Abstract
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder, characterised by the build-up of amyloid beta (Aβ) plaques and neurofibrillary tangles comprising hyper-phosphorylated tau. Increasing evidence indicates that in the early stages of AD, elevated levels of oligomeric forms of Aβ and phosphorylated tau (p-tau) gives rise to impaired synaptic function which ultimately drives AD-associated cognitive abnormalities. Thus, developing drugs that can limit the synaptic impairments that occur early in AD may have therapeutic benefits. Clinical evidence increasingly supports a link between lifestyle choices and AD risk. Indeed, there is an association between the circulating levels of the metabolic hormone leptin, mid-life obesity and disease risk, which has in turn stimulated interest in targeting the leptin system to treat AD. It is well-established that leptin readily accesses the brain, with the hippocampus, a key region that degenerates in AD, identified as a prime target for this hormone. Within the hippocampus, leptin has cognitive enhancing properties as it markedly influences the cellular events underlying hippocampal-dependent learning and memory, with significant impact on synaptic plasticity and trafficking of glutamate receptors at hippocampal excitatory CA1 synapses. Moreover, studies using a range of cell-based systems and animal models of disease indicate not only that leptin has powerful pro-cognitive effects, but also that leptin protects against the unwanted synapto-toxic effects of Aβ and tau, as well as enhancing neuronal cell viability. Moreover, recent studies have demonstrated that smaller leptin-based molecules replicate the full repertoire of protective features of whole leptin. Here we review the evidence that the leptin system is a potential novel avenue for drug discovery in AD.
1. Introduction
Leptin, the obese (ob) gene product, is an important metabolic hormone that is primarily made in white adipocytes. The circulating levels of leptin are directly proportional to body fat stores [1], however, daily fluctuations in leptin levels arise in response to altered feeding status. Peripherally derived leptin is readily transported across the blood–brain barrier into the brain where it targets the key hypothalamic nuclei involved in regulating energy homeostasis. Leptin receptors (LepRs), which comprise six distinct LepR isoforms (LepRa-f), mediate the effects of leptin. LepRb, which is known as the long form due to its extended C-terminal, is the main isoform with signalling capacity. Like other class I cytokines [2], leptin binding to LepRs drives the homo-dimerization of the receptor, leading to phosphorylation and activation of janus tyrosine kinase 2 (JAK2), which in turn stimulates different signalling pathways. In neurons, the activation of LepRs drives JAK2-signal transducers and activators of transcription (STAT3) as well as phosphoinositide 3-kinase (PI 3-kinase) and ERK signalling [3].
It is well-documented that LepRs are distributed throughout the brain. In line with leptin’s role in regulating food intake and body weight, LepRs are highly concentrated in hypothalamic regions that control energy homeostasis [4,5]. However, high levels of LepR expression have been detected in limbic regions, particularly within the hippocampus [4,6]; a brain region that is critically involved in learning and memory processes [7]. Using dual-labelling immunocytochemical techniques, Shanley et al. [8] were the first to probe the subcellular distribution of LepRs and verified LepR expression at hippocampal synapses. Subsequent studies also identified LepRs expression at cerebellar synapses [9]. There is good evidence for the expression of LepRs at excitatory synapses as LepR co-localises with GluN2A-containing NMDA receptors (NMDARs) [10], which points towards leptin playing a potential regulatory role at hippocampal excitatory synapses. Indeed, widespread evidence supports this notion, as leptin has been shown to markedly influence synaptic transmission at hippocampal CA1 synapses [3].
2. Regulation of Hippocampal Excitatory Synaptic Function by Leptin
Activity-dependent changes in the strength of hippocampal excitatory synapses are known as synaptic plasticity. Two of the most well-documented forms of synaptic plasticity are long-term potentiation (LTP) and long-term depression (LTD); processes that are critically involved in hippocampal-dependent learning and memory [7]. Numerous reports have identified that alterations in the functioning of leptin significantly impact hippocampal synaptic plasticity. Indeed, impairments in both LTP and LTD have been observed in obese rodents (db/db mice; fa/fa rats) that are leptin-insensitive due to lepR mutations [11,12]. In line with the role of synaptic plasticity in memory function, db/db mice and fa/fa rats also exhibit deficits in their ability to perform hippocampal-dependent memory tasks [13,14]. Thus, studies in db/db mice and fa/fa rats have identified impaired performance in the Morris water maze, indicating deficits in hippocampal-dependent spatial memory. Moreover, obese leptin-insensitive (fa/fa) rats exhibit diminished ability to undertake the variable-interval delayed alternation task, suggesting deficits in hippocampal-dependent memory. Direct hippocampal administration of leptin in wild type rodents is also reported to result in the facilitation of synaptic plasticity, and it improves performance in hippocampal-dependent memory tasks [15,16]. Thus, a bi-lateral administration of leptin into the hippocampus facilitates performance in the T-maze footshock avoidance task [15], whereas an intravenous application of leptin into wild-type rodents leads to improved performance in passive avoidance and spatial memory tasks [17]. Collectively, these findings indicate that leptin boosts hippocampal learning and memory and therefore leptin acts as a potential cognitive enhancer.
Within the hippocampus, two distinct excitatory inputs converge on CA1 pyramidal neurons. The most well-studied is the Schaffer-collateral (SC) input which forms part of the classical tri-synaptic hippocampal circuitry. The other input to CA1 neurons is the temporoammonic (TA) input, which involves direct innervation by the entorhinal cortex. Activity-dependent synaptic plasticity at TA-CA1 synapses is implicated in episodic memory, as well as spatial memory [18,19]. Evidence is growing that leptin can modify the efficacy of excitatory synaptic transmission at both SC-CA1 and TA-CA1 synapses [20,21,22].
Electrophysiological studies by Shanley et al. [23] first showed that the direct application of leptin to hippocampal slices drives the conversion of short-term potentiation (STP) into LTP. Subsequent studies established that acute exposure to leptin resulted in the induction of a novel form of LTD, and it can reverse (or de-potentiate) LTP at juvenile SC-CA1 synapses [24,25]. Conversely, at adult SC-CA1 synapses, leptin induces a sustained increase in synaptic efficacy (leptin-induced LTP) that persists after leptin washout [22]. A common feature in these studies is that NMDA receptor activation is pivotal for leptin’s effects on synaptic efficacy at SC-CA1 synapses. Thus, the ability of leptin to regulate synaptic transmission is completely inhibited following the blockade of NMDA receptors with D-AP5 [20,22,26]. Interestingly, the polarity of leptin’s effects on hippocampal synaptic efficacy is highly dependent on the subunit composition of NMDA receptors. For instance, the leptin-driven depression of synaptic transmission at SC-CA1 synapses early in postnatal development involves the activation of GluN2B-containing NMDA receptors as the effects are blocked by ifenprodil [26]. In contrast, in an adult hippocampus, the persistent increase in synaptic transmission induced by leptin depends on GluN2A subunits as prior treatment with NVP-AAM077, an NMDA receptor antagonist with preferential selectivity for GluN2A subunits [26,27]. The activation of NMDA receptors with distinct molecular identity also mediates the effects of leptin at TA-CA1 synapses. Thus, in a juvenile hippocampus (P14–21), the ability of leptin to induce LTP at TA-CA1 synapses requires stimulation of GluN2B-containing NMDA receptors, whereas at adult TA-CA1 synapses GluN2A-containing NMDA receptors are pivotal for the persistent synaptic depression induced by leptin [21,28].
