Abstract
Alzheimer’s disease (AD) is the most prevalent cause of dementia, characterized by progressive cognitive decline, amyloid-β (Aβ) plaques, and neurofibrillary tangles composed of hyperphosphorylated tau protein. Other tauopathies, including frontotemporal lobar degeneration (FTLD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) share pathological hallmarks centered on abnormal tau biology. Increasing evidence highlights the role of post-translational modifications in modulating these pathogenic processes. Among these, glycosylation, the enzymatic attachment of glycans to proteins or lipids, has emerged as a critical regulator of protein folding, trafficking, aggregation, and clearance. Both N-linked glycosylation (N-glycosylation) and O-linked glycosylation (O-glycosylation) influence tau stability, Aβ processing, receptor signaling, synaptic integrity, and neuroinflammation. Dysregulated glycosylation patterns have been documented in brains and cerebrospinal fluid (CSF) of AD patients, suggesting biomarker potential and novel therapeutic targets. Moreover, glycosyltransferases and glycosidases show altered expression in neurodegeneration, linking metabolic and inflammatory pathways to tauopathy progression. This review synthesizes current evidence on the implications and consequences of glycosylation in AD and other tauopathies, integrating mechanistic, pathological, and clinical findings. We also discuss advances in glycoproteomics, the interplay between glycosylation and phosphorylation, and the translational potential of targeting glycosylation pathways for diagnosis and therapy.
1. Introduction
Alzheimer’s disease (AD) is the leading cause of dementia affecting over 50 million individuals worldwide, with prevalence projected to rise sharply as populations age. Clinically, AD is defined by progressive cognitive decline, memory impairment, and eventual loss of autonomy [1]. Neuropathologically, it is characterized by extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein [2]. While these lesions form the cornerstone of the amyloid and tau hypotheses, AD is increasingly recognized as a multifactorial disorder, integrating immune, vascular, metabolic, and protein homeostasis pathways.
A crucial layer of regulation in these processes is post-translational modification (PTM), which fine-tunes protein structure, stability, and localization. Among PTMs, tau phosphorylation has been extensively studied, but glycosylation has gained momentum as a determinant of disease progression [3]. Glycosylation refers to the enzymatic addition of glycans to proteins or lipids. Two major classes relevant to AD and tauopathies are N-glycosylation, where glycans attach to asparagine residues and O-glycosylation, where glycans attach to serine or threonine residues [4].
Variants such as O-GlcNAcylation—the dynamic attachment of N-acetylglucosamine—play pivotal roles in regulating protein interactions and preventing aggregation. Glycosylation influences protein folding, trafficking, degradation, and cell–cell signaling, processes that are fundamental to neuronal viability [5].
Tauopathies, a group of neurodegenerative diseases characterized by abnormal aggregation of the microtubule-associated protein tau include AD, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Pick’s disease. These disorders share tau accumulation but differ in isoform composition, filament morphology, and regional pathology [4,5].
In tauopathies such as frontotemporal lobar degeneration (FTLD), PSP, and CBD, glycosylation abnormalities intersect with tau phosphorylation, altering aggregation dynamics and neuronal toxicity [4]. Similarly, amyloid precursor protein (APP) processing and Aβ secretion are influenced by N-glycosylation, linking glycan biology to amyloid pathology [5]. Moreover, glycosylation affects receptor signaling, immune activation, and synaptic function, positioning it as a central regulator of neurodegeneration [3].
Advances in glycoproteomics now enable precise mapping of glycosylation changes in AD brain tissue, cerebrospinal fluid (CSF), and serum, revealing disease-specific signatures with potential diagnostic and prognostic value [1]. Furthermore, enzymes such as glycosyltransferases and glycosidases emerge as candidate drug targets while metabolic pathways regulating the hexosamine biosynthetic pathway (HBP) link systemic glucose handling to neuronal glycosylation patterns [4].
This review synthesizes current knowledge on the implications and consequences of glycosylation in AD and other tauopathies, with focus on tau biology, amyloid processing, immune and synaptic modulation, and translational potential for biomarker discovery and therapy.
