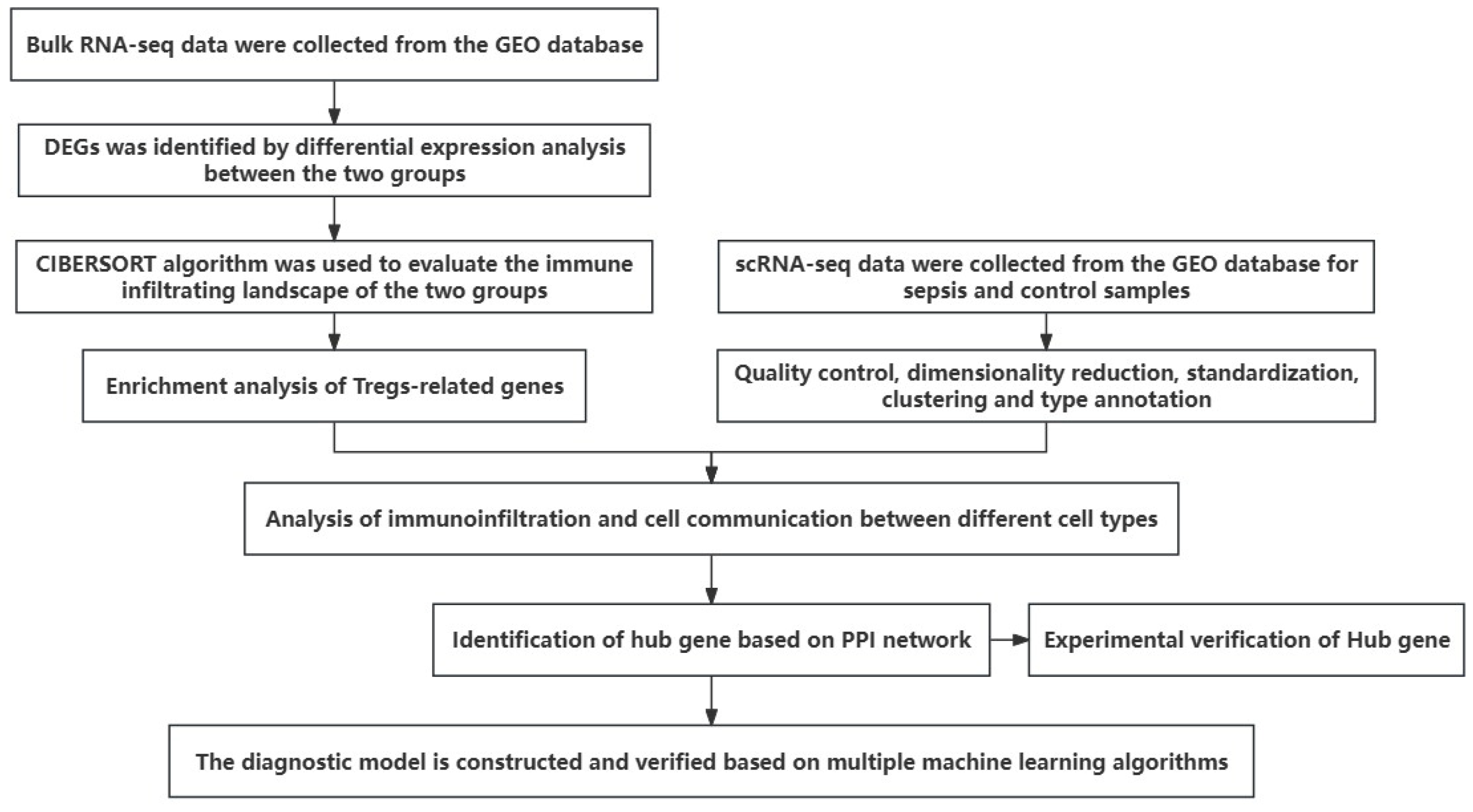
2. Materials and Methods
2.1. Data Acquisition
2.2. Differential Expression Analysis
2.3. Immune Cell Abundance Calculation
2.4. Enrichment Analysis
2.5. Protein Interaction Analysis
2.6. Machine Learning Algorithms Used to Screen Diagnosis-Related Genes and Diagnostic Model Construction
2.6.1. Random Forest
2.6.2. Support Vector Machine
2.6.3. Logistic Regression
2.6.4. Deep Neural Network
2.6.5. Self-Encoder
2.6.6. Noise Reduction Self-Encoder
2.7. Drug Network Analysis
2.8. Methods for scRNA-Seq Data Analysis
2.9. qRT-PCR
3. Results
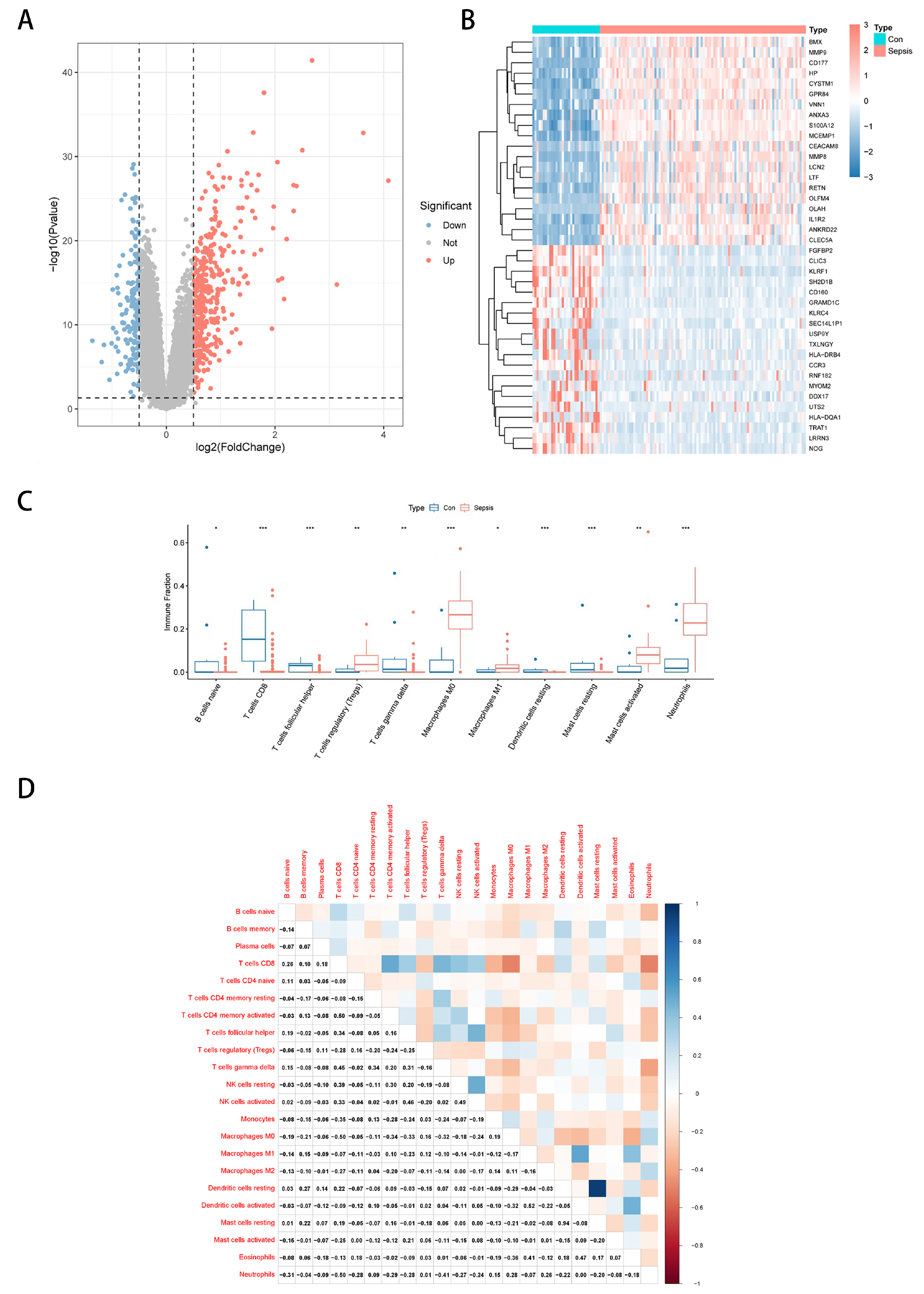
3.1. Differential Expression Analysis and Immune Landscape of Sepsis Transcriptome Data
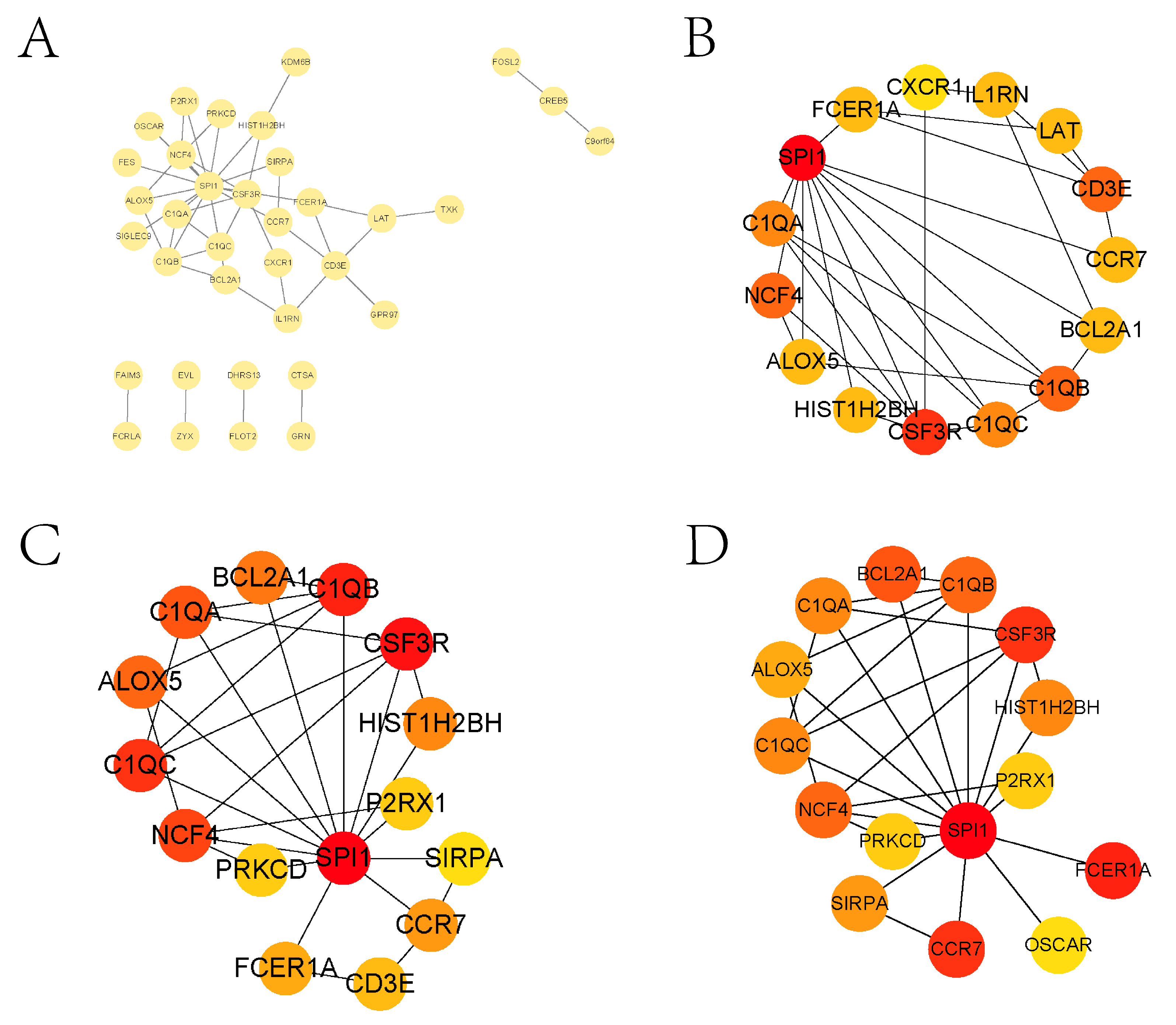
3.2. Enrichment Analysis and PPI Analysis of Treg-Related Genes
3.3. Construction of Diagnostic Models Based on Multiple Machine Learning Algorithms
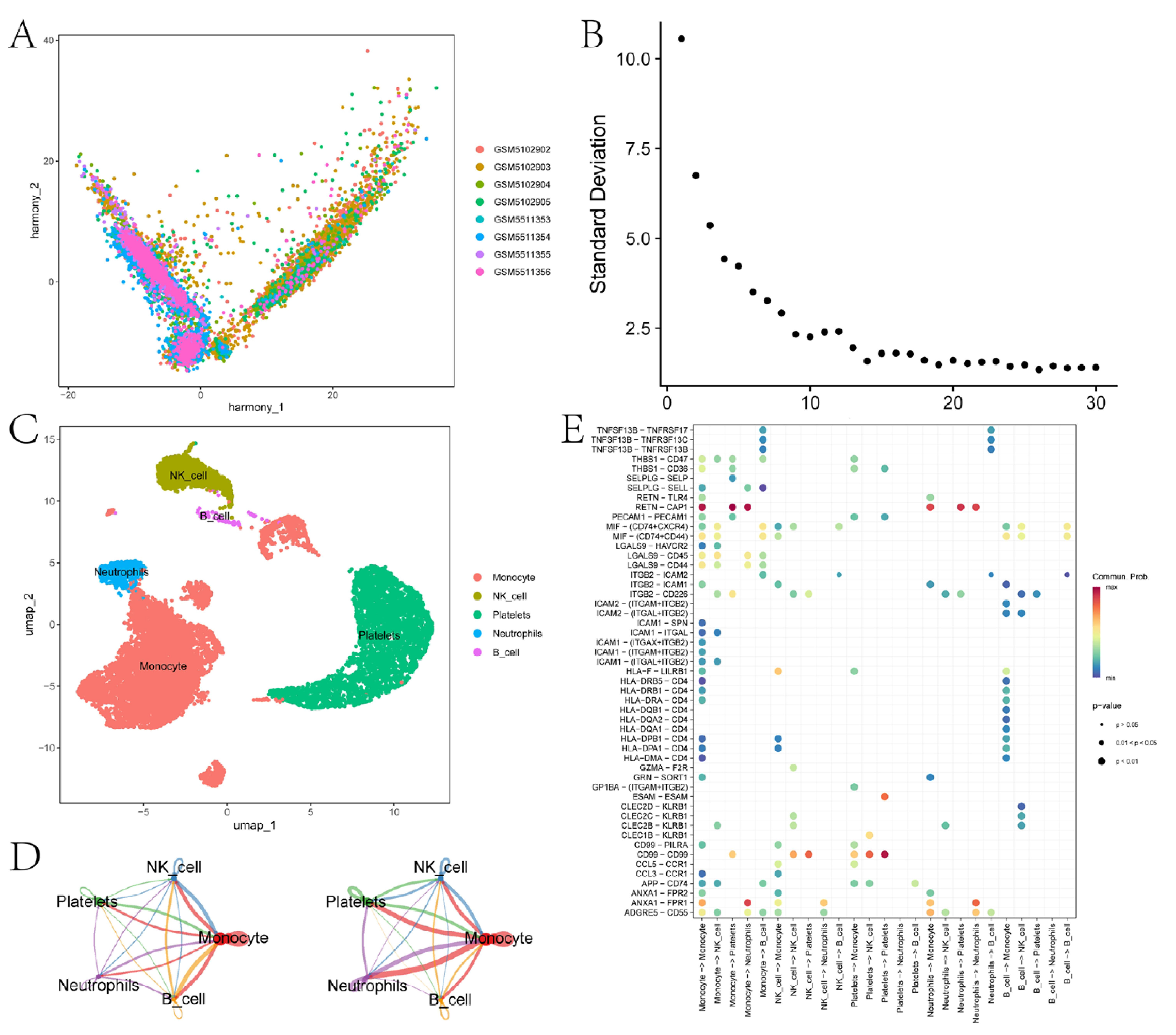
3.4. Cell Communication Analysis Identifies Key Intercellular Pathways
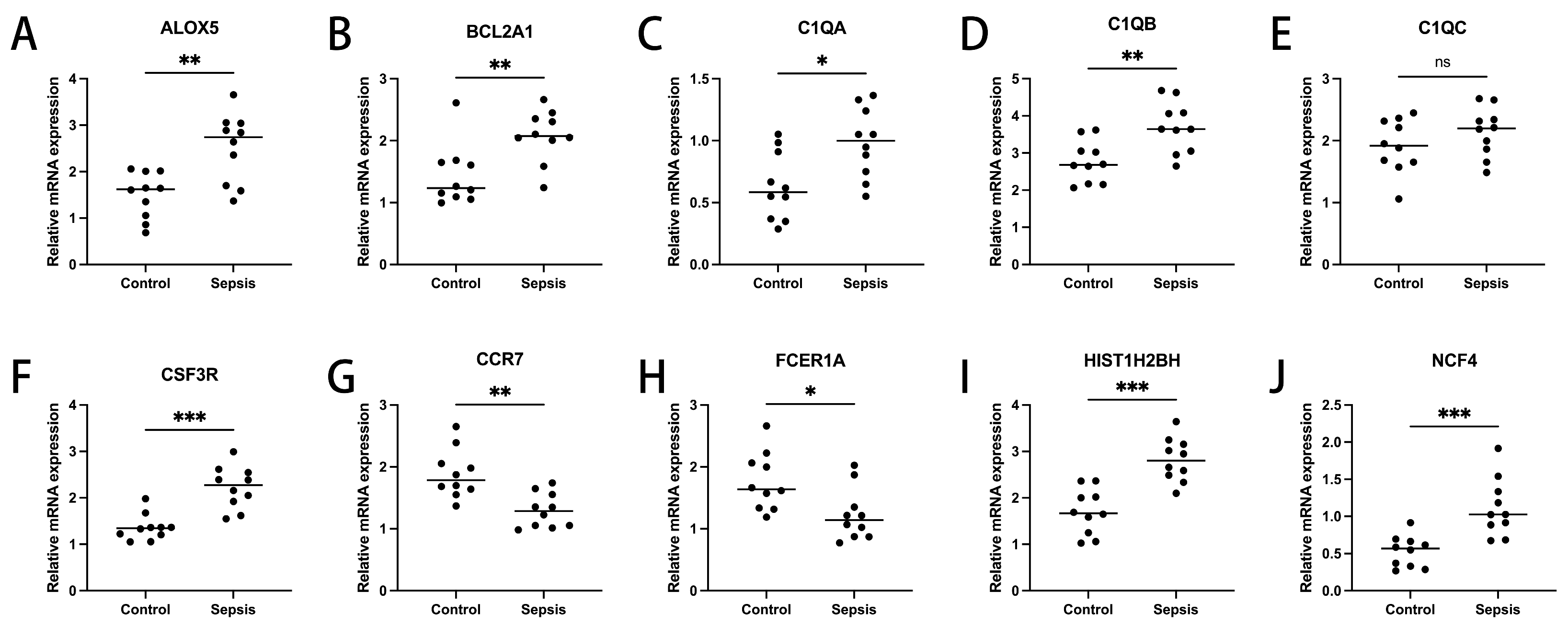
3.5. Expression Validation of Diagnosis-Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Hellman, J.; Myatra, S.N.; Ostermann, M.; Prescott, H.C.; Talmor, D.; Antonelli, M.; Pontes Azevedo, L.C.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Pierrakos, C.; Velissaris, D.; Bisdorff, M.; Marshall, J.C.; Vincent, J.L. Biomarkers of sepsis: Time for a reappraisal. Crit. Care 2020, 24, 287. [Google Scholar] [CrossRef]
