1. Introduction
In the developed world, epithelial ovarian cancer is the fourth most common cause of cancer-related death in women and has the highest mortality rate amongst all gynecological tumors [
1]. In the USA it is estimated that there were ~20,000 new cases of ovarian carcinoma diagnosed in 2024, with ~13,000 women predicted to die from the disease [
2]. The overall 5-year survival is ~50% depending on the type of ovarian cancer and the stage at which it is diagnosed. The reason for the high mortality rate is that many women are diagnosed at late stages and have inadequate treatment options, in particular, for platinum-resistant ovarian cancer [
3]. Types of ovarian carcinoma are distinguished by the origin of the cancer, with epithelial adenocarcinoma being ~90% of cases. The most prevalent types consist of serous, endometrioid tumors and borderline and unspecified adenocarcinoma [
4]. Ovarian cancer most commonly occurs in women after menopause [
5].
Chemerin is a small protein that behaves as an adipokine and immune modulator [
6,
7]. It binds and signals via two receptors from the G-protein coupled receptor family, chem1 (chemokine-like receptor 1; CMKLR1) and chem2 (G protein-coupled receptor 1; GPR) as well as binding to a non-signaling receptor, CCRL2 before subsequent presentation to chem1 or chem2 [
8,
9,
10,
11].
In many tumors, chemerin is downregulated in the tumor tissue while elevated levels are observed in circulation [
12]. In breast cancer, chemerin and chem1 are downregulated in the tissue, resulting in changes in the tumor micro-environment for persistent tumor growth while over-expression of chemerin reverses these effects [
13]. In contrast, chemerin has been shown to promote the growth of several tumors, including clear-cell renal carcinoma and ovarian cancer [
14,
15,
16]. Expression of chemerin correlated with progesterone receptor and estrogen receptor β when investigated by anti-chemerin antibodies staining of histological sections of ovarian carcinoma tissue. The level of chemerin determined by ELISA was higher in ascitic fluid than in serum in 12 ovarian carcinoma patients [
17]. High levels of chemerin in ascitic fluid samples was also found to be high in two other studies [
10,
18].
Translation of chemerin mRNA results in a 163-amino-acid chain that has its signal peptide removed during secretion, resulting in a secreted form, chem163S, that is inactive [
19]. Subsequent proteolytic processing of the C-terminus of the protein chain generates partially active chem158K, fully active chem157S, and chem156F before forming inactive chem155A and smaller forms. We specifically developed ELISAs for these varied forms of chemerin to detect both human and the homologous mouse versions to allow analysis of biopsy samples that always contain mixtures of these forms. In contrast, the commercial ELISAs used in most other publications do not distinguish between the different chemerin forms and have not shown that all chemerin forms have parallel dose response curves with equipotent signal for the different forms.
In this study, we hypothesized that ascitic fluid from ovarian carcinoma patients would contain high levels of active chemerin forms in comparison to the levels present in plasma from control individuals.
4. Discussion
Chemerin is secreted from cells as an inactive precursor (chem163S) that requires proteolytic cleavage into functional forms that can further activate its specific receptors, chem1 and chem2 [
33]. In addition to the two active forms, chem157S and chem156F, several other forms have been identified in biopsy samples, including partially active chem158K and inactive chem155A and chem144D [
34]. Here, we show that conversion of plasma to serum in individuals with <25 BMI results in changes in total chemerin as well as many of the different chemerin forms (
Table 2). This is explained by the activation of enzymes in the coagulation cascade that can also process chemerin as well as by release of chemerin from platelets [
33,
35,
36]. Because we did not measure the chemerin content of the platelets, we are unable to quantitate how much chemerin or which chemerin forms contributed to the levels in serum.
In this study, we identified a higher fraction of cleaved chemerin in plasma than in our earlier reports [
20,
30]. Since these prior studies, we have developed ELISAs for other forms, in particular, chem156F, which account for the increase we observed in proteolytic processing.