2. Tau Glycosylation in Alzheimer’s Disease and Related Tauopathies
Tau protein is a microtubule-associated protein essential for stabilizing cytoskeletal architecture and supporting axonal transport [4,5]. In AD and other tauopathies, tau undergoes extensive PTMs that alter its biochemical and biophysical properties. While hyperphosphorylation has been studied extensively, evidence increasingly demonstrates that glycosylation is a critical determinant of tau structure, aggregation, and toxicity [6].
2.1. N-Glycosylation of Tau
Early studies revealed that tau is aberrantly N-glycosylated in AD brains, whereas normal adult tau is typically not glycosylated [7]. The addition of N-linked glycans occurs at asparagine residues and alters the conformation of tau, reducing its affinity for microtubules while increasing aggregation propensity [8]. N-glycosylated tau has been detected within paired helical filaments and neurofibrillary tangles, supporting a pathogenic role [4].
Importantly, aberrant N-glycosylation often precedes or facilitates hyperphosphorylation. Glycosylated tau shows increased susceptibility to kinases such as glycogen synthase kinase-3β (GSK3β), leading to pathogenic phosphorylation pattern. This sequence highlights glycosylation as an upstream event in tauopathy pathogenesis [7]. Glycosyltransferases—enzymes that catalyze the transfer of sugar moieties to proteins or lipids—mediate the covalent addition of glycan structures that can modulate tau folding, stability, and aggregation [6,7].
2.2. O-GlcNAcylation of Tau
Another critical modification is O-GlcNAcylation, the addition of O-linked N-acetylglucosamine to serine or threonine residues. Unlike N-glycosylation, O-GlcNAcylation appears protective. Increased O-GlcNAcylation reduces tau phosphorylation and aggregation by competing with phosphorylation sites [8]. Animal studies show that enhancing O-GlcNAcylation stabilizes tau, improves neuronal survival, and ameliorates memory deficits [5].
However, AD brains consistently show reduced O-GlcNAcylation, correlating with increased phosphorylation and tangle burden [9]. This reduction may result from impaired glucose metabolism and flux through the HBP [3].
2.3. Glycosylation in Other Tauopathies
In FTLD, PSP, and CBD, tau aggregates share common modifications. Abnormal glycosylation of tau has been demonstrated across these conditions, despite disease-specific differences in glycosylation site occupancy and glycan composition [10]. In PSP, tau shows increased high-mannose N-glycans, while in CBD, complex-type N-glycans are enriched, suggesting distinct enzymatic dysregulation [11].
Moreover, O-GlcNAcylation deficits are not restricted to AD but also appear in FTLD brain tissue, further linking impaired glucose metabolism to tau pathology across tauopathies [9].
While all tauopathies share abnormal tau aggregation, key differences exist in isoform composition, filament structure, and regional vulnerability. In AD, neurofibrillary tangles contain both 3-repeat (3R) and 4-repeat (4R) tau isoforms that assemble into paired helical filaments (PHFs), typically distributed from the entorhinal cortex to the neocortex in a hierarchical pattern [9]. In contrast, PSP and CBD predominantly accumulate 4R-tau, forming straight filaments and involving basal ganglia, brainstem, and motor cortex [8,9]. FTLD-tau subtypes vary—Pick’s disease features 3R-tau inclusions (Pick bodies) within frontotemporal regions, whereas globular glial tauopathies exhibit mixed isoforms with astrocytic or oligodendroglial predominance [10]. These distinctions suggest that the biochemical context of tau, including PTMs such as glycosylation, may differ by isoform and brain region. Evidence indicates that AD-associated tau tends to be more extensively N-glycosylated and exhibits reduced O-GlcNAcylation, while PSP and CBD display more restricted N-glycan complexity and relatively conserved O-GlcNAc profiles [11]. Thus, differential tau glycosylation may contribute to disease-specific aggregation patterns, cellular tropism, and progression rates among tauopathies [9,10,11].
2.4. Interplay Between Glycosylation and Phosphorylation
Perhaps the most critical insight is the interplay between glycosylation and phosphorylation. N-glycosylated tau is a better substrate for phosphorylation, while O-GlcNAcylation prevents phosphorylation at adjacent residues [12]. This creates a pathogenic imbalance, reduced O-GlcNAcylation and increased N-glycosylation synergistically drive tau hyperphosphorylation and aggregation [13].