- Font, M.D.; Thyagarajan, B.; Khanna, A.K. Sepsis and Septic Shock—Basics of diagnosis, pathophysiology and clinical decision making. Med. Clin. 2020, 104, 573–585. [Google Scholar] [CrossRef]
- Pan, S.; Lv, Z.; Wang, R.; Shu, H.; Yuan, S.; Yu, Y.; Shang, Y. Sepsis-Induced Brain Dysfunction: Pathogenesis, Diagnosis, and Treatment. Oxid. Med. Cell. Longev. 2022, 2022, 1328729. [Google Scholar] [CrossRef] [PubMed]
- Plesa-Furda, P.; Lancu, L.; Cioltean, C.; Breaza, C.; Brinzaila, S.; Odaini, A.; Bocse, H.; Herdean, A.; Bartos, D.; Turculet, C.; et al. Abdominal Sepsis—Current Definitions and Practice. Chirurgia 2021, 116, S16–S27. [Google Scholar]
- Kessel, A.; Bamberger, E.; Masalha, M.; Toubi, E. The role of T regulatory cells in human sepsis. J. Autoimmun. 2009, 32, 211–215. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Venet, F.; Chung, C.S.; Kherouf, H.; Geeraert, A.; Malcus, C.; Poitevin, F.; Bohe, J.; Lepape, A.; Ayala, A.; Monneret, G. Increased circulating regulatory T cells (CD4+CD25+CD127−) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. 2009, 35, 678–686. [Google Scholar] [CrossRef]
- Wisnoski, N.; Chung, C.; Chen, Y.; Huang, X.; Ayala, A. The contribution of CD4+ CD25+ T-regulatory-cells to immune suppression in sepsis. Shock 2007, 27, 251–257. [Google Scholar] [CrossRef]
- Qin, J.M.; Liu, C.; Chen, S.B.; Guo, Y.; Arcucci, R. Freeze The Backbones: A Paramaeter-efficientcontrastive a pproach to robust medical vision-language pre-training. In Proceedings of the IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSSP), Seoul, Republic of Korea, 14–19 April 2024; pp. 1686–1690. [Google Scholar] [CrossRef]