There is now good evidence that NMDA-dependent synaptic plasticity at TA-CA1 synapses is pivotally involved in learning and memory [18,19]. Moreover, increasing evidence indicates that TA-CA1 synapses are regulated by various neuromodulators, which in turn influences not only synaptic function and memory, but also other higher brain functions. Indeed, TA-CA1 synapses are regulated by corticosterone and serotonin, and this plays a pivotal role in mediating the synaptic alterations associated with chronic stress [29,30]. In line with TA-CA1 synapses being a target for modulation, leptin regulates the functioning of TA-CA1 synapses at different stages of development. In juvenile tissue, the application of leptin results in a novel form of NMDA-dependent TA-CA1 LTP [20] whereas in adult tissue, leptin gives rise to a persistent synaptic depression [21,28]. Furthermore, divergent cellular mechanisms underlie the opposing effects of leptin at TA-CA1 and SC-CA1 synapses, as leptin-induced TA-CA1 LTP requires PI3-kinase, whereas ERK signalling is implicated in synaptic depression induced by leptin at SC-CA1 synapses [3]. Interestingly, in occlusion studies, activity-dependent synaptic plasticity at TA-CA1 synapses has been found to display similar expression mechanisms to the persistent synaptic changes induced by leptin [20,21].
3. Leptin Regulates the Trafficking of Glutamate Receptors
In line with the pivotal role of NMDA receptors in mediating leptin’s effects, there is now significant evidence from cellular studies in primary neurons and brain slices, that exposure to leptin facilitates NMDA-induced responses in the hippocampus [23,26]. Moreover, in two electrode voltage clamp studies performed in Xenopus oocytes expressing a combination of LepRs and GluN1/GluN2A NMDA receptors, NMDA-induced currents are significantly larger after treatment with leptin [23,31]. Interestingly, in Xenopus oocyte studies, leptin increased the magnitude of the inward currents induced by maximal concentrations of NMDA. This suggests that leptin rapidly alters the density of plasma membrane NMDA receptors and therefore leptin may regulate the trafficking of NMDA receptors [31]. In agreement with this, recent studies by Bland et al. [32] have revealed that the formation of glutamatergic synapses is regulated by leptin, and that the capacity of leptin to boost trafficking of NMDA receptor contributes to synaptogenesis.
In addition to controlling the NMDA receptor trafficking, the mobility of AMPA receptors to and away from synapses is also modified by leptin. Thus, in adult hippocampal slices, the ability of leptin to induce SC-CA1 LTP is coupled to an increase in the rectification of synaptic AMPA receptors. This suggests that the leptin-driven persistent increase in synaptic strength is due to the synaptic insertion of GluA2-lacking AMPA receptors [22]. Prior treatment with philanthotoxin, an inhibitor of GluA2-lacking AMPA receptors, prevented the effects of leptin, thereby supporting the notion that leptin drives the delivery of these AMPA receptors to synapses. These findings are consistent with the pivotal role that AMPA receptor trafficking plays in hippocampal synaptic plasticity [33], but there are also parallels with the transient alterations in the molecular composition of synaptic AMPA receptors observed after the induction of NMDA-dependent LTP [34]. Studies in primary hippocampal neurons [22] support these electrophysiological findings, as the exposure to leptin augments the plasma membrane and synaptic expression of the AMPA receptor subunit, GluA1. Moult et al. [22] further explored the cellular mechanisms involved downstream of LepRs and identified a key role for the inhibition of the phosphatase PTEN. As PTEN promotes dephosphorylation of PIP3 to PIP2, the inhibition of the PTEN function would ultimately lead to an increase in intracellular PIP3 levels. Consequently, it is likely that leptin-driven inhibition of PTEN leads to an elevation in intracellular PIP3 levels, which in turns promotes the insertion of GluA2-lacking AMPA receptors into synapses [22]. Collectively, the ability of leptin to modify glutamate receptor trafficking, combined with its persistent effects on excitatory synaptic efficacy at CA1 synapses, suggests that it has potential cognitive enhancing actions.
4. Leptin and Neurodegenerative Disease
Evidence from clinical studies indicates that key features of human cases of Alzheimer’s disease (AD) and related dementias are deficits in cognition and memory. Although most AD cases are sporadic, increasing evidence indicates that lifestyle choices contribute significantly to AD risk later in life [35,36]. Recent studies indicate an association between circulating leptin levels and AD. Moreover, clinical findings indicate that obesity in mid-life markedly influences AD risk [37,38]. The association of body weight with the development of AD indicates the possible involvement of leptin and other adipokines, as adipose tissue is the prominent site for generation of various hormones involved in energy homeostasis control [39]. Consequently, as an obese phenotype is caused by leptin resistance, it is likely that alterations in the functioning of leptin and/or leptin resistance later in life contribute to the development of AD. Indeed, attenuated plasms leptin levels are common in AD patients [40], and clinical evidence has identified correlations between weight loss and the progression of AD [41]. A prospective study of the Framingham cohort observed much lower incidence of AD in non-obese individuals with high leptin levels, than those with reduced leptin levels [42]. Recent studies have also identified a correlation between plasma leptin levels and Aβ levels, suggesting molecular interplay between the leptin metabolism and brain amyloid deposition [43]. Furthermore, functional imaging studies of brain atrophy in healthy middle-aged adults observed that the higher bioavailability of leptin was associated with greater protection of brain white matter, indicating that in mid-life, elevated blood leptin bioavailability may protect against the risk of dementia [44]. However, not all studies have observed an association between leptin and impaired cognitive function or have found any change in plasma leptin levels in AD patients [45,46], indicating that leptin’s role is liable to be complex. It is likely that the overall AD risk involves the consideration of a combination of potential causative factors, including interactions between leptin and other metabolic hormones, as well as any underlying metabolic disease.