This dynamic crosstalk suggests that therapies aimed at restoring O-GlcNAcylation or preventing aberrant N-glycosylation could rebalance tau homeostasis and slow disease progression. Indeed, small-molecule inhibitors of O-GlcNAcase (OGA), the enzyme removing O-GlcNAc, are under investigation as disease-modifying treatments [6].
2.5. Comparative Analysis of Tau Glycosylation and Phosphorylation Sites
N-glycosylation appears to promote tau hyperphosphorylation and aggregation, whereas O-GlcNAcylation exerts a protective role [11]. However, the relationship between glycosylation and phosphorylation is highly dependent on site specificity and structural context. Tau protein contains over 80 potential phosphorylation sites and at least 10 experimentally confirmed glycosylation sites, many of which are spatially proximal or overlapping [12]. N-glycosylation typically occurs on asparagine residues (e.g., Asn167, Asn359), whereas O-GlcNAcylation affects serine and threonine residues (e.g., Ser400, Thr123, Thr245, Ser356) that often coincide with major phosphorylation motifs recognized by kinases such as GSK3β, cyclin-dependent kinase 5 (CDK5), and microtubule affinity-regulating kinase 2 (MARK2) [13]. These modifications act in a competitive and regulatory manner. N-glycosylation changes the tertiary structure of tau, reducing its microtubule affinity and exposing neighboring serine and threonine residues to kinases, thereby facilitating hyperphosphorylation. In contrast, O-GlcNAcylation directly competes with phosphorylation at the same or adjacent residues, inhibiting excessive phosphorylation and stabilizing tau–microtubule interactions [12]. Consequently, decreased O-GlcNAcylation in AD—often linked to impaired glucose metabolism—removes this protective brake, leading to pathological phosphorylation and tangle formation [11,12,13]. In AD, both 3R and 4R tau isoforms exhibit extensive N-glycosylation and reduced O-GlcNAcylation, driving PHF assembly [10]. In other tauopathies such as PSP and CBD, tau aggregates are composed predominantly of 4R isoforms with more restricted glycosylation patterns and distinct phosphorylation site usage (e.g., Ser262, Ser396) [10,11]. These biochemical differences suggest that glycosylation and phosphorylation site occupancy contribute to the diversity of filament morphology and regional vulnerability observed across tauopathies [10,11,13]. Future work should address these site-specific interactions, emphasizing that N-glycosylation (Asn167/359) acts as an early structural modifier that primes tau for pathogenic phosphorylation, while O-GlcNAcylation (Ser400, Thr245, Ser356) counteracts aggregation by masking phosphorylation motifs [11,13]. This comparative framework could enhance understanding of tau pathology across AD and non-AD tauopathies and guide the design of site-directed therapeutic interventions.
3. Glycosylation and Amyloid-β Pathology
The amyloid cascade hypothesis proposes that abnormal processing of APP initiates a series of pathogenic events culminating in AD [8]. Glycosylation profoundly influences APP trafficking, secretase accessibility, and Aβ generation [5].
3.1. Amyloid Precursor Protein N-Glycosylation and Trafficking
APP is a heavily N-glycosylated type I transmembrane protein. N-glycans at asparagine residues near its luminal domain regulate APP folding, trafficking through the secretory pathway, and stability at the cell surface [14]. Disruption of these N-glycans alters APP sorting, diverting it toward endosomal compartments enriched in β-site β-secretase enzyme, APP-cleaving enzyme 1 (BACE1), thereby increasing amyloidogenic cleavage [15].
Experimental studies confirm that loss of APP N-glycosylation enhances Aβ production, whereas stabilizing N-glycan structures reduces amyloidogenic processing [16]. These findings indicate that altered N-glycosylation in the AD brains may bias APP processing toward Aβ generation.
3.2. O-Glycosylation and Secretase Regulation
In addition to N-glycosylation, APP and its secretases undergo O-glycosylation, particularly O-GalNAc modification, which influences protein trafficking and enzymatic activity. O-glycosylation of APP modulates its endocytosis rate, thereby controlling access to β- and γ-secretases [5]. Reduced O-glycosylation correlates with increased amyloidogenic cleavage and elevated extracellular Aβ accumulation [8].