- Liu, C.; OuYang, C.; Cheng, S.B.; Shah, A.; Bai, W.; Arcucci, R. G2D: From global to dense radiography representation Learning via vision-language pre-training. arXiv 2023. [Google Scholar] [CrossRef]
- Wang, T.; Shao, W.; Huang, Z.; Tang, H.; Zhang, J.; Ding, Z.; Huang, K. MOGONET integrates multi-omics data using graph convolutional networks allowing patient classification and biomarker identification. Nat. Commun. 2021, 12, 3445. [Google Scholar] [CrossRef]
- Jiao, C.-N.; Gao, Y.-L.; Ge, D.-H.; Shang, J.; Liu, J.-X. Multi-modal imaging genetics data fusion by deep auto-encoder and self-representation network for Alzheimer’s disease diagnosis and biomarkers extraction. Eng. Appl. Artif. Intell. 2024, 130, 107782. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Huang, S.; Liu, D.; Han, L.; Deng, J.; Wang, Z.; Jiang, J.; Zeng, L. Decoding the potential role of regulatory T cells in sepsis-induced immunosuppression. Eur. J. Immunol. 2024, 54, e2350730. [Google Scholar] [CrossRef]
- Li, R.; Li, H.; Yang, X.; Hu, H.; Liu, P.; Liu, H. Crosstalk between dendritic cells and regulatory T cells: Protective effect and therapeutic potential in multiple sclerosis. Front. Immunol. 2022, 13, 970508. [Google Scholar] [CrossRef]
