
Relevance of Cellular Homeostasis-Related Gene Expression Signatures in Distinct Molecular Subtypes of Breast Cancer

 , , , , , , , , , and
, , , , , , , , , and
Abstract
1. Background
2. Methods
2.1. Patient Population and Samples
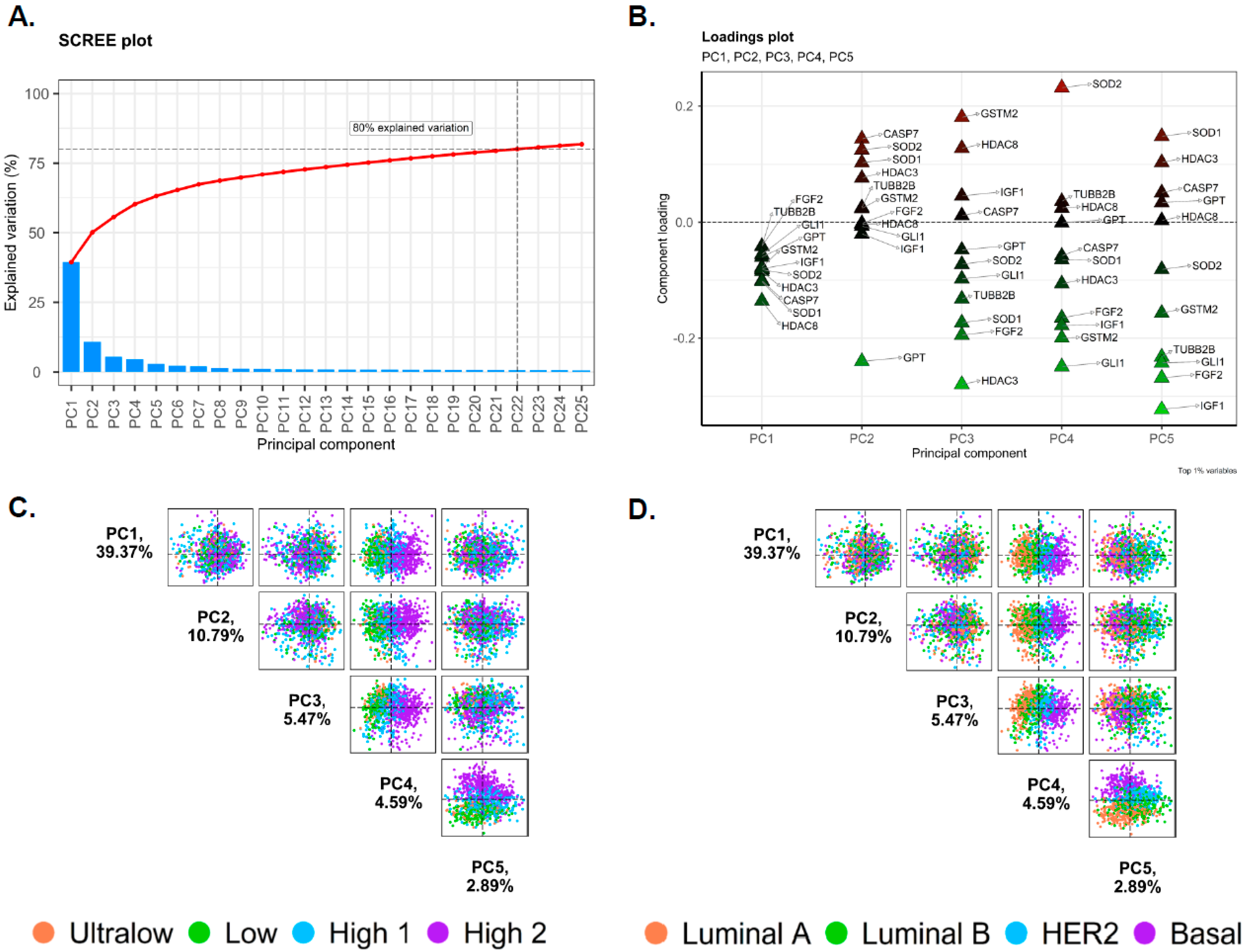
2.2. Principal Component Analysis of Gene Expression Data
2.3. Differential Gene Expression Analysis
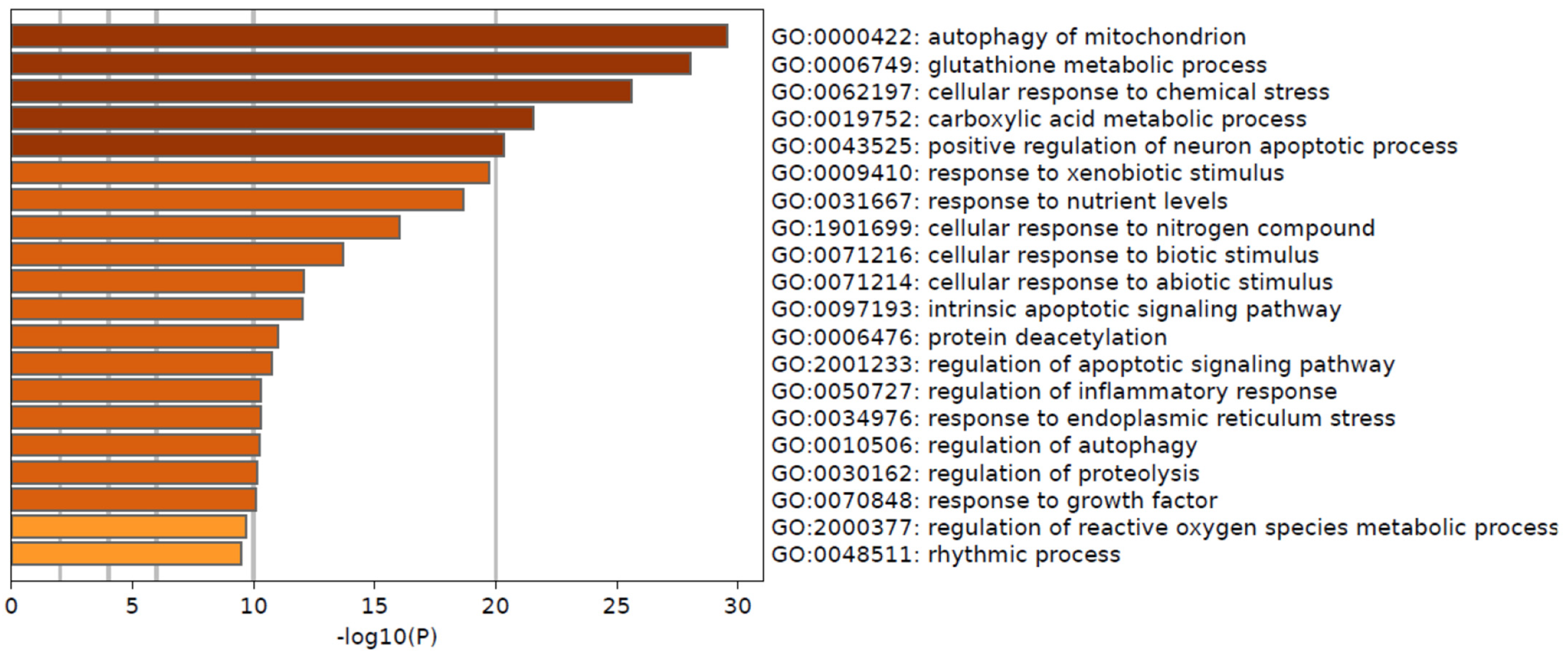
2.4. Gene Ontology (GO) and KEGG Pathway Enrichment Analysis
2.5. Protein–Protein Interaction (PPI) Network Analysis
3. Results
3.1. Study Population
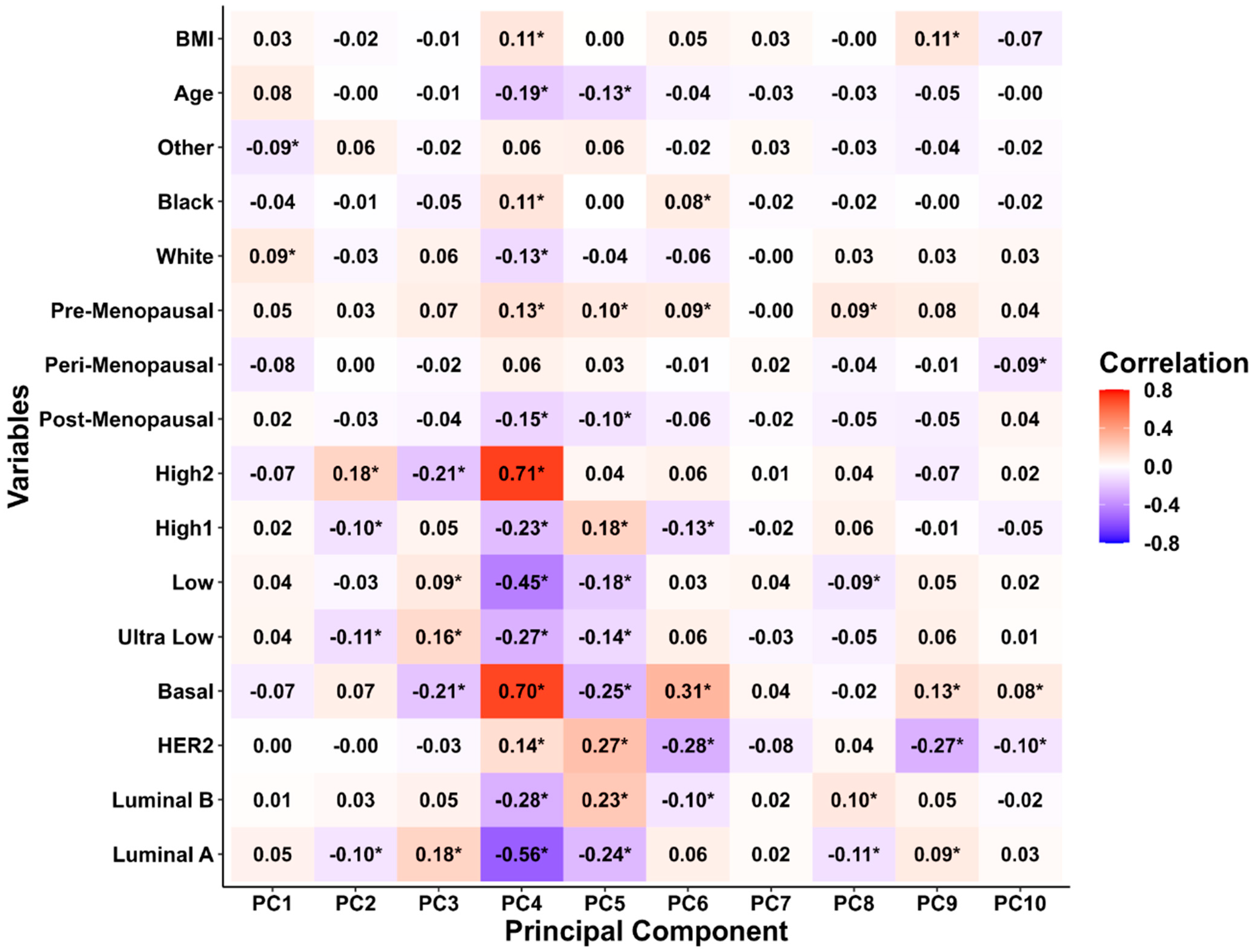
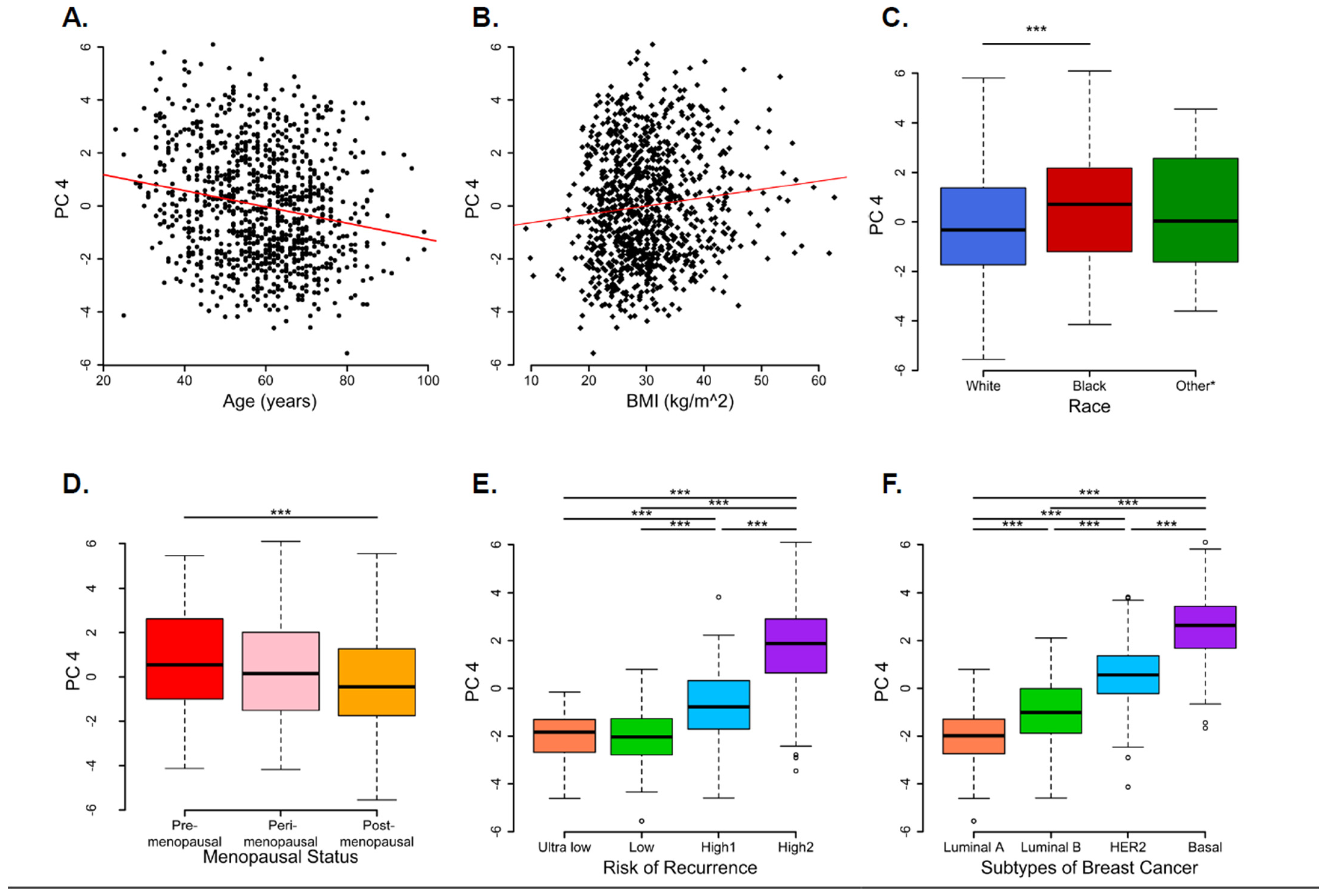
3.2. Principal Component Analysis
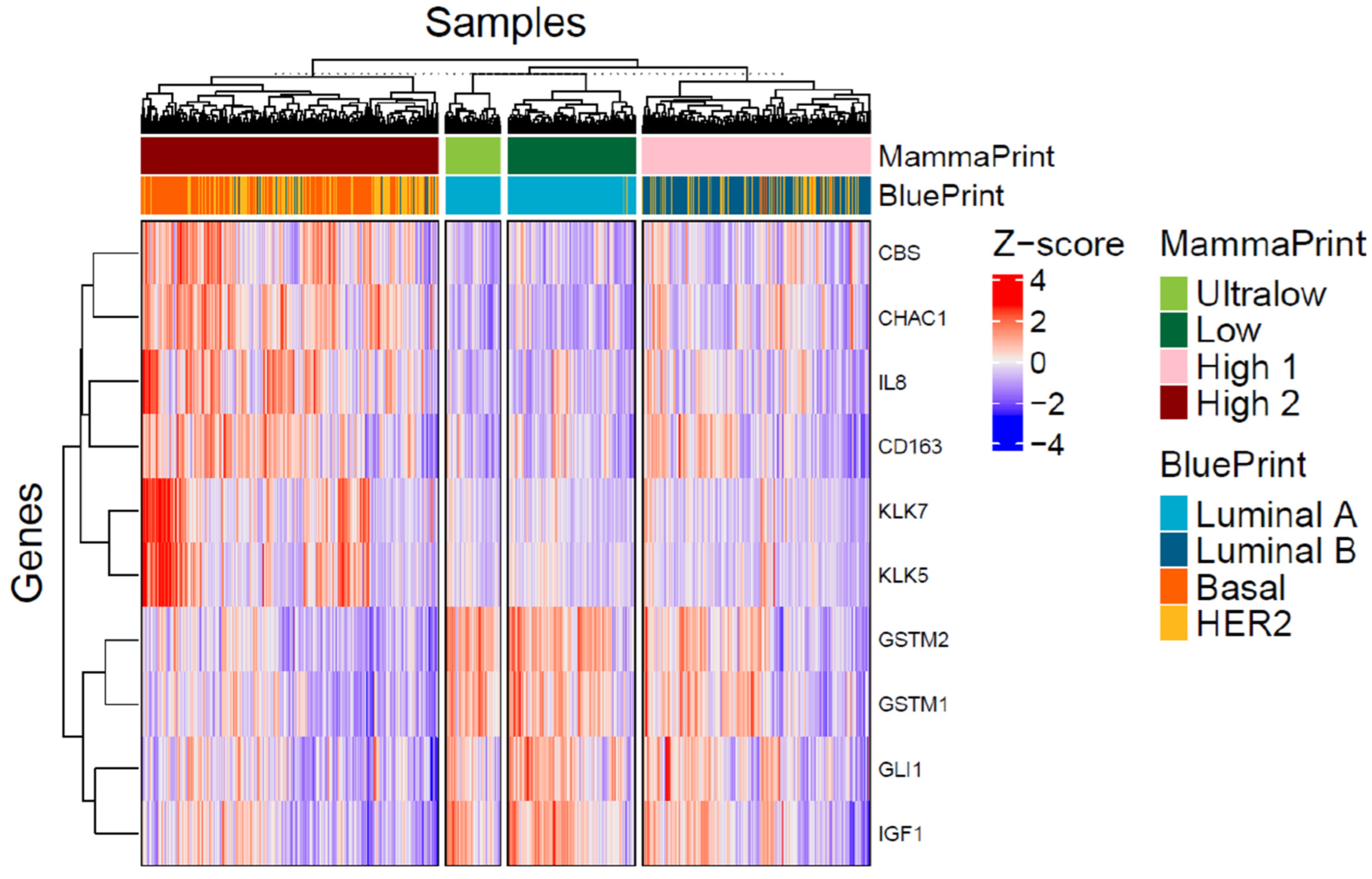
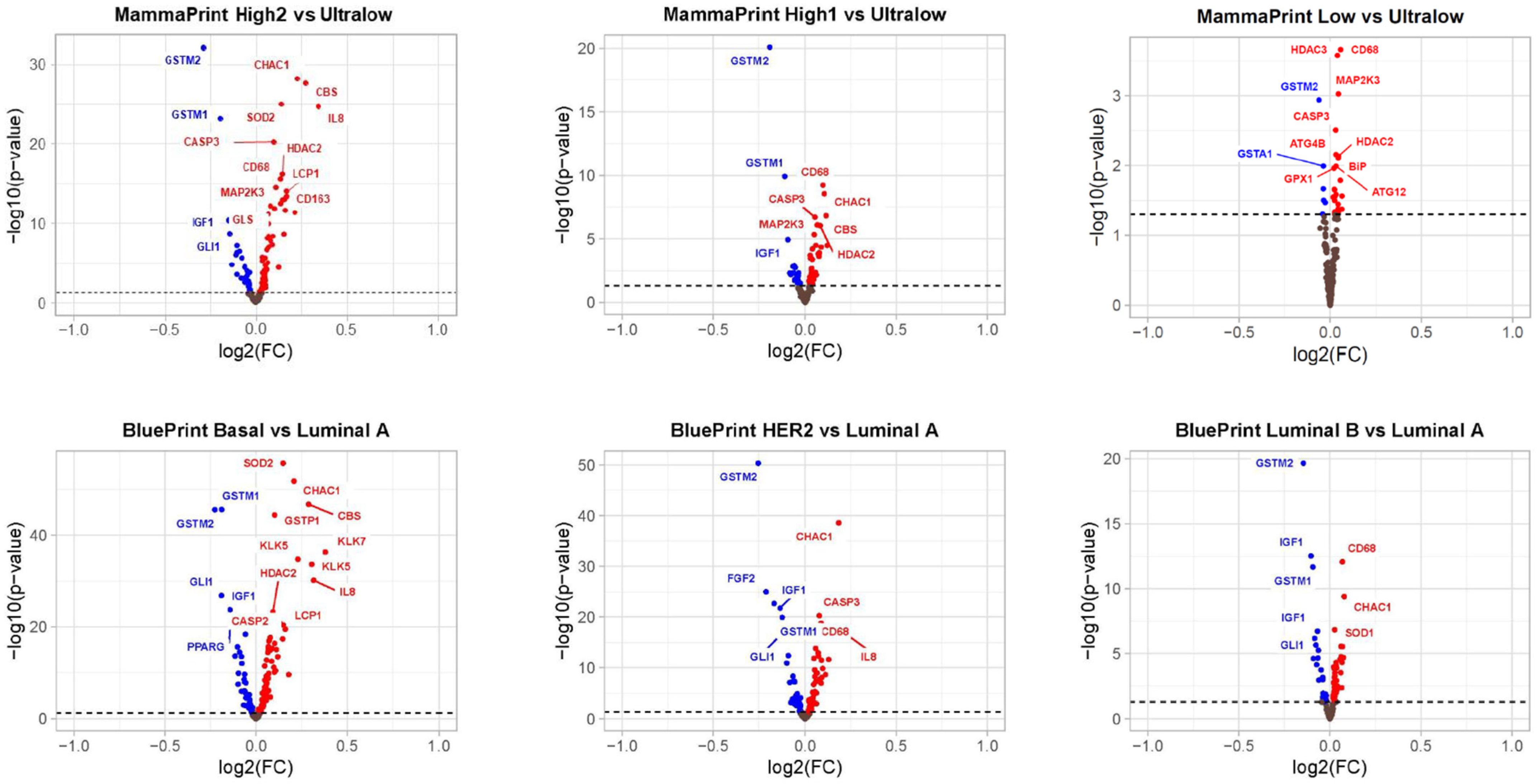
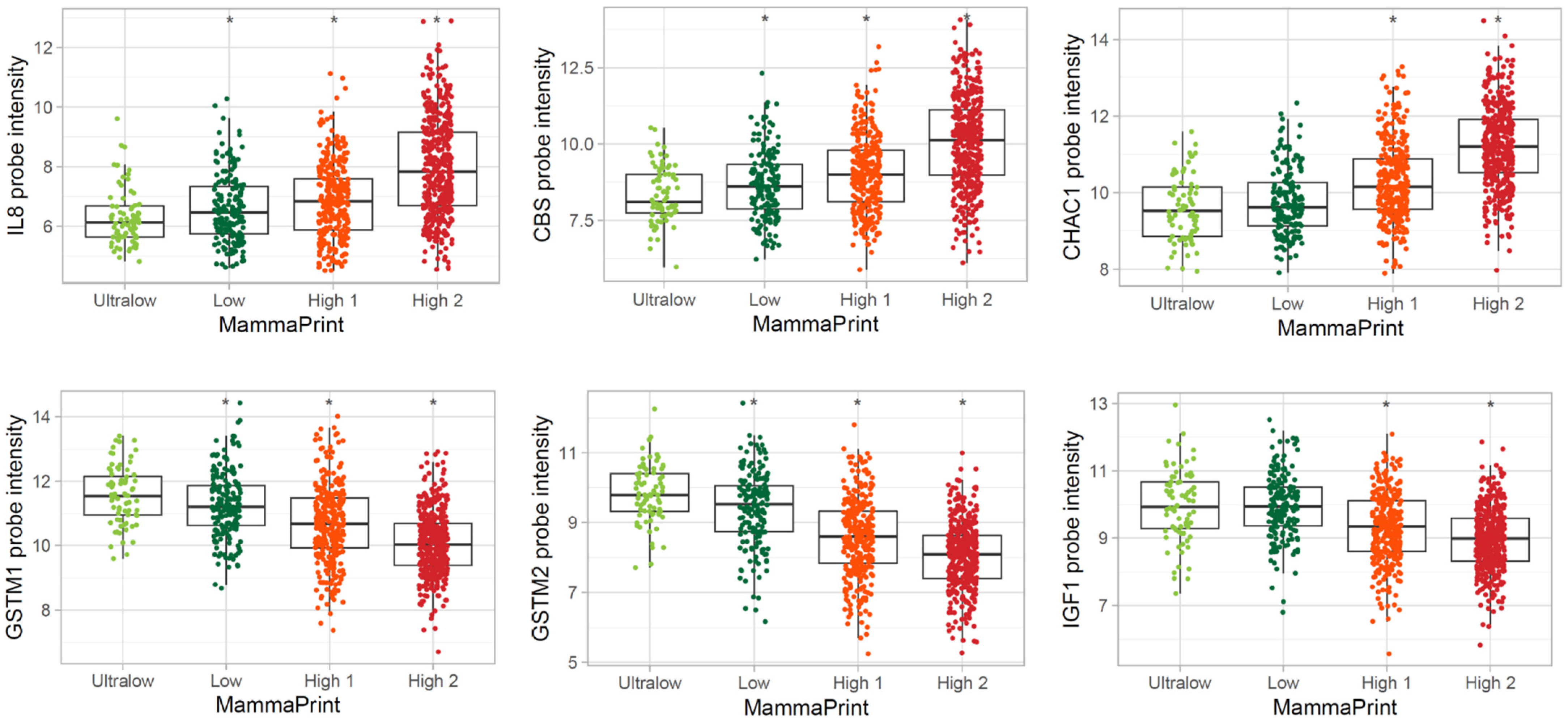
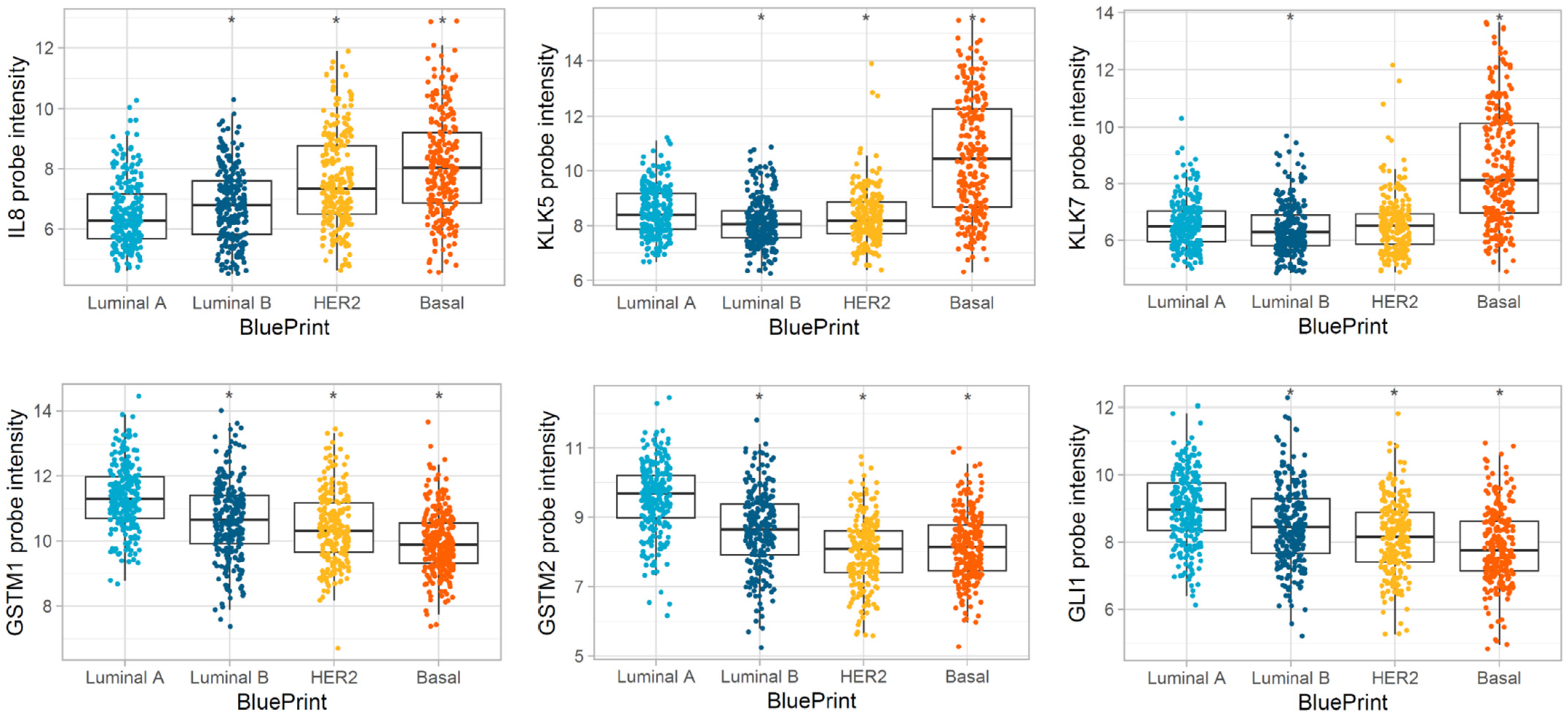
3.3. Differentially Expressed Genes in MP Risk Categories and BP Molecular Subtypes
3.4. Pathways Differentially Modulated in MP and BP Subgroups
3.5. Protein–Protein Interaction Network Associated with Functional Enrichment Analysis of DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Shockney, L.D. Breast Cancer Facts & Stats 2024. Available online: https://www.nationalbreastcancer.org/breast-cancer-facts/ (accessed on 15 October 2024).
- Masood, S. Breast cancer subtypes: Morphologic and biologic characterization. Womens Health 2016, 12, 103–119. [Google Scholar] [CrossRef]