Secretases themselves are glycoproteins. BACE1 contains multiple N-glycans that regulate folding, transport, and stability. Inhibition of BACE1 glycosylation reduces its enzymatic activity and impairs Aβ generation [17]. Similarly, γ-secretase components such as nicastrin are highly glycosylated. The glycan structures near the substrate-binding pocket determine APP cleavage patterns. These modifications fine-tune Aβ species ratios, including the pathogenic Aβ42/40 balance [18].
3.3. Aberrant Glycosylation of Amyloid-β Peptides
Emerging evidence suggests that Aβ peptides themselves can undergo glycosylation or interact with glycans. Modified Aβ shows altered aggregation kinetics and toxicity. Glycosylated Aβ peptides aggregate faster and form more neurotoxic oligomers compared to unmodified Aβ [6]. Additionally, glycans on neuronal membranes interact with Aβ, promoting deposition and impairing clearance [1].
Glycosylation also modulates Aβ clearance via receptor-mediated endocytosis. For instance, the receptor for advanced glycation end products (RAGE) binds both glycated proteins and Aβ, facilitating their transcytosis across the blood–brain barrier [2]. Enhanced RAGE glycosylation increases its affinity for Aβ, promoting deposition in the brain parenchyma [19].
The interplay between glycosylation and Aβ toxicity extends beyond processing and clearance. Aberrant glycosylation of synaptic proteins, receptors, and ion channels exacerbates Aβ-induced dysfunction. For example, N-methyl-D-aspartate (NMDA) receptors require N-glycosylation for proper surface expression as loss of this modification sensitizes neurons to Aβ-mediated excitotoxicity [20]. Similarly, glycosylation of prion protein (PrPC) modulates its interaction with Aβ oligomers, influencing synaptotoxic signaling cascades [17,21].
Furthermore, microglial and astrocytic receptors involved in Aβ clearance—such as triggering receptor expressed on myeloid cells 2 (TREM2) and cluster of differentiation 33 (CD33)—are heavily glycosylated. Altered glycan structures modulate receptor affinity and downstream inflammatory responses [22]. This suggests that glycosylation contributes to the balance between protective clearance and harmful neuroinflammation.
3.4. Implications for Therapy
Given the centrality of glycosylation in APP metabolism and Aβ biology, targeting glycosylation pathways represents a promising therapeutic approach. Experimental strategies include inhibiting BACE1 glycosylation, enhancing protective O-glycosylation, and modulating glycosyltransferases that remodel glycans on APP and its processing enzymes [16,17]. Additionally, blocking RAGE glycosylation has been proposed to limit Aβ transport into the brain and reduce Aβ deposition within the cerebral parenchyma and vasculature [2,19,23,24,25].
4. Glycosylation, Synaptic Function, and Neuroinflammation
Synaptic dysfunction and chronic neuroinflammation are central drivers of neurodegeneration in AD and tauopathies. Both processes are intimately modulated by glycosylation, which shapes receptor signaling, immune cell activation, and glial responses [22,26,27].
4.1. Glycosylation of Synaptic Receptors and Adhesion Molecules
Neuronal communication depends on the proper glycosylation of neurotransmitter receptors and cell adhesion proteins. NMDA receptors, critical for synaptic plasticity, require N-glycosylation for assembly, trafficking, and surface expression. In AD models, aberrant glycosylation disrupts NMDA receptor localization, sensitizing neurons to Aβ-induced excitotoxicity [20].
Similarly, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors rely on N-glycans for stability and gating properties. Altered glycosylation reduces synaptic strength and long-term potentiation, processes essential for learning and memory [15].
Adhesion molecules such as neural cell adhesion molecule (NCAM) and L1CAM are highly glycosylated, and polysialylation of NCAM regulates neurite outgrowth and synaptic remodeling. In addition, reduced polysialylation has been detected in AD hippocampus, correlating with impaired synaptic connectivity [28].
4.2. Immune Receptor Glycosylation and Microglial Activation
Microglia, the brain’s resident immune cells, rely on glycosylated receptors for recognition and clearance of pathological proteins [22]. TREM2 contains multiple N-glycosylation sites that regulate folding and surface expression [26]. Mutations altering TREM2 glycosylation reduce receptor stability, impair microglial phagocytosis of Aβ, and increase AD risk [22,26,27].