In the bariatric surgery patients with >40 BMI the changes upon converting plasma to serum were larger than in individuals with BMI < 25 (
Table 3 and
Table 4). This is consistent with the hypothesis that inflammation is a part of the mechanism by which obesity develops and, thus, a low level of activation of inflammatory proteases is present in the circulation of individuals with high BMI, leading to increased proteolytic cleavage of chem163S [
37].
The differences in levels of the chemerin forms detected in circulation are easiest to explain as a consequence of differences in metabolism between people with BMI < 25 and those with BMI > 40 as well as other differences between the groups that might confound the results. The race/ethnicity in the BMI < 25 cohort included a higher fraction of Asians and fewer whites than the BMI > 40 cohort that might affect the comparison.
Both race and age are potential confounding factors to the results here. And the size of the groups is too small for a definitive analysis of race and age. To our knowledge, there have been no studies of the effects of race on chemerin levels, nor have there been any longitudinal studies on chemerin levels. When a group of healthy centenarians was compared to a group that was 70–80-year-olds, the centenarians had a lower total chemerin level than the younger group [
38]. In contrast, chemerin was found to increase with age in a study on the use of chemerin levels as a predictor of acute coronary syndromes [
39]. In a comparison of chemerin levels in serum to those in cerebrospinal fluid (CSF) in patients with neurological disorders, no correlation with age was found in serum but was in CSF [
40]. The mean age was similar in the two cohorts of people with BMI > 40 (47 ± 1.6) and BMI < 25 (47 ± 0.4), suggesting that chemerin differences due to age was not affecting the comparison between these two groups but was significantly older in the ovarian cancer patient cohort (63 ± 1.6).
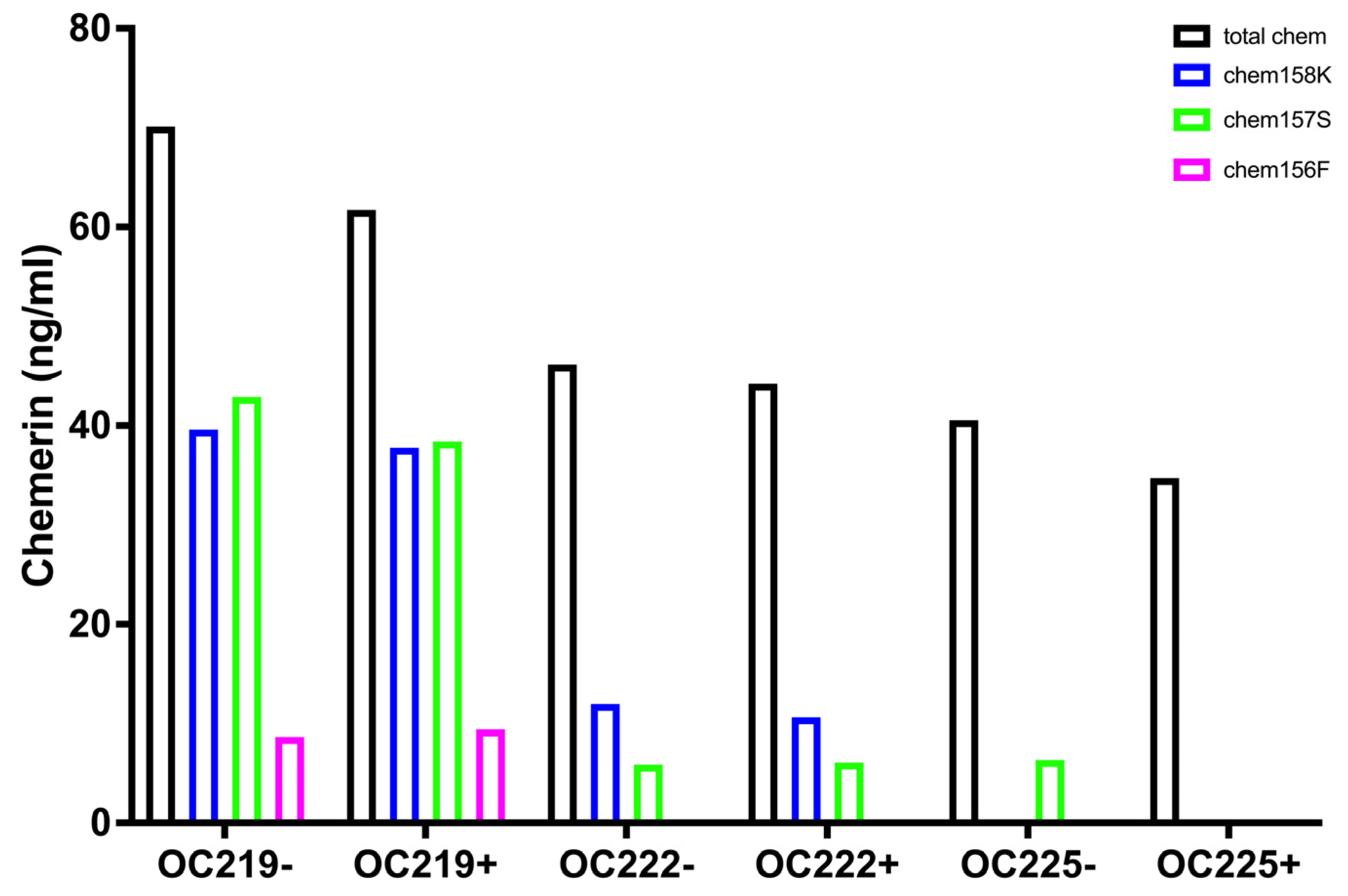
The fractions of the different chemerin forms found in ovarian carcinoma ascitic samples with a low proportion of intact precursor chem163S plus high levels of active chemerin, chem157S and chem156F, differ from those in circulation irrespective of whether they were plasma or serum and if they were from participants with BMI < 25 or patients with BMI > 40 (