- Erasmo Orrantia-Borunda, P.A.-N.; Acuña-Aguilar, L.E.; Gómez-Valles, F.O.; Ramírez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer (Internet); HN, M., Ed.; Exon Publications: Brisbane, Australia, 2022. [Google Scholar]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Santonja, A.; Sánchez-Muñoz, A.; Lluch, A.; Chica-Parrado, M.R.; Albanell, J.; Chacón, J.I.; Antolín, S.; Jerez, J.M.; de la Haba, J.; de Luque, V.; et al. Triple negative breast cancer subtypes and pathologic complete response rate to neoadjuvant chemotherapy. Oncotarget 2018, 9, 26406–26416. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Hill, L.; Artiga, S. Racial Disparities in Cancer Outcomes, Screening, and Treatment 2022. Available online: https://www.kff.org/racial-equity-and-health-policy/issue-brief/racial-disparities-in-cancer-outcomes-screening-and-treatment/ (accessed on 15 October 2024).
- American Cancer Society. Breast-Cancer-Facts-and-Figures-2017–2018. 2019. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/breast-cancer-facts-and-figures/breast-cancer-facts-and-figures-2017-2018.pdf (accessed on 15 October 2024).
- Scott, L.C.; Mobley, L.R.; Kuo, T.M.; Il’yasova, D. Update on triple-negative breast cancer disparities for the United States: A population-based study from the United States Cancer Statistics database, 2010 through 2014. Cancer 2019, 125, 3412–3417. [Google Scholar] [CrossRef] [PubMed]
- Parab, A.Z.; Kong, A.; Lee, T.A.; Kim, K.; Nutescu, E.A.; Malecki, K.C.; Hoskins, K.F.; Calip, G.S. Socioecologic Factors and Racial Differences in Breast Cancer Multigene Prognostic Scores in US Women. JAMA Netw. Open 2024, 7, e244862. [Google Scholar] [CrossRef]
- Germani, A.; Petrucci, S.; De Marchis, L.; Libi, F.; Savio, C.; Amanti, C.; Bonifacino, A.; Campanella, B.; Capalbo, C.; Lombardi, A.; et al. Beyond BRCA1 and BRCA2: Deleterious Variants in DNA Repair Pathway Genes in Italian Families with Breast/Ovarian and Pancreatic Cancers. J. Clin. Med. 2020, 9, 3003. [Google Scholar] [CrossRef]