Another example is CD33, a sialic acid-binding receptor implicated in AD genetic risk [6,8,19]. CD33 glycosylation governs ligand binding and inhibitory signaling while aberrant glycosylation enhances its inhibitory effect, reducing Aβ clearance and exacerbating inflammation [12].
Astrocytes also display glycosylated receptors that influence neuroinflammation. For instance, glial fibrillary acidic protein (GFAP)-interacting proteins undergo glycan remodeling in reactive astrocytes, altering cytokine release and glial scar formation [5,29].
4.3. Cytokines, Chemokines, and Glycosylation
Inflammatory mediators in the central nervous system (CNS) are heavily glycosylated. Interleukin-6 (IL-6), a pro-inflammatory cytokine elevated in AD, requires N-glycosylation for secretion and receptor binding [29]. Altered IL-6 glycosylation patterns modulate its bioactivity and half-life in the extracellular space [29,30,31]. Similarly, tumor necrosis factor-alpha (TNF-α) and interleukin-1β (IL-1β) undergo glycosylation changes in AD, influencing receptor affinity and downstream signaling [32,33,34].
Chemokine receptors such as CCR5 and CX3CR1 are also glycosylated, with changes in glycan branching modulating microglial migration and response to injury [11]. In AD, aberrant glycosylation of these receptors contributes to chronic non-resolving inflammation [11,28].
4.4. Glycosylation and the Complement System
The complement cascade, a major mediator of synaptic pruning and neuroinflammation, is highly influenced by glycosylation [7]. C1q, the initiator of the classical pathway, carries sialylated glycans that regulate binding to immune complexes and synaptic elements [7,11]. Loss of sialylation enhances complement activation and drives synapse elimination in AD models [9,35].
Other complement proteins, including C3 and factor H, show altered glycosylation in AD brain and CSF, correlating with disease severity [5,8,14,23,26]. This suggests that glycosylation defects amplify maladaptive immune responses.
4.5. Neuroinflammation Beyond Amyloid and Tau
Glycosylation also extends its influence to pattern recognition receptors (PRRs) that detect damage-associated molecular patterns (DAMPs) [2]. RAGE is heavily glycosylated, and its modification enhances binding to glycated proteins and Aβ, facilitating inflammatory cascades [2,26]. Glycosylated RAGE amplifies NF-κB signaling, perpetuating neuroinflammation [3].
In addition, toll-like receptors (TLRs) rely on N-glycans for stability and ligand recognition [35]. Dysregulated TLR glycosylation biases microglia toward pro-inflammatory phenotypes, further fueling pathology [35,36,37,38,39].
4.6. Therapeutic Implications
Given its central role in receptor biology and inflammatory signaling, modulating glycosylation could rebalance immune responses. Potential strategies include glycoengineering of anti-inflammatory cytokines, inhibition of aberrant receptor glycosylation, or restoring protective glycosylation patterns on complement regulators [2,12,40].
5. Advances in Glycoproteomics and Biomarker Discovery
Biomarker discovery remains a critical frontier in AD and tauopathies. While classical markers such as CSF amyloid-β42 (Aβ42), total tau (t-tau), and phosphorylated tau (p-tau) have clinical utility, their specificity and predictive power are limited [32]. Glycoproteomics, the large-scale study of glycosylated proteins, has emerged as a powerful approach to identify novel biomarkers by leveraging disease-specific glycosylation changes [34,41,42,43]. Glycoproteomics provides detailed mapping of glycan sites and structures, enabling the identification of disease-specific tau modifications [34].
5.1. Glycoproteomic Alterations in Alzheimer’s Disease
Proteomic studies demonstrate profound changes in glycosylation within AD brains. Comparative analyses of postmortem cortical tissue show altered N-glycan branching, sialylation, and fucosylation across multiple glycoproteins [1,44,45]. Specific glycosylation signatures distinguish AD from age-matched controls, implicating disrupted glycosyltransferase and glycosidase activity [3,33].