Although there are complexities around the causative role of leptin in human cases of AD, several studies have explored the role of leptin in various animal models of AD. Thus, in APPSwe and CRND8 mice, the circulating levels of leptin are markedly less than their wild type littermates at the same age [15,47], suggesting an impaired metabolic function in AD. Moreover, treatment with leptin reversed the AD-related cognitive impairments in the AD mouse models [15], indicating not only that these deficits are leptin-dependent but also that it is feasible that dysfunctions in the leptin system contribute to the development of AD in rodents. Recent studies have detected altered leptin and LepR brain expression in the 5XFAD rodent model of AD [48], which also supports the motion that impaired leptin and/or lepR-driven signalling may be involved in AD in rodents.
5. Leptin Has Protective Actions in the CNS
Since the early studies that uncovered that leptin deficiencies resulted in reduced brain weight due to decreased neuronal viability [49], it is now well-established that leptin has protective actions in the CNS. Indeed, numerous reports have confirmed that leptin can promote neuronal survival as well as limiting the degree of neuronal cell death [50,51]. Recent evidence has implicated dynamic alterations in mitochondrial structure and function in mediating leptin’s protective actions. Thus, leptin protects the hippocampal neurons against glutamate-induced excitotoxicity by stabilising the mitochondrial membrane [52]. Furthermore, the treatment of hippocampal cells with leptin prevents mitochondrial membrane depolarisation and mitochondrial fragmentation induced by toxic Aβ1–42, via a process involving the altered expression of mitochondrial fusion and fission proteins [53]. Interestingly, mitochondrial dysfunction is a common feature in neurodegenerative disorders, and correcting mitochondrial dysfunction is a growing avenue for developing novel therapies for these CNS disorders [54]. Additionally, there is increasing evidence that leptin displays protective actions in other brain disorders. Indeed, exposure to leptin protects against the aberrant brain changes that occur in various animal models of ischaemic stroke. The pathological injury that occurs following ischaemic stroke has been linked to mitochondrial damage and production of oxygen free radicals [55,56]. Leptin treatment not only limits infarct size but also reduces the levels of oxygen free radicals by enhancing the expression of antioxidants and superoxide dismutase [56]. The leptin-driven protective mechanisms have been well-characterised in a variety of models of neuronal cell death. Two key signalling cascades activated downstream of LepRs, namely JAK-STAT3 and PI3-kinase, have been identified as fundamental for leptin-driven neuroprotection [52,57,58,59]. Indeed, in a model of cerebral ischaemic/reperfusion injury, leptin inhibits neuronal apoptosis and other pathological changes via a PI3-kinase-dependent mechanism [60]. Similarly, leptin-driven stimulation of PI3-kinase is vital for leptin’s ability to limit MPP+-induced cell death in SH-SY5Y cells [61], and to prevent Aβ1–42-induced apoptosis in hippocampal neurons [57]. Furthermore, JAK-STAT3 signalling contributes to leptin’s ability to reduce injury in models of cerebral ischaemic [62,63]. Recent studies have also implicated leptin-driven regulation of microglia in the neuroprotective actions of leptin [64], suggesting that leptin may limit neuronal damage via controlling the pro-inflammatory factors from microglia. However, further studies are required to uncover the precise role of microglia, and the interplay between microglia and the other neuroprotective mechanisms driven by leptin.
6. Leptin Has Protective Effects at Synapses in Early AD
Accumulation of amyloid beta (Aβ) and formation of amyloid plaques are well-established pathological features of AD. Treatment with leptin has been shown to decrease expression of β- and γ secretase, two key enzymes that catalyse the cleavage of amyloid precursor protein (APP) into the toxic forms of Aβ [65,66]. Thus, in wild-type H4 cells, treatment with leptin attenuated the expression of several key parts of the γ-secretase enzyme complex, including PS1, PEN2, nicastrin, and APH1B, by up to 50% [66]. Moreover, in the same study it was observed that leptin reduced the levels of Aβ by directly inhibiting the activity of β-secretase, the key enzyme that catalyses production of toxic Aβ1–42. Other studies performed in neuronal cells and in AD models that overexpress Aβ [67] found that exposure to leptin decreased the extracellular levels of Aβ by increasing LRP1-dependent uptake of Aβ, thereby reducing the overall amyloid load in the brain. Additionally, leptin reportedly increases the expression of the insulin-degrading enzyme which facilitates the degradation of Aβ [65].
Build-up of neurofibrillary tau tangles is another well-documented pathological feature that occurs in AD. Tau is a microtubule-associated protein that becomes hyper-phosphorylated in AD, due to actions of the serine/threonine kinase, glycogen synthase kinase 3β (GSK3β). Recent reports indicate that the treatment of PC12 neuronal cells with leptin stimulates PI3-kinase which drives the inhibition of GSK3β, ultimately leading to attenuated levels of phosphorylated tau [68,69]. In line with leptin regulating the levels of phosphorylated tau (p-tau), insensitivity or resistance to leptin is linked to enhanced neuronal expression of p-tau in rodents [57]. Moreover, markedly reduced circulating levels of leptin have been detected in rodents with progressive tau-related pathology [70].
In the early stages of AD, exposure to oligomeric Aβ gives rise to impaired functioning of excitatory synapses. In acute hippocampal slices, application of oligomeric Aβ markedly influences hippocampal synaptic plasticity as Aβ blocks the induction of LTP, and it facilitates long-term depression (LTD) [71,72]. Furthermore, in accordance with the pivotal role of AMPA receptor trafficking in synaptic plasticity [33], acute treatment of hippocampal neurons with oligomeric Aβ drives the synaptic removal of AMPA receptors [71]. Studies by Doherty et al. [57] demonstrated that leptin protects against the aberrant synapto-toxic effects of Aβ as the ability of Aβ to block LTP induction is prevented in brain slices treated with leptin. Similarly, treatment with leptin prevented the facilitation of hippocampal LTD induced by Aβ [57]. Moreover, in parallel studies utilising primary hippocampal neurons, the ability of Aβ to remove the AMPA receptor subunit, GluA1 from synapses was blocked after treatment with leptin [57]. In line with the known protective actions of the PI3-kinase signalling cascade, the ability of leptin to prevent the aberrant effects of Aβ on synaptic plasticity and AMPA receptor trafficking involves the stimulation of a PI 3-kinase-dependent pathway [57]. Previous studies have identified that GSK3β inhibitors not only mirror but also occlude the effects of leptin [58,68]. Moreover, as PI 3-kinase activity drives inhibition of GSK3β, it is feasible that GSK3β is a crucial component in leptin’s protective effects against the aberrant effects of Aβ on synaptic function.