- Nahleh, Z.; Otoukesh, S.; Dwivedi, A.; Mallawaarachchi, I.; Sanchez, L.; Saldivar, J.; Cataneda, K.; Heydarian, R. Clinical and pathological characteristics of Hispanic BRCA-associated breast cancers in the American-Mexican border city of El Paso, TX. Am. J. Cancer Res. 2015, 5, 466–471. [Google Scholar]
- Takahashi, M.; Cortés, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Denkert, C.; Park, Y.H.; Im, S.A.; Ahn, J.H.; et al. Pembrolizumab Plus Chemotherapy Followed by Pembrolizumab in Patients With Early Triple-Negative Breast Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e2342107. [Google Scholar] [CrossRef]
- Haan, J.C.; Bhaskaran, R.; Ellappalayam, A.; Bijl, Y.; Griffioen, C.J.; Lujinovic, E.; Audeh, W.M.; Penault-Llorca, F.; Mittempergher, L.; Glas, A.M. MammaPrint and BluePrint comprehensively capture the cancer hallmarks in early-stage breast cancer patients. Genes. Chromosomes Cancer 2022, 61, 148–160. [Google Scholar] [CrossRef]
- Barrón-Gallardo, C.A.; Garcia-Chagollán, M.; Morán-Mendoza, A.J.; Delgadillo-Cristerna, R.; Martínez-Silva, M.G.; Aguilar-Lemarroy, A.; Jave-Suárez, L.F. Transcriptomic Analysis of Breast Cancer Patients Sensitive and Resistant to Chemotherapy: Looking for Overall Survival and Drug Resistance Biomarkers. Technol. Cancer Res. Treat. 2022, 21, 15330338211068965. [Google Scholar] [CrossRef]
- Frost, H.R. Analyzing cancer gene expression data through the lens of normal tissue-specificity. PLoS Comput. Biol. 2021, 17, e1009085. [Google Scholar] [CrossRef]