Figure 1) in the low proportion of intact precursor chem163S. They are also different from the levels in synovial fluid and cerebrospinal fluid where chem158K, the inactive partially processed form, is the major form and in plasma, where the intact precursor predominates [
29].
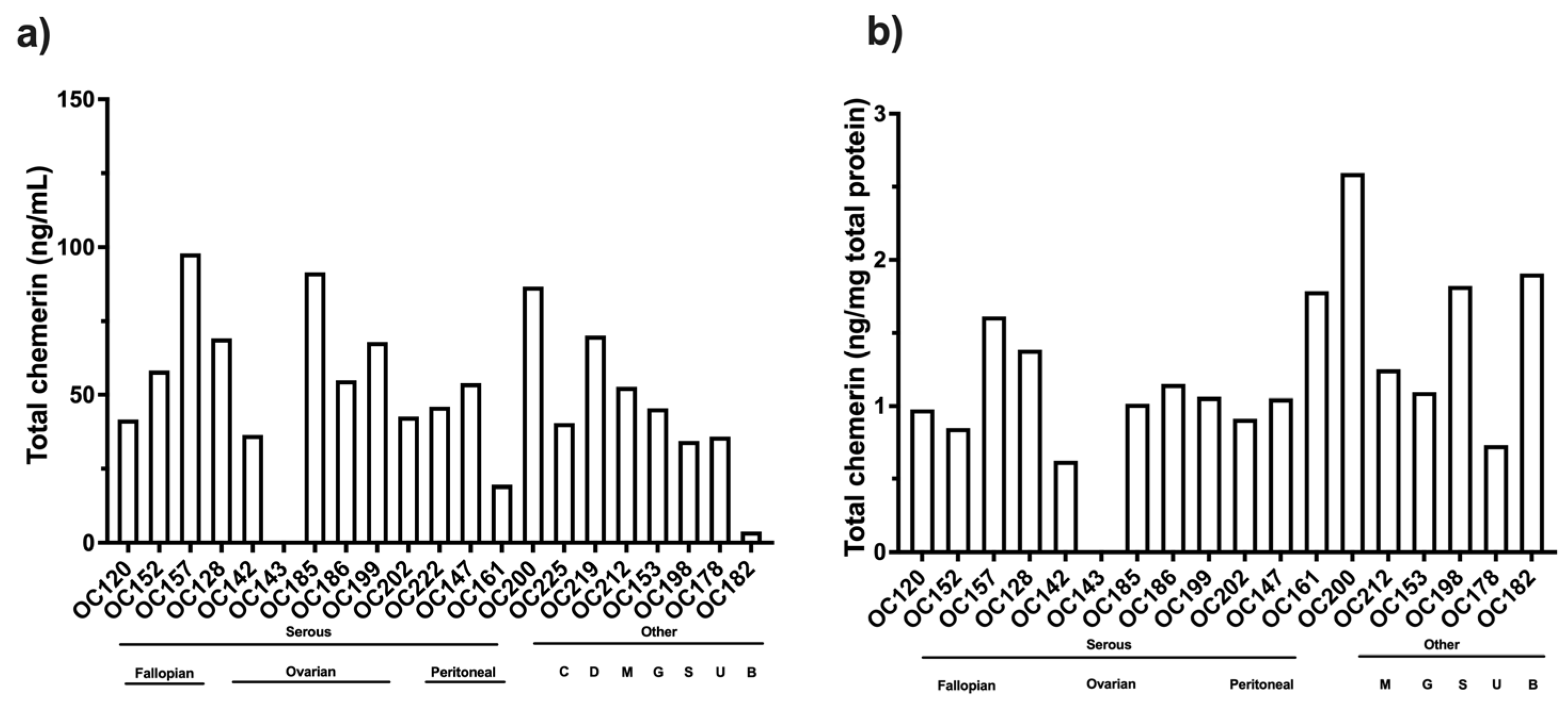
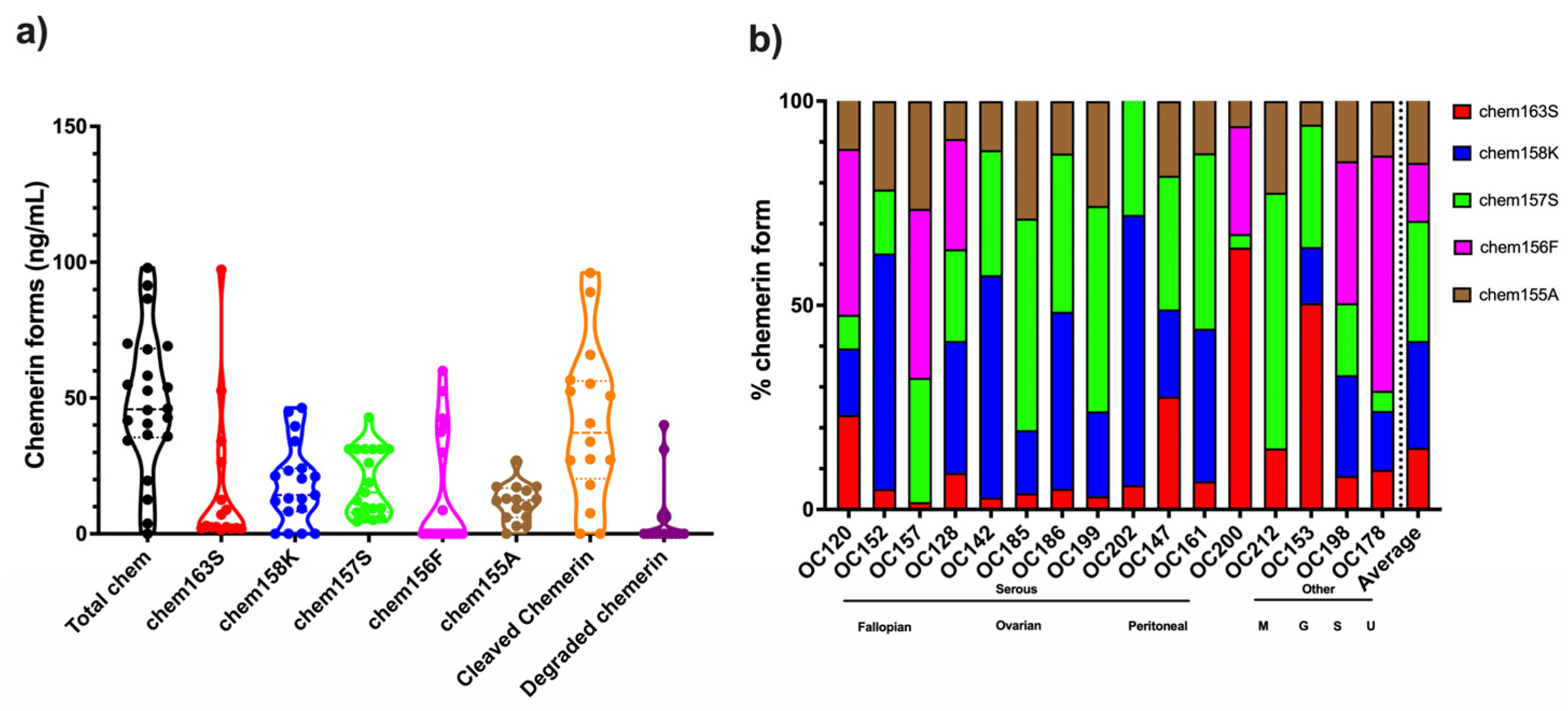
Analysis of the levels of different chemerin forms in ascitic fluid from ovarian carcinoma patients showed high levels of proteolytic processing of chemerin in all samples with an average of >80% cleavage (
Figure 7 and
Table 6). This is much higher than in the plasma and serum samples, where the maximum cleavage is <40% found in plasma samples from people with obesity (
Figure 2c). By comparing samples of ascitic fluid collected in the presence or absence of protease inhibitors with a broad spectrum of inhibition, we showed no differences in levels of chemerin forms showing that the chemerin forms had not been generated by ex vivo processing (
Figure 3). The level of chemerin proteolytic cleavage found suggests that the ovarian carcinoma is a highly inflammatory environment [
41,
42,
43]. Previously, we have found such high levels of chemerin cleavage in biopsy samples from other indications with inflammation, such as synovial fluid from arthritis patients or cerebrospinal fluid from glioblastoma patients [
25,
29]. In addition, various classes of proteases including kallikreins, coagulation, and fibrinolytic enzymes that can process chemerin have been identified in ascites [
44,
45,
46,
47].
The active chemerin forms, chem157S and chem156F, have ED
50s around 5 nM on chem1 and chem 2 [
30,
31]. In ovarian carcinoma ascitic fluid chem157S and chem156F are present at levels that would cause activation of the chemerin receptors and downstream chemerin effects and is consistent with chemerin modulating the disease. The volume of ascites fluid in ovarian carcinoma can reach >2 L, which would contain ~100 mg of total chemerin [
48].
Most patients in this study were diagnosed with high-grade serous ovarian carcinoma, but no differences were found with respect to their sites of origin. A few samples from different types of ovarian carcinoma were included in this study as well as ascites from patients with other cancers, but no differences were observed in the levels of chemerin forms. This is a very tentative conclusion because the numbers of patients were low.
Id8 cells are a well-accepted model of ovarian carcinoma that has been widely used [
49,
50]. When injected intra peritoneally into mice, ID8 cells form ascites similar to human ovarian carcinoma ascites [
23,
51]. We measured the levels of different chemerin forms in ascitic fluid from this murine model of ovarian carcinoma, finding that there were high levels of proteolytic cleavage of chemerin, including the active forms, mchem156S and mchem155F (
Figure 8,
Table 7). The levels of the active forms were sufficient to trigger responses at the mouse chemerin receptors. The distribution of the different chemerin forms was similar to that found in ascites from human ovarian carcinoma patients, suggesting that this model would be suitable for testing the role of chemerin.
Unsurprisingly, the coefficient of variance (CV) of cleaved chemerin and degraded chemerin was high because they were calculated from the results of multiple ELISAs. Overall CVs were lowest in the data sets of chemerin levels in the ascites fluid from the mouse ID8 model, which were calculated to provide a comparator for the human data. The CVs in chemerin ELISAs determined on samples from the Id8 model would be expected to be more consistent as the genetic background of the mice was identical, the mice were housed and fed identically, and ID8 cells are a cloned cell line with very little variation. In general, the CV was higher in serum samples than in plasma samples, suggesting that, when possible, chemerin and its forms should be analyzed in plasma. The CV was similar in samples from both <25 and >40 BMI cohorts of participants irrespective of their BMI. The CV was highest in the ascitic fluid samples from ovarian cancer patients possibly as this a more heterogeneous cohort than the other two cohorts with different types of ovarian cancer at various stages of the disease with differing treatments.
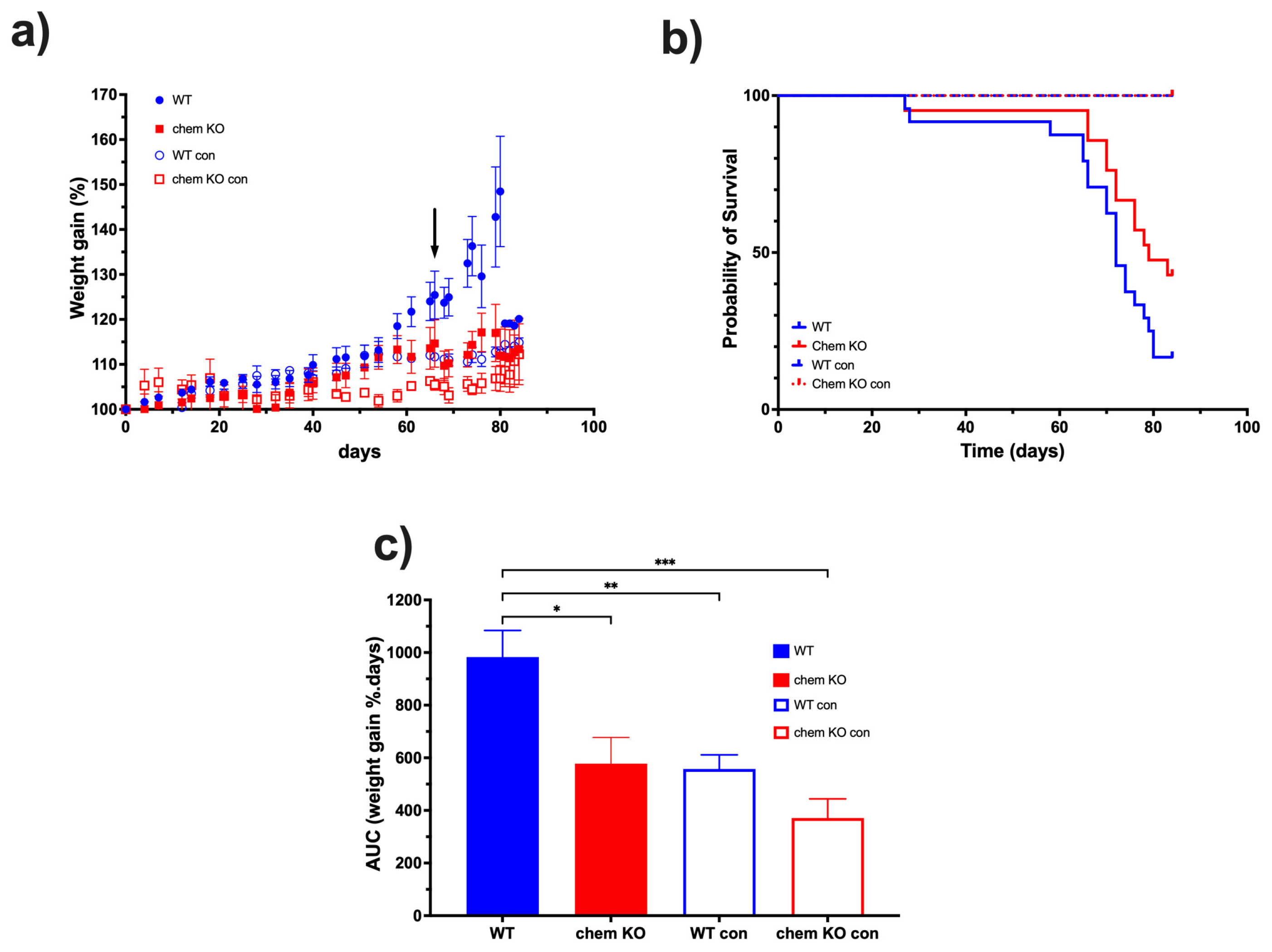
Based on the data on the high levels of proteolytic cleavage of chemerin in ascites from the ID8 model, we investigated the role of chemerin in ID8 tumor growth by comparing chemerin deficient to WT mice. ID8 tumor growth was slower in chemerin KO mice than in WT mice, leading to their prolonged survival (