Tau is a microtubule associated protein that plays a pivotal role in preserving the stability and flexibility of microtubules, which in turn helps to sustain neuronal structure [73]. Tau is usually highly expressed in axons, but in AD tau becomes hyper-phosphorylated which uncouples tau from microtubules and drives tau from its usual locus in axons into synapses [74]. This in turn ultimately results in impaired excitatory synaptic function and loss of synapses [75]. It is known that the detrimental effects of tau on hippocampal excitatory synapses correlate well with the early cognitive deficits reported in AD [76,77]. Consequently, identifying ways to prevent the synapto-toxic effects of tau in the early pre-clinical stages of AD is likely to have beneficial effects in AD. Indeed, our recent immunocytochemical studies support this, as the treatment of a cellular model of tau-related synaptic dysfunction with leptin prevented tau mis-location to dendrites and synapses [58]. In this model, chronic treatment with Aβ oligomers drives the activation of GSK-3β, which in turn promotes the phosphorylation of tau [78] and the subsequent delivery of tau to synapses [74,75]. There is good evidence that GSK-3β-dependent phosphorylation of tau at serine 396 (Ser396) is significantly elevated in AD and that phosphorylation at the tau Ser396 site is crucial for the aberrant movement of tau to synapses [58]. Our recent studies demonstrated that leptin prevented tau phosphorylation at Ser396, thereby inhibiting the synaptic insertion of tau [58]. In line with previous work that uncovered that leptin-driven inhibition of GSK-3β reduces tau phosphorylation in neuronal cells [68], the capacity of leptin to restrict tau movement to hippocampal synapses also involves PI 3-kinase-driven inhibition of GSK-3β [58].
Previous studies have identified that tau phosphorylation and its resulting delivery to synapses leads to impaired synaptic function due to the synaptic removal of AMPA receptors [74]. In accordance with this, trafficking of tau to synapses is correlated with attenuated synaptic expression of GluA1-containing AMPA receptors, thereby confirming that tau phosphorylation prompts AMPA receptor endocytosis and removal from synapses [58]. Consistent with this, the direct application of oligomeric tau to hippocampal neurons also reduced the surface expression of GluA1, supporting the notion that oligomeric tau drives the internalisation of AMPA receptors [58]. Our recent studies have shown that leptin protects against these tau-dependent synaptic impairments, as AMPA receptor removal from synapses induced by either tau phosphorylation or the direct administration of oligomeric tau is prevented by prior treatment with leptin [58]. Moreover, in acute brain slices, exposure to leptin blocked the oligomeric tau-driven inhibition of activity-dependent LTP at hippocampal SC-CA1 synapses [58]. Thus, leptin not only attenuates GSK3β-driven phosphorylation of tau, which in turn limits tau trafficking to synapses, but leptin also restricts the tau-driven synaptic abnormalities by preventing synaptic removal of AMPA receptors and inhibiting hippocampal synaptic plasticity. Increasing evidence supports a pivotal role for abnormal build-up of tau in the cognitive decline associated with AD [76]. Consequently, leptin’s capacity to restrict the aberrant synapto-toxic effects of tau has important implications for leptin’s protective role in neurodegenerative disorders like AD.
7. Leptin Has Pro-Cognitive Effects in AD Models
There is now good evidence from studies using obese rodents (db/db mice; fa/fa rats) that leptin plays a crucial role in learning and memory processes, as impaired spatial memory is observed in rodents with LepR mutations which result in leptin insensitivity [14]. In addition to enhancing memory in wild-type rodents, significant evidence now indicates that exposure to leptin has pro-cognitive actions in rodent models of AD. Thus, improvements in novel object recognition tasks, and the cue fear conditioning test were detected in CRND8 transgenic mice (TgCRND8) following treatment with leptin for 8 weeks [47]. Intra-cerebroventricular administration of a leptin viral gene into APP/PS1 transgenic mice also rescued Aβ-associated memory deficits [67], whereas spatial memory impairments induced in rats by ICV administration of Aβ were alleviated following chronic leptin treatment [79]. On the flip side, deficiencies and/or loss of leptin are reported to exacerbate cognitive decline in AD, as cognitive function is reportedly much poorer in double-mutant (APP(+)-ob/ob) mice compared to APP(+) mice [80].
8. Therapeutic Potential of Leptin and Leptin-Derived Peptides
Clinical evidence has identified that treating leptin-deficient patients with leptin results in improved cognitive function [81,82]. Although it is well-documented that leptin-based therapies can be safely used in humans, there has been a paucity of clinical studies evaluating the effectiveness of leptin in AD patients. However, what is clear is that it is unlikely that all AD patients would benefit from leptin-based therapies. Indeed, individuals with leptin-resistance due to mid-life obesity are unlikely to see any benefit, whereas those with low circulating leptin levels may be highly responsive to leptin replacement. Indeed, metreleptin, which is a modified synthetic form of human leptin, has already been approved for treatment of lipodystrophy [82]. Moreover, clinical studies have identified beneficial effects of metreleptin on cognitive function in individuals with low leptin levels due to lipodystrophy [83,84]. Marked improvements in cognition have also been observed following metreleptin treatment in anorexia patients, who also display low circulating levels of leptin [85,86]. Consequently, as there is good evidence that leptin-based therapies have therapeutic efficacy in individuals with low leptin levels, it feasible that the use of leptin-based therapies will have beneficial effects in AD, but the usefulness may be limited to a specific cohort of AD patients.
As leptin has wide-spread neuronal actions, it is key that steps are taken to circumvent or reduce the likelihood of potential unwanted side effects. Accordingly, developing leptin-based drugs that are modified to target the specific brain regions, such as the hippocampus and cortex, that degenerate in AD is one potential way forward. Additionally, fragments of the whole leptin molecule (eg Leptin116–130) are not only bioactive [87] but also recapitulate the hippocampal actions of leptin [88], indicating that small molecule leptin mimetics could have benefits in AD. Indeed, our recent work has identified that leptin116–130 prevents the aberrant effects of Aβ on hippocampal synaptic plasticity, AMPA receptor trafficking, and neuronal toxicity [88]. Moreover, peripheral administration of leptin116–130 mirrored the pro-cognitive actions of whole leptin as mice treated with leptin116–130 displayed improved performance in behavioural tests of episodic-like memory [88]. Our more recent studies have observed similar results using smaller leptin-based hexamers, such that four out of the eight hexamers derived from leptin116–130 were found to replicate the full spectrum of pro-cognitive and neuroprotective actions of leptin [89]. Interestingly, a small molecule leptin mimetic, MA-[D-Leu-4]-OB3 has also been reported to enhance episodic memory in rodent models of diabetes [90]. Collectively, this confirms not only that leptin116–130 and the bioactive hexamers can readily access the brain and influence hippocampal function, but these leptin-based molecules also offer potential novel avenues for developing new drugs to treat AD in the future. Although the identification of leptin-based molecules as potential therapeutic targets is a promising advance, there are still some challenges to be overcome before these agents could be used in AD patients. Significant pre-clinical studies are still needed, before leptin-derived agents could be used in clinical trials. Indeed, it is vital that in vivo studies are undertaken to determine the potential efficacy of the various bioactive leptin fragments/hexamers in rodent models of AD. Further studies are required to evaluate the pharmacokinetic properties of the leptin-based molecules, as it is not clear to what extent these agents are metabolised after peripheral or oral administration, and what proportion of the parent molecule reaches the brain target sites.