- Ma, S.; Kosorok, M.R. Identification of differential gene pathways with principal component analysis. Bioinformatics 2009, 25, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Dai, Y. Principal component analysis based methods in bioinformatics studies. Brief. Bioinform. 2011, 12, 714–722. [Google Scholar] [CrossRef]
- Tian, J.; Wang, V.; Wang, N.; Khadang, B.; Boudreault, J.; Bakdounes, K.; Ali, S.; Lebrun, J.J. Identification of MFGE8 and KLK5/7 as mediators of breast tumorigenesis and resistance to COX-2 inhibition. Breast Cancer Res. 2021, 23, 23. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Berry, J.A.; Vincent, L.E.; Lucci, A. Involvement of IL-8 in COX-2-Mediated Bone Metastases from Breast Cancer. J. Surg. Res. 2006, 134, 44–51. [Google Scholar] [CrossRef]
- Yousef, G.M.; Scorilas, A.; Kyriakopoulou, L.G.; Rendl, L.; Diamandis, M.; Ponzone, R.; Biglia, N.; Giai, M.; Roagna, R.; Sismondi, P.; et al. Human kallikrein gene 5 (KLK5) expression by quantitative PCR: An independent indicator of poor prognosis in breast cancer. Clin. Chem. 2002, 48, 1241–1250. [Google Scholar] [CrossRef]
- Talieri, M.; Diamandis, E.P.; Gourgiotis, D.; Mathioudaki, K.; Scorilas, A. Expression analysis of the human kallikrein 7 (KLK7) in breast tumors: A new potential biomarker for prognosis of breast carcinoma. Thromb. Haemost. 2004, 91, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Alateyah, N.; Gupta, I.; Rusyniak, R.S.; Ouhtit, A. SOD2, a Potential Transcriptional Target Underpinning CD44-Promoted Breast Cancer Progression. Molecules 2022, 27, 811. [Google Scholar] [CrossRef]