- Roquilly, A.; McWilliam, H.E.G.; Jacqueline, C.; Tian, Z.; Cinotti, R.; Rimbert, M.; Wakim, L.; Caminschi, I.; Lahoud, M.H.; Belz, G.T.; et al. Local Modulation of Antigen-Presenting Cell Development after Resolution of Pneumonia Induces Long-Term Susceptibility to Secondary Infections. Immunity 2017, 47, 135–147.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, H.; Qu, M.; Nan, K.; Cao, H.; Cata, J.P.; Chen, W.; Miao, C. Review: The Emerging Role of Neutrophil Extracellular Traps in Sepsis and Sepsis-Associated Thrombosis. Front. Cell. Infect. Microbiol. 2021, 11, 653228. [Google Scholar] [CrossRef]
- Halbgebauer, R.; Schmidt, C.Q.; Karsten, C.M.; Ignatius, A.; Lang-Huber, M. Janus face of complement-driven neutrophil activation during sepsis. Semin. Immunol. 2018, 37, 12–20. [Google Scholar] [CrossRef]
- Gallo, P.M.; Gallucci, S. The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front. Immunol. 2013, 4, 138. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, N.; Shi, Y.; Zhou, B.; Sun, D.; Ma, B.; Xu, Z.; Yang, J.; Li, C. Astaxanthin Protects Dendritic Cells from Lipopolysaccharide-Induced Immune Dysfunction. Mar. Drugs 2021, 19, 346. [Google Scholar] [CrossRef]
- Jia, P.; Xu, S.; Wang, X.; Wu, X.; Ren, T.; Zou, Z.; Zeng, Q.; Shen, B.; Ding, X. Chemokine CCL2 from proximal tubular epithelial cells contributes to sepsis-induced acute kidney injury. Am. J. Physiol.-Ren. Physiol. 2022, 323, F107–F119. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Thatoi, P.K.; Pandey, A.D.; Das, B.K.; Ravindran, B.; Bhattachearjee, S.; Mohapatra, S.K. Transcriptomic meta-analysis reveals up-regulation of gene expression functional in osteoclast differentiation in human septic shock. PLoS ONE 2017, 12, e0171689. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, G.A.; Dionisi, T.; Tarli, C.; Augello, G.; Mirijello, A.; Cosmo, D.S.; Gasbarromo, A.; Addolorato, G. Alcohol-related Liver Disease and sepsis. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4563–4569. [Google Scholar] [PubMed]
- Ivanov, I.; Golovanov, A.B.; Ferretti, C.; Canyelles-Nino, M.; Heydeck, D.; Stehling, S.; Liuch, J.M.; Gonzalez-Lafont, A.; Kuhn, H. Mutations of Triad Determinants Changes the Substrate Alignment at the Catalytic Center of Human ALOX5. ACS Chem. Biol. 2019, 14, 2768–2782. [Google Scholar] [CrossRef]
- Lu, X.; Xue, L.; Sun, W.; Ye, J.; Zhu, Z.; Mei, H. Identification of key pathogenic genes of sepsis based on the Gene Expression Omnibus database. Mol. Med. Rep. 2018, 17, 3042–3054. [Google Scholar] [CrossRef]
- Li, J.; Zhou, M.; Feng, J.Q.; Hong, S.M.; Yang, S.Y.; Zhi, L.X.; Lin, W.Y.; Zhu, C.; Yu, Y.T.; Lu, J.L. Bulk RNA Sequencing With Integrated Single-Cell RNA Sequencing Identifies BCL2A1 as a Potential Diagnostic and Prognostic Biomarker for Sepsis. Front. Public Health 2022, 10, 937303. [Google Scholar] [CrossRef]
- Behairy, M.Y.; Abdelrahman, A.A.; Abdallah, H.Y.; Ibrahim, E.E.D.A.; Sayed, A.A.; Azab, M.M. In silico analysis of missense variants of the C1qA gene related to infection and autoimmune diseases. J. Taibah Univ. Med. Sci. 2022, 17, 1074–1082. [Google Scholar] [CrossRef]