9. Conclusions
The hippocampus is now established as a key target site for the hormone leptin in the CNS, with significant evidence indicating that leptin has pro-cognitive properties. It is well-documented that leptin can modify excitatory synaptic strength at SC-CA1 and TA-CA1 synapses, which has important implications for its role in hippocampal learning and memory. Moreover, as the hippocampus is a key site for degeneration in AD, the regulatory actions of leptin are also crucial in age-related neurodegenerative disorders associated with cognitive deficits. Indeed, clinical evidence increasingly supports a link between brain metabolic health and AD, with an identified association between the circulating levels of leptin and the risk of developing AD later in life. Furthermore, significant evidence from studies using cellular and rodent models of AD indicates that leptin-based treatments have pro-cognitive and neuroprotective actions, thereby signifying that targeting the leptin system may be advantageous in the treatment of AD (see Figure 1). However, the use of leptin therapeutically is liable to be limited by metabolic status, as leptin is unlikely to be effective in individuals with resistance to leptin. Consequently, further clinical evaluation of the potential feasibility and efficacy of using leptin-based treatments in AD is needed.
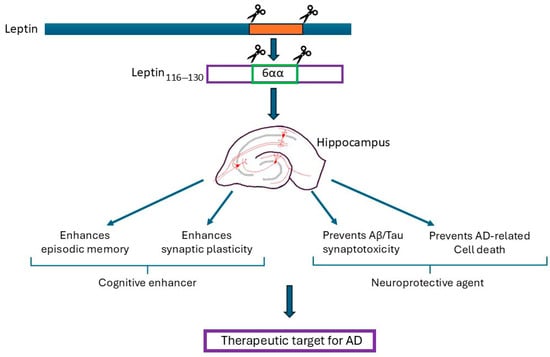
Figure 1.
The summary of the potential therapeutic benefits of leptin and leptin-derived fragments. The hippocampus is a key brain target for the actions of leptin. Exogenous application of leptin results in pro-cognitive effects as leptin facilitates synaptic plasticity and AMPA receptor trafficking; the key cellular changes required for hippocampal-dependent learning and memory. The administration of leptin into rodents also results in enhanced performance in various types of memory, including episodic-like memory which is the first memory that is lost in AD. In various models of AD, treatment with leptin prevents the unwanted toxic synaptic effects of β-amyloid (Aβ) and p-tau, thereby protecting against the acute synaptotoxic effects that occur in early AD. Moreover, leptin limits the chronic neurotoxic effects of Aβ and tau, with enhanced neuronal viability observed after treatment with leptin. Collectively, studies using cellular and rodent models of AD have identified that leptin-based treatments have pro-cognitive and neuroprotective actions, and therefore have potential therapeutic benefit in AD. Moreover, the potential cognitive enhancing and neuroprotective actions of whole leptin are replicated by smaller leptin fragments (leptin116–130), and leptin hexamers (eg leptin116–121).
Funding
This research was funded by Alzheimer’s Society (Grant 449: AS-PhD-18-007).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The author declares no conflict of interest.
References
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef]
- Ihle, J.N. Cytokine receptor signalling. Nature 1995, 377, 591–594. [Google Scholar] [CrossRef]
- Irving, A.; Harvey, J. Regulation of hippocampal synaptic function by the metabolic hormone leptin: Implications for health and disease. Prog. Lipid Res. 2021, 82, 101098. [Google Scholar] [CrossRef]
- Elmquist, J.K.; Bjørbaek, C.; Ahima, R.S.; Flier, J.S.; Saper, C.B. Distributions of leptin receptor mRNA isoforms in the rat brain. J. Comp. Neurol. 1998, 395, 535–547. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Campfield, L.A.; Burn, P.; Baskin, D.G. Identification of targets of leptin action in rat hypothalamus. J. Clin. Investig. 1996, 98, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.G.; Hoggard, N.; Williams, L.M.; Lawrence, C.B.; Hannah, L.T.; Trayhurn, P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996, 387, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Shanley, L.J.; Irving, A.J.; Rae, M.G.; Ashford, M.L.; Harvey, J. Leptin inhibits rat hippocampal neurons via activation of large conductance calcium-activated K+ channels. Nat. Neurosci. 2002, 5, 299–300. [Google Scholar] [CrossRef]
- Irving, A.J.; Wallace, L.; Durakoglugil, D.; Harvey, J. Leptin enhances NR2B-mediated N-methyl-D-aspartate responses via a mitogen-activated protein kinase-dependent process in cerebellar granule cells. Neuroscience 2006, 138, 1137–1148. [Google Scholar] [CrossRef]
- O’Malley, D.; MacDonald, N.; Mizielinska, S.; Connolly, C.N.; Irving, A.J.; Harvey, J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol. Cell. Neurosci. 2007, 35, 559–572. [Google Scholar] [CrossRef]
- Gerges, N.Z.; Aleisa, A.M.; Alkadhi, K.A. Impaired long-term potentiation in obese zucker rats: Possible involvement of presynaptic mechanism. Neuroscience 2003, 120, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Ramakers, G.M.; Gispen, W.H.; Biessels, G.J. Hyperinsulinemia in rats causes impairment of spatial memory and learning with defects in hippocampal synaptic plasticity by involvement of postsynaptic mechanisms. Exp. Brain Res. 2013, 226, 45–51. [Google Scholar] [CrossRef]
- Manuel Sánchez, D.M.; Limón, D.; Silva Gómez, A.B. Obese male Zucker rats exhibit dendritic remodeling in neurons of the hippocampal trisynaptic circuit as well as spatial memory deficits. Hippocampus 2022, 32, 828–838. [Google Scholar] [CrossRef]