- Liao, Z.Q.; Ye, M.; Yu, P.G.; Xiao, C.; Lin, F.Y. Glioma-Associated Oncogene Homolog1 (Gli1)-Aquaporin1 pathway promotes glioma cell metastasis. BMB Rep. 2016, 49, 394–399. [Google Scholar] [CrossRef]
- Hashemi, M.; Paskeh, M.D.A.; Orouei, S.; Abbasi, P.; Khorrami, R.; Dehghanpour, A.; Esmaeili, N.; Ghahremanzade, A.; Zandieh, M.A.; Peymani, M.; et al. Towards dual function of autophagy in breast cancer: A potent regulator of tumor progression and therapy response. Biomed. Pharmacother. 2023, 161, 114546. [Google Scholar] [CrossRef]
- Győrffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sharma, D. Autophagy and Breast Cancer: Connected in Growth, Progression, and Therapy. Cells 2023, 12, 1156. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Metur, S.P.; Lei, Y.; Zhang, Z.; Klionsky, D.J. Regulation of autophagy gene expression and its implications in cancer. J. Cell Sci. 2023, 136, jcs260631. [Google Scholar] [CrossRef]
- Gomez, M.L.; Shah, N.; Kenny, T.C.; Jenkins, E.C., Jr.; Germain, D. SOD1 is essential for oncogene-driven mammary tumor formation but dispensable for normal development and proliferation. Oncogene 2019, 38, 5751–5765. [Google Scholar] [CrossRef]
- Mira, E.; Carmona-Rodríguez, L.; Pérez-Villamil, B.; Casas, J.; Fernández-Aceñero, M.J.; Martínez-Rey, D.; Martín-González, P.; Heras-Murillo, I.; Paz-Cabezas, M.; Tardáguila, M.; et al. SOD3 improves the tumor response to chemotherapy by stabilizing endothelial HIF-2α. Nat. Commun. 2018, 9, 575. [Google Scholar] [CrossRef]
- Rebbeck, T.R. Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol. Biomark. Prev. 1997, 6, 733–743. [Google Scholar]