- Kikutani, K.; Hosokawa, K.; Giga, H.; Ota, K.; Matsumata, M.; Zhu, M.; Takemoto, H.; Ji, B.; Ohshimo, S.; Shime, N.; et al. Genetic deletion of translocator protein exacerbates post-sepsis syndrome with activation of the C1Q pathway in septic mouse model. Shock 2023, 59, 82–90. [Google Scholar] [CrossRef]
- Fan, Y.; Han, Q.; Li, J.; Ye, G.; Zhang, X.; Xu, T.; Li, H. Revealing potential diagnostic gene biomarkers of septic shock based on machine learning analysis. BMC Infect. Dis. 2022, 22, 65. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Chen, J.; Pan, J. Constructing a 10-core genes panel for diagnosis of pediatric sepsis. J. Clin. Lab. Anal. 2021, 35, e23680. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Rong, Y.; Wang, Y.; Guo, Y.; Shan, L.; Yu, X.; Li, L.; Si, J.; LI, X.; Ma, K. A Systematic Study of the Mechanism of Acacetin Against Sepsis Based on Network Pharmacology and Experimental Validation. Front. Pharmacol. 2021, 12, 683645. [Google Scholar] [CrossRef]
- Li, G.; Yang, Z.; Yang, C.; Xie, Y.; Gong, S.; Lv, S.; Xiao, B.; Wang, J.; Weng, Q.; Wang, J.; et al. Single-cell RNA sequencing reveals cell-cell communication and potential biomarker in sepsis and septic shock patients. Int. Immunopharmacol. 2024, 132, 111938. [Google Scholar] [CrossRef] [PubMed]
- Buss, N.A.; Gavins, F.N.; Cover, P.O.; Terron, A.; Buckingham, J.C. Targeting the annexin 1-formyl peptide receptor 2/ALX pathway affords protection against bacterial LPS-induced pathologic changes in the murine adrenal cortex. FASEB J. 2015, 29, 2930–2942. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Walsh, G.; Ashley, S.; Chua, S.; Agarwal, R.; O’Brien, M.; Johnston, S.; Smith, I.E. A randomised pilot Phase II study of doxorubicin and cyclophosphamide (AC) or epirubicin and cyclophosphamide (EC) given 2 weekly with pegfilgrastim (accelerated) vs 3 weekly (standard) for women with early breast cancer. Br. J. Cancer 2009, 100, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Choudhery, M.S.; Arif, T.; Mahmood, R.; Harris, D.T. CAR-T-Cell-Based Cancer Immunotherapies: Potentials, Limitations, and Future Prospects. J. Clin. Med. 2024, 13, 3202. [Google Scholar] [CrossRef]
- Mirmozaffari, M.; Kamal, N. The Application of Data Envelopment Analysis to Emergency Departments and Management of Emergency Conditions: A Narrative Review. Healthcare 2023, 11, 2541. [Google Scholar] [CrossRef]
- Mirmozaffari, M.; Yazdani, R.; Shadkam, E.; Khalili, S.M.; Tavassoli, L.S.; Boskabadi, A. A Novel Hybrid Parametric and Non-Parametric Optimisation Model for Average Technical Efficiency Assessment in Public Hospitals during and Post-COVID-19 Pandemic. Bioengineering 2021, 9, 7. [Google Scholar] [CrossRef]
- Mirmozaffari, M.; Shadkam, E.; Khalili, S.M.; Yazdani, M. Developing a Novel Integrated Generalised Data Envelopment Analysis (DEA) to Evaluate Hospitals Providing Stroke Care Services. Bioengineering 2021, 8, 207. [Google Scholar] [CrossRef]
- Xu, J.; Zhu, M.; Luo, P.; Gong, Y. Machine Learning Screening and Validation of PANoptosis-Related Gene Signatures in Sepsis. J. Inflamm. Res. 2024, 17, 4765–4780. [Google Scholar] [CrossRef]
- Chen, Z.; Wei, S.; Yuan, Z.; Chang, R.; Chen, X.; Fu, Y.; Wu, W. Machine learning reveals ferroptosis features and a novel ferroptosis classifier in patients with sepsis. Immun. Inflamm. Dis. 2024, 12, e1279. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xu, N.; Guo, H.; Li, J. Identification of the Diagnostic Signature of Sepsis Based on Bioinformatic Analysis of Gene Expression and Machine Learning. Comb. Chem. High Throughput Screen. 2022, 25, 21–28. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Guo, Z.; Wang, X.; Wang, Z. Construction of Diagnostic Model for Regulatory T Cell-Related Genes in Sepsis Based on Machine Learning. Biomedicines 2025, 13, 1060. https://doi.org/10.3390/biomedicines13051060
Wang X, Guo Z, Wang X, Wang Z. Construction of Diagnostic Model for Regulatory T Cell-Related Genes in Sepsis Based on Machine Learning. Biomedicines. 2025; 13(5):1060. https://doi.org/10.3390/biomedicines13051060
Chicago/Turabian StyleWang, Xuesong, Zhe Guo, Xinrui Wang, and Zhong Wang. 2025. "Construction of Diagnostic Model for Regulatory T Cell-Related Genes in Sepsis Based on Machine Learning" Biomedicines 13, no. 5: 1060. https://doi.org/10.3390/biomedicines13051060
APA StyleWang, X., Guo, Z., Wang, X., & Wang, Z. (2025). Construction of Diagnostic Model for Regulatory T Cell-Related Genes in Sepsis Based on Machine Learning. Biomedicines, 13(5), 1060. https://doi.org/10.3390/biomedicines13051060