- Winocur, G.; Greenwood, C.E.; Piroli, G.G.; Grillo, C.A.; Reznikov, L.R.; Reagan, L.P.; McEwen, B.S. Memory impairment in obese Zucker rats: An investigation of cognitive function in an animal model of insulin resistance and obesity. Behav. Neurosci. 2005, 119, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Banks, W.A.; Morley, J.E. Effects of leptin on memory processing. Peptides 2006, 27, 1420–1425. [Google Scholar] [CrossRef]
- Wayner, M.J.; Armstrong, D.L.; Phelix, C.F.; Oomura, Y. Orexin-A (Hypocretin-1) and leptin enhance LTP in the dentate gyrus of rats in vivo. Peptides 2004, 25, 991–996. [Google Scholar] [CrossRef]
- Oomura, Y.; Hori, N.; Shiraishi, T.; Fukunaga, K.; Takeda, H.; Tsuji, M.; Matsumiya, T.; Ishibashi, M.; Aou, S.; Li, X.; et al. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides 2006, 27, 2738–2749. [Google Scholar] [CrossRef] [PubMed]
- Brun, V.H.; Leutgeb, S.; Wu, H.Q.; Schwarcz, R.; Witter, M.P.; Moser, E.I.; Moser, M.B. Impaired spatial representation in CA1 after lesion of direct input from entorhinal cortex. Neuron 2008, 57, 290–302. [Google Scholar] [CrossRef]
- Stokes, J.; Kyle, C.; Ekstrom, A.D. Complementary roles of human hippocampal subfields in differentiation and integration of spatial context. J. Cogn. Neurosci. 2015, 27, 546–559. [Google Scholar] [CrossRef]
- Luo, X.; McGregor, G.; Irving, A.J.; Harvey, J. Leptin Induces a Novel Form of NMDA Receptor-Dependent LTP at Hippocampal Temporoammonic-CA1 Synapses. eNeuro 2015, 2, ENEURO.0007-15.2015. [Google Scholar] [CrossRef]
- McGregor, G.; Clements, L.; Farah, A.; Irving, A.J.; Harvey, J. Age-dependent regulation of excitatory synaptic transmission at hippocampal temporoammonic-CA1 synapses by leptin. Neurobiol. Aging 2018, 69, 76–93. [Google Scholar] [CrossRef]
- Moult, P.R.; Cross, A.; Santos, S.D.; Carvalho, A.L.; Lindsay, Y.; Connolly, C.N.; Irving, A.J.; Leslie, N.R.; Harvey, J. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J. Neurosci. 2010, 30, 4088–4101. [Google Scholar] [CrossRef]
- Shanley, L.J.; Irving, A.J.; Harvey, J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 2001, 21, RC186. [Google Scholar] [CrossRef]
- Durakoglugil, M.; Irving, A.J.; Harvey, J. Leptin induces a novel form of NMDA receptor-dependent long-term depression. J. Neurochem. 2005, 95, 396–405. [Google Scholar] [CrossRef]
- Moult, P.R.; Milojkovic, B.; Harvey, J. Leptin reverses long-term potentiation at hippocampal CA1 synapses. J. Neurochem. 2009, 108, 685–696. [Google Scholar] [CrossRef]
- Moult, P.R.; Harvey, J. NMDA receptor subunit composition determines the polarity of leptin induced synaptic plasticity. Neuropharmacology 2011, 61, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Auberson, Y.P.; Allgeier, H.; Bischoff, S.; Lingenhoehl, K.; Moretti, R.; Schmutz, M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg. Med. Chem. Lett. 2002, 12, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- McGregor, G.; Harvey, J. Leptin Regulation of Synaptic Function at Hippocampal TA-CA1 and SC-CA1 Synapses: Implications for Health and Disease. Neurochem. Res. 2019, 44, 650–660. [Google Scholar] [CrossRef]
- Kallarackal, A.J.; Kvarta, M.D.; Cammarata, E.; Jaberi, L.; Cai, X.; Bailey, A.M.; Thompson, S.M. Chronic stress induces a selective decrease in AMPA receptor-mediated synaptic excitation at hippocampal temporoammonic-CA1 synapses. J. Neurosci. 2013, 33, 15669–15674. [Google Scholar] [CrossRef] [PubMed]
- Kvarta, M.D.; Bradbrook, K.E.; Dantrassy, H.M.; Bailey, A.M.; Thompson, S.M. Corticosterone mediates the synaptic and behavioral effects of chronic stress at rat hippocampal temporoammonic synapses. J. Neurophysiol. 2015, 114, 1713–1724. [Google Scholar] [CrossRef]
- Harvey, J.; Shanley, L.J.; O’Malley, D.; Irving, A.J. Leptin: A potential cognitive enhancer? Biochem. Soc. Trans. 2005, 33, 1029–1032. [Google Scholar] [CrossRef]
- Bland, T.; Zhu, M.; Dillon, C.; Sahin, G.S.; Rodriguez-Llamas, J.L.; Appleyard, S.M.; Wayman, G.A. Leptin Controls Glutamatergic Synaptogenesis and NMDA-Receptor Trafficking via Fyn Kinase Regulation of NR2B. Endocrinology 2020, 161, bqz030. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Isaac, J.T.; Wang, Y.T. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 2004, 5, 952–962. [Google Scholar] [CrossRef]
- Plant, K.; Pelkey, K.A.; Bortolotto, Z.A.; Morita, D.; Terashima, A.; McBain, C.J.; Collingridge, G.L.; Isaac, J.T. Transient Incorporation of Native GluR2-Lacking AMPA Receptors during Hippocampal Long-Term Potentiation. Nat. Neurosci. 2006, 9, 602–604. [Google Scholar] [CrossRef]
- Businaro, R.; Ippoliti, F.; Ricci, S.; Canitano, N.; Fuso, A. Alzheimer’s disease promotion by obesity: Induced mechanisms molecular links and perspectives. Curr. Gerontol. Geriatr. Res. 2012, 2012, 986823. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Khemka, V.K.; Banerjee, A.; Chatterjee, G.; Ganguly, A.; Biswas, A. Metabolic risk factors of sporadic Alzheimer’s disease: Implications in the pathology, pathogenesis and treatment. Aging Dis. 2015, 6, 282–299. [Google Scholar] [CrossRef] [PubMed]
- Flores-Cordero, J.A.; Pérez-Pérez, A.; Jiménez-Cortegana, C.; Alba, G.; Flores-Barragán, A.; Sánchez-Margalet, V. Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin. Int. J. Mol. Sci. 2022, 23, 5202. [Google Scholar] [CrossRef] [PubMed]
- Wondmkun, Y.T. Obesity, insulin resistance, and type 2 diabetes: Associations and therapeutic implications. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 3611–3616. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef]