- Tian, X.; Wu, L.; Jiang, M.; Zhang, Z.; Wu, R.; Miao, J.; Liu, C.; Gao, S. Downregulation of GLYAT Facilitates Tumor Growth and Metastasis and Poor Clinical Outcomes Through the PI3K/AKT/Snail Pathway in Human Breast Cancer. Front. Oncol. 2021, 11, 641399. [Google Scholar] [CrossRef] [PubMed]
- Dehesh, T.; Fadaghi, S.; Seyedi, M.; Abolhadi, E.; Ilaghi, M.; Shams, P.; Ajam, F.; Mosleh-Shirazi, M.A.; Dehesh, P. The relation between obesity and breast cancer risk in women by considering menstruation status and geographical variations: A systematic review and meta-analysis. BMC Womens Health 2023, 23, 392. [Google Scholar] [CrossRef]
- Abdelaziz, S.A.; Hussein, M.T.E.-S.; Hamza, M.M.; Mahmoud, M.M. CD68 and CD163 expressions can predict high grade and spread in breast carcinoma: A histopathologic and immunohistochemical study. Egypt. J. Pathol. 2021, 41, 24–33. [Google Scholar] [CrossRef]
- Wang, Z.; Ni, F.; Yu, F.; Cui, Z.; Zhu, X.; Chen, J. Prognostic significance of mRNA expression of CASPs in gastric cancer. Oncol. Lett. 2019, 18, 4535–4554. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Mean ± SD [n (%)] |
|---|---|
| Age | 58.84 ± 13.04 |
| BMI | 30.04 ± 7.34 |
| Race | |
| White | 799 (81.7) |
| Black | 123 (12.6) |
| Other * | 56 (5.7) |
| Menopausal status | |
| Post | 717 (73.3) |
| Peri | 128 (13.1) |
| Pre | 133 (13.6) |
| ER status | |
| Negative | 265 (27.1) |
| Positive | 713 (72.9) |
| PR status | |
| Negative | 265 (27.1) |
| Positive | 590 (60.5) |
| HER2 status | |
| Negative | 721 (73.7) |
| Positive | 257 (26.3) |
| Risk of recurrence | |
| Ultralow | 76 (7.8) |
| Low | 176 (18) |
| High 1 | 315 (32.2) |
| High 2 | 411 (42) |