Figure 9). Therefore, we confirmed that the production and activation of chemerin drives tumorigenesis in the ID8 model which is consistent with reports in some other models but is in contrast to others [
12]. This conclusion that murine ovarian carcinoma growth is enhanced by chemerin is consistent with the surprisingly high levels of active chemerin found in ovarian carcinoma ascites, suggesting that chemerin is also exacerbating tumor growth. Although the Id8 mouse tumor model represents well one type of ovarian cancer, it does not represent the various other types and also, has all the well-documented limitations of mouse models for translation to humans. In particular, the Id8 model uses a mouse cell line that does not represent the genetic heterogeneity found in all human tumors while the mouse immune system has differences from the human immune system [
52].
In addition to chemerin’s regulation of the immune system regulating the host anti-tumor response [
6,
7,
18,
53], chemerin also regulates tumor growth via its effects on energy balance and glucose and lipid metabolism [
54,
55,
56]. The tumor micro-environment contains immune cells, endothelial cells, fibroblasts, and adipocytes amongst cells mobilized by the tumor cells. Ovarian carcinoma reprograms adipocytes to mobilize their energy stores by changing their energy balance [
57]. These changes in energy metabolism, which chemerin could be regulating, contribute to the Warburg effect, possibly leading to resistance and more aggressive growth [
58,
59,
60].
Chemerin can both enhance or inhibit cancer growth, which has been reviewed for ovarian carcinoma amongst other cancers [
12,
61]. In a mouse model of clear cell renal carcinoma, for example, inhibition of chemerin by an anti-chemerin antibody improves outcomes via both chem1 and chem2 that, in turn, regulates lipid metabolism [
14,
62]. Pharmacological or genetic suppression of either chem1 or chem2 improved outcomes although the mechanistic pathways differed. In patients with oral squamous cell carcinoma, higher serum levels of chemerin correlate with worse outcomes [
63]. In squamous cell carcinoma cell lines, chemerin activates STAT-3 signaling, increasing production of IL-6 and TNF-α, leading to infiltration of neutrophils promoting tumor growth [
64].
Chemerin can also inhibit tumor growth, as demonstrated in studies including melanoma and hepatocellular carcinoma. In the B16 cell mouse model of melanoma, chemerin expression reduced tumor growth in vivo but not in vitro, showing that the effect required the host immune system or other cells present in the tumor micro-environment [
65]. Similarly, in hepatocellular carcinoma chemerin had a protective role by reducing the number of myeloid-derived suppressor cells [
65,
66]. Overall, the contradictory effects of studies on the role of chemerin in different cancers may be because the composition of the tumor micro-environment is different in different cancers. The importance of adipocyte reprogramming and changes in energy metabolism as well as in the host anti-tumor immune response varies among cancers, suggesting that chemerin’s modulation of these elements may result in the apparently contradictory data.
Obesity is recognized as a risk factor for cancer with adipose tissue providing a source of secreted factors referred to as adipokines in addition to the role of adipose tissue in metabolism [
67]. As well as regulating energy balance and other metabolic parameters, adipokines modulate the immune system and inflammation and affect ovarian carcinoma [
61,
68]. In addition to its role as an adipokine, chemerin, like other adipokines, regulates the immune system and uniquely as an adipokine can increase blood pressure [
69,
70].
In cancer, chemerin has both tumor-promoting via direct effects on the cancer cells and the tumor microenvironments and tumor-suppressing roles mostly via the immune system [
18]. Chemerin treatment enhances migration and invasion of ovarian carcinoma cell lines by activation of chem1 leading to epithelial to mesenchymal transition (EMT) in cell culture experiments [
71]. In two ovarian carcinoma cell lines, one with high metastatic potential and the other as the parental cell line, the parental cell line produces less chemerin, suggesting that higher levels of chemerin enhances metastasis [
17]. In these two cell lines, treatment with chemerin promotes proliferation and induces expression of PD-L1 inhibiting host anti-cancer immune reactions. In different ovarian carcinoma cell lines, treatment with chemerin inhibited proliferation-induced bisphenol-A and its derivatives [
72].
Ovarian carcinoma uses active chemerin to enhance growth, suggesting that chemerin antagonism is a therapeutic target. In particular, the chemerin receptors, chem 1 and chem2, as GPCRs are in a class of proteins for which many other drugs have been developed [
73,
74]. One small molecule antagonist reported to date of chem1 is 2-(alpha-naphthoyl) ethyltrimethylammonium iodide (alpha-NETA), which does not have good specificity [
75]. Other small molecules and peptide analogs that are antagonists for chem1 or chem2 have been described [
76,
77,
78]. In addition, other approaches to antagonizing chemerin signaling have been evaluated, including antibodies directed against chem1 and anti-sense oligonucleotides to inhibit chemerin production [
79,
80]. Although these approaches have shown promising data in animal models, so far none have advanced into the clinic, and no trials are listed in clinicaltrials.gov.
Ovarian carcinoma cells increase interferon alpha (IFN-α) production while in patients overall survival correlates with increases in IFN-α and interferon-induced genes [
81]. In cell lines chem1 but not chem 2 is well-expressed on all ovarian carcinoma cell lines investigated, suggesting that active chemerin binds to chem1 to signal into the cells. In tumor samples, immunohistochemistry showed the presence of both chemerin and chem1 and an association with steroid hormone receptors [
16]. Chemerin reduces IGF1 production by human granulosa cells as well as activating the MAPK pathway and AKT [
82]. Additionally, patients with high chemerin mRNA had lower overall survival than those with low chemerin mRNA. Studies of the chemerin signaling mechanism via chem1 in ovarian carcinoma cells are required to improve understanding of the reported chemerin effects. Future studies also should focus on the role of chemerin in regulating different immune cells in ovarian carcinoma as well as the effects of chemerin on the cells that form the tumor micro-environment, including fibroblasts, adipocytes, and endothelial cells.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}