In CSF, glycoproteomics reveals increased bisected N-glycans and altered glycoforms of tau and APP fragments, correlating with disease severity [46,47]. Serum analyses have also detected disease-specific glycosylation patterns, including reduced sialylation of acute-phase proteins and altered IgG Fc (Immunoglobulin G—fragment crystallizable) glycosylation, linking systemic inflammation to neurodegeneration [10,14,24,32,47,48].
5.2. Glycosylation as a Diagnostic and Prognostic Biomarker
The diagnostic potential of glycosylation is underscored by studies demonstrating that altered glycosylation of tau precedes overt phosphorylation changes [49]. Detection of glycosylated tau in CSF or plasma could thus provide early disease biomarkers. Likewise, O-GlcNAcylation status of tau correlates inversely with disease progression, suggesting its utility as a prognostic marker [46,50,51].
Moreover, glycan-based profiling has shown promise in differentiating AD from other dementias. For instance, patients with FTLD and PSP display distinct glycosylation patterns compared to AD, enabling differential diagnosis [16,20].
5.3. Mass Spectrometry and Glycoproteomic Technologies
Recent advances in MS have revolutionized glycoproteomics. Methods such as electron-transfer dissociation (ETD) and higher-energy collisional dissociation (HCD) provide site-specific glycan characterization with high sensitivity [42]. Enrichment strategies using lectins, hydrophilic interaction chromatography (HILIC), and nanoporous materials further enhance glycopeptide detection [5,12,52].
Novel probes and nanocomposites allow in situ visualization of glycosylation in brain tissue, thereby bridging biochemical analyses with histopathology. These tools have revealed regional differences in glycosylation within hippocampus, cortex, and subcortical structures, aligning with clinical phenotypes of memory loss, executive dysfunction, and motor impairment [53,54].
Given the complexity of tau PTMs, including phosphorylation and glycosylation, the development of specific and validated antibodies remains a significant challenge. Most commercially available anti-tau antibodies are designed against phosphorylated epitopes (e.g., pSer202, pThr231, pSer396), many of which have been extensively validated for immunohistochemistry (IHC), Western blot, and enzyme-linked immunosorbent assay (ELISA) applications in brain and CSF samples [12]. In contrast, antibodies specifically targeting glycosylated tau are scarce and often lack full validation due to the structural diversity and low abundance of glycoforms [53]. Only a limited number of monoclonal antibodies raised against N-glycosylated or O-GlcNAc-modified tau have been reported, with most data derived from research-grade reagents used in immunoprecipitation or lectin-based assays rather than standardized diagnostic kits [12,53].
Validation of tau glycosylation-specific antibodies for clinical biofluids such as CSF or serum remains preliminary, primarily constrained by low antigen concentration and conformational masking of glycan epitopes [52,53]. While recent studies have employed antibody–lectin hybrid assays and antibody enrichment followed by liquid chromatography (LC)–MS/MS to enhance specificity, no single antibody currently demonstrates broad cross-platform reliability [5,12]. For IHC detection, phospho-tau antibodies remain highly reliable and routinely used to stage pathology (e.g., AT8, PHF-1, CP13), whereas glycosylation-targeting antibodies are still in experimental phases [54]. As glycoproteomic characterization advances, development of rigorously validated monoclonal antibodies recognizing specific glycoepitopes on tau will be critical for translating glycosylation research into clinical biomarker applications [5,12,52,53,54].
5.4. Immunoglobulin Glycosylation and Systemic Biomarkers
Beyond CNS, peripheral immune system glycosylation offers insights into neurodegeneration. Altered IgG Fc glycosylation has been documented in AD, with reduced galactosylation and sialylation associated with pro-inflammatory phenotypes [20,47,55]. These changes may reflect systemic inflammation or feedback from neuroinflammatory cascades.
Additionally, plasma glycoproteomic profiling identified altered glycosylation of acute-phase reactants such as haptoglobin and α1-acid glycoprotein, linking metabolic stress to disease progression [56,57,58].
5.5. Integration with Multimodal Biomarkers
Glycosylation-based biomarkers should not be considered in isolation but integrated with imaging, fluid, and genetic markers. Combining CSF tau glycosylation profiles with positron emission tomography (PET) amyloid imaging improves diagnostic accuracy over either modality alone [10,51]. Similarly, serum glycoproteomic signatures complement plasma phosphorylated tau (phospho-tau