| Intrinsic molecular subtypes | |
| Luminal A | 250 (25.6) |
| Luminal B | 250 (25.6) |
| HER2 | 228 (23.3) |
| Basal | 250 (25.6) |
| GO | Description | Count | % | −Log10(P) | −Log10(q) |
|---|---|---|---|---|---|
| GO:0000422 | autophagy of mitochondrion | 18 | 16.22 | 29.56 | 25.39 |
| GO:0006749 | glutathione metabolic process | 17 | 15.32 | 28.03 | 24.15 |
| GO:0062197 | cellular response to chemical stress | 24 | 21.62 | 25.58 | 22.19 |
| GO:0019752 | carboxylic acid metabolic process | 30 | 27.03 | 21.57 | 18.47 |
| GO:0043525 | positive regulation of neuron apoptotic process | 14 | 12.61 | 20.36 | 17.35 |
| GO:0009410 | response to xenobiotic stimulus | 23 | 20.72 | 19.73 | 16.82 |
| GO:0031667 | response to nutrient levels | 23 | 20.72 | 18.64 | 15.82 |
| GO:1901699 | cellular response to nitrogen compound | 23 | 20.72 | 16.02 | 13.33 |
| GO:0071216 | cellular response to biotic stimulus | 15 | 13.51 | 13.68 | 11.12 |
| GO:0071214 | cellular response to abiotic stimulus | 15 | 13.51 | 12.09 | 9.69 |
| GO:0097193 | intrinsic apoptotic signaling pathway | 12 | 10.81 | 12.04 | 9.66 |
| GO:0006476 | protein deacetylation | 6 | 5.41 | 10.99 | 8.64 |
| GO:2001233 | regulation of apoptotic signaling pathway | 15 | 13.51 | 10.73 | 8.4 |
| GO:0050727 | regulation of inflammatory response | 15 | 13.51 | 10.3 | 8.01 |
| GO:0034976 | response to endoplasmic reticulum stress | 12 | 10.81 | 10.29 | 8.01 |
| GO:0010506 | regulation of autophagy | 14 | 12.61 | 10.24 | 7.97 |
| GO:0030162 | regulation of proteolysis | 17 | 15.32 | 10.14 | 7.88 |
| GO:0070848 | response to growth factor | 16 | 14.41 | 10.08 | 7.84 |
| GO:2000377 | regulation of reactive oxygen species metabolic process | 10 | 9.01 | 9.69 | 7.47 |
| GO:0048511 | rhythmic process | 12 | 10.81 | 9.49 | 7.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.P.; Dhanasekara, C.S.; Melkus, M.W.; Bose, C.; Khan, S.Y.; Sardela de Miranda, F.; Mahecha, M.F.; Gukhool, P.J.; Tonk, S.S.; Jun, S.-R.; et al. Relevance of Cellular Homeostasis-Related Gene Expression Signatures in Distinct Molecular Subtypes of Breast Cancer. Biomedicines 2025, 13, 1058. https://doi.org/10.3390/biomedicines13051058
Singh SP, Dhanasekara CS, Melkus MW, Bose C, Khan SY, Sardela de Miranda F, Mahecha MF, Gukhool PJ, Tonk SS, Jun S-R, et al. Relevance of Cellular Homeostasis-Related Gene Expression Signatures in Distinct Molecular Subtypes of Breast Cancer. Biomedicines. 2025; 13(5):1058. https://doi.org/10.3390/biomedicines13051058
Chicago/Turabian StyleSingh, Sharda P., Chathurika S. Dhanasekara, Michael W. Melkus, Chhanda Bose, Sonia Y. Khan, Flavia Sardela de Miranda, Maria F. Mahecha, Prrishti J. Gukhool, Sahil S. Tonk, Se-Ran Jun, and et al. 2025. "Relevance of Cellular Homeostasis-Related Gene Expression Signatures in Distinct Molecular Subtypes of Breast Cancer" Biomedicines 13, no. 5: 1058. https://doi.org/10.3390/biomedicines13051058
APA StyleSingh, S. P., Dhanasekara, C. S., Melkus, M. W., Bose, C., Khan, S. Y., Sardela de Miranda, F., Mahecha, M. F., Gukhool, P. J., Tonk, S. S., Jun, S.-R., Uygun, S., & Layeequr Rahman, R. (2025). Relevance of Cellular Homeostasis-Related Gene Expression Signatures in Distinct Molecular Subtypes of Breast Cancer. Biomedicines, 13(5), 1058. https://doi.org/10.3390/biomedicines13051058