- Power, D.A.; Noel, J.; Collins, R.; O’Neill, D. Circulating leptin levels and weight loss in Alzheimer’s disease patients. Dement. Geriatr. Cogn. Disord. 2001, 12, 167–170. [Google Scholar] [CrossRef]
- White, H.; Pieper, C.; Schmader, K. The association of weight change in Alzheimer’s disease with severity of disease and mortality: A longitudinal analysis. J. Am. Geriatr. Soc. 1998, 46, 1223–1227. [Google Scholar] [CrossRef]
- Lieb, W.; Beiser, A.S.; Vasan, R.S.; Tan, Z.S.; Au, R.; Harris, T.B.; Roubenoff, R.; Auerbach, S.; DeCarli, C.; Wolf, P.A.; et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 2009, 302, 2565–2572. [Google Scholar] [CrossRef]
- Lilamand, M.; Bouaziz-Amar, E.; Dumurgier, J.; Cognat, E.; Hourregue, C.; Mouton-Liger, F.; Sanchez, M.; Troussière, A.C.; Martinet, M.; Hugon, J.; et al. Plasma Leptin Is Associated with Amyloid CSF Biomarkers and Alzheimer’s Disease Diagnosis in Cognitively Impaired Patients. J. Gerontol. A Biol. Sci. Med. Sci. 2023, 78, 645–652. [Google Scholar] [CrossRef]
- Charisis, S.; Short, M.I.; Bernal, R.; Kautz, T.F.; Treviño, H.A.; Mathews, J.; Dediós, A.G.V.; Muhammad, J.A.S.; Luckey, A.M.; Aslam, A.; et al. Leptin bioavailability and markers of brain atrophy and vascular injury in the middle age. Alzheimers Dement. 2024, 20, 5849–5860. [Google Scholar] [CrossRef]
- Oania, R.; McEvoy, L.K. Plasma leptin levels are not predictive of dementia in patients with mild cognitive impairment. Age Ageing 2015, 44, 53–58. [Google Scholar] [CrossRef]
- Teunissen, C.E.; van der Flier, W.M.; Scheltens, P.; Duits, A.; Wijnstok, N.; Nijpels, G.; Dekker, J.M.; Blankenstein, R.M.; Heijboer, A.C. Serum leptin is not altered nor related to cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.J.; Bryan, K.J.; Sarkar, S.; Zhu, X.; Smith, M.A.; Ashford, J.W.; Johnston, J.M.; Tezapsidis, N.; Casadesus, G. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Pratap, A.A.; Holsinger, R.M.D. Altered Brain Leptin and Leptin Receptor Expression in the 5XFAD Mouse Model of Alzheimer’s Disease. Pharmaceuticals 2020, 13, 401. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Bjorbaek, C.; Osei, S.; Flier, J.S. Regulation of neuronal and glial proteins by leptin: Implications for brain development. Endocrinology 1999, 140, 2755–2762. [Google Scholar] [CrossRef]
- Davis, C.; Mudd, J.; Hawkins, M. Neuroprotective effects of leptin in the context of obesity and metabolic disorders. Neurobiol. Dis. 2014, 72 Pt A, 61–71. [Google Scholar] [CrossRef]
- Doherty, G.H.; Oldreive, C.; Harvey, J. Neuroprotective actions of leptin on central and peripheral neurons in vitro. Neuroscience 2008, 154, 1297–1307. [Google Scholar] [CrossRef]
- Guo, Z.; Jiang, H.; Xu, X.; Duan, W.; Mattson, M.P. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008, 283, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Buchan, M.; Vitanova, K.; Aitken, L.; Gunn-Moore, F.J.; Ramsay, R.R.; Doherty, G. Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem. 2020, 155, 191–206. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, J. Leptin protects hippocampal CA1 neurons against ischemic injury. J. Neurochem. 2008, 107, 578–587. [Google Scholar] [CrossRef]
- Zhang, W.F.; Jin, Y.C.; Li, X.M.; Yang, Z.; Wang, D.; Cui, J.J. Protective effects of leptin against cerebral ischemia/reperfusion injury. Exp. Ther. Med. 2019, 17, 3282–3290. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.H.; Beccano-Kelly, D.; Yan, S.D.; Gunn-Moore, F.J.; Harvey, J. Leptin prevents hippocampal synaptic disruption and neuronal cell death induced by amyloid beta. Neurobiol. Aging 2013, 34, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.; Morrow, K.; Markantoni, E.; Harvey, J. Leptin prevents aberrant targeting of tau to hippocampal synapses via PI 3 kinase driven inhibition of GSK3beta. J. Neurochem. 2023, 167, 520–537. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Liu, S.; Feng, Y.; Zhang, T.; Wu, Z.; Huang, J.; Zhao, W. Hypo-methylated leptin receptor reduces cerebral ischaemia-reperfusion injury by activating the JAK2/STAT3 signalling pathway. J. Int. Med. Res. 2024, 52, 3000605241261912. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, D.; Jie, C.; Liu, T.; Huang, S.; Hu, S. leptin alleviates endoplasmic reticulum stress induced by cerebral ischemia/reperfusion injury via the PI3K/Akt signaling pathway. Biosci. Rep. 2022, 42, BSR20221443. [Google Scholar] [CrossRef]
- Lu, J.; Park, C.S.; Lee, S.K.; Shin, D.W.; Kang, J.H. Leptin inhibits 1-methyl-4-phenylpyridinium-induced cell death in SH-SY5Y cells. Neurosci. Lett. 2006, 407, 240–243. [Google Scholar] [CrossRef]
- Amantea, D.; Tassorelli, C.; Russo, R.; Petrelli, F.; Morrone, L.A.; Bagetta, G.; Corasaniti, M.T. Neuroprotection by leptin in a rat model of permanent cerebral ischemia: Effects on STAT3 phosphorylation in discrete cells of the brain. Cell Death Dis. 2011, 2, e238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jin, Y.; Wang, D.; Cui, J. Neuroprotective effects of leptin on cerebral ischemia through JAK2/STAT3/PGC-1-mediated mitochondrial function modulation. Brain Res. Bull. 2020, 156, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hou, Y.H.; Liao, Z.Y.; Ma, Z.; Zhang, X.X.; Wang, J.L.; Zhu, Y.B.; Shan, H.L.; Wang, P.Y.; Li, C.B.; et al. Neuroprotective Effects of Leptin on the APP/PS1 Alzheimer’s Disease Mouse Model: Role of Microglial and Neuroinflammation. Degener. Neurol. Neuromuscul. Dis. 2023, 13, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Dasari, B.; Prasanthi, J.R.; Schommer, J.; Ghribi, O. Leptin reduces the accumulation of Aβ and phosphorylated tau induced by 27-hydroxycholesterol in rabbit organotypic slices. J. Alzheimers Dis. 2010, 19, 1007–1019. [Google Scholar] [CrossRef]
- Niedowicz, D.M.; Studzinski, C.M.; Weidner, A.M.; Platt, T.L.; Kingry, K.N.; Beckett, T.L.; Bruce-Keller, A.J.; Keller, J.N.; Murphy, M.P. Leptin regulates amyloid β production via the γ-secretase complex. Biochim. Biophys. Acta 2013, 1832, 439–444. [Google Scholar] [CrossRef]
- Pérez-González, R.; Alvira-Botero, M.X.; Robayo, O.; Antequera, D.; Garzón, M.; Martín-Moreno, A.M.; Brera, B.; de Ceballos, M.L.; Carro, E. Leptin gene therapy attenuates neuronal damages evoked by amyloid-β and rescues memory deficits in APP/PS1 mice. Gene Ther. 2014, 21, 298–308. [Google Scholar] [CrossRef]
- Greco, S.J.; Sarkar, S.; Johnston, J.M.; Tezapsidis, N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 2009, 380, 98–104. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, M.; Zhang, J.; Du, C.; Xing, Y. Leptin Regulates Tau Phosphorylation through Wnt Signaling Pathway in PC12 Cells. Neurosignals. 2016, 24, 95–101. [Google Scholar] [CrossRef]
- Cente, M.; Zorad, S.; Smolek, T.; Fialova, L.; Paulenka Ivanovova, N.; Krskova, K.; Balazova, L.; Skrabana, R.; Filipcik, P. Plasma Leptin Reflects Progression of Neurofibrillary Pathology in Animal Model of Tauopathy. Cell. Mol. Neurobiol. 2022, 42, 125–136. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef]
- Miller, E.C.; Teravskis, P.J.; Dummer, B.W.; Zhao, X.; Huganir, R.L.; Liao, D. Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 2014, 39, 1214–1224. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Schonhaut, D.R.; Scholl, M.; Lockhart, S.N.; Ayakta, N.; Baker, S.L.; O’neil, J.P.; Janabi, M.; Lazaris, A.; Cantwell, A.; et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016, 139, 1551–1567. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.Q.; Zhang, J.; Hao, M.; Yang, J.; Han, Y.F.; Liu, X.J.; Shi, H.; Wu, M.N.; Liu, Q.S.; Qi, J.S. Leptin attenuates the detrimental effects of beta-amyloid on spatial memory and hippocampal later-phase long term potentiation in rats. Horm. Behav. 2015, 73, 125–130. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [PubMed]
- Matochik, J.A.; London, E.D.; Yildiz, B.O.; Ozata, M.; Caglayan, S.; DePaoli, A.M.; Wong, M.L.; Licinio, J. Effect of leptin replacement on brain structure in genetically leptin-deficient adults. J. Clin. Endocrinol. Metab. 2005, 90, 2851–2854. [Google Scholar] [CrossRef]
- Paz-Filho, G.J.; Babikian, T.; Asarnow, R.; Delibasi, T.; Esposito, K.; Erol, H.K.; Wong, M.L.; Licinio, J. Leptin replacement improves cognitive development. PLoS ONE 2008, 3, e3098. [Google Scholar] [CrossRef]
- Cook, K.; Adamski, K.; Gomes, A.; Tuttle, E.; Kalden, H.; Cochran, E.; Brown, R.J. Effects of Metreleptin on Patient Outcomes and Quality of Life in Generalized and Partial Lipodystrophy. J. Endocr. Soc. 2021, 5, bvab019. [Google Scholar] [CrossRef]
- Schlögl, H.; Villringer, A.; Miehle, K.; Fasshauer, M.; Stumvoll, M.; Mueller, K. Metreleptin Robustly Increases Resting-state Brain Connectivity in Treatment-naive Female Patients with Lipodystrophy. J. Endocr. Soc. 2023, 7, bvad072. [Google Scholar] [CrossRef] [PubMed]
- Hebebrand, J.; Hildebrandt, T.; Schlögl, H.; Seitz, J.; Denecke, S.; Vieira, D.; Gradl-Dietsch, G.; Peters, T.; Antel, J.; Lau, D.; et al. The role of hypoleptinemia in the psychological and behavioral adaptation to starvation: Implications for anorexia nervosa. Neurosci. Biobehav. Rev. 2022, 141, 104807. [Google Scholar] [CrossRef] [PubMed]
- Milos, G.; Antel, J.; Kaufmann, L.K.; Barth, N.; Koller, A.; Tan, S.; Wiesing, U.; Hinney, A.; Libuda, L.; Wabitsch, M.; et al. Short-term metreleptin treatment of patients with anorexia nervosa: Rapid on-set of beneficial cognitive, emotional, and behavioral effects. Transl. Psychiatry 2020, 10, 303. [Google Scholar] [CrossRef]
- Grasso, P.; Leinung, M.C.; Ingher, S.P.; Lee, D.W. In vivo effects of leptin-related synthetic peptides on body weight and food intake in female ob/ob mice: Localization of leptin activity to domains between amino acid residues 106–140. Endocrinology 1997, 138, 1413–1418. [Google Scholar] [CrossRef]
- Malekizadeh, Y.; Holiday, A.; Redfearn, D.; Ainge, J.A.; Doherty, G.; Harvey, J. A Leptin Fragment Mirrors the Cognitive Enhancing and Neuroprotective Actions of Leptin. Cereb. Cortex 2017, 27, 4769–4782. [Google Scholar] [CrossRef]
- Doherty, G.; Holiday, A.; Malekizadeh, Y.; Manolescu, C.; Duncan, S.; Flewitt, I.; Hamilton, K.; MacLeod, B.; Ainge, J.A.; Harvey, J. Leptin-based hexamers facilitate memory and prevent amyloid-driven AMPA receptor internalisation and neuronal degeneration. J. Neurochem. 2023, 165, 809–826. [Google Scholar] [CrossRef]
- Hirschstein, Z.; Vanga, G.R.; Wang, G.; Novakovic, Z.M.; Grasso, P. MA-[D-Leu-4]-OB3, a small molecule synthetic peptide leptin mimetic, improves episodic memory, and reduces serum levels of tumor necrosis factor-alpha and neurodegeneration in mouse models of Type 1 and Type 2 Diabetes Mellitus. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129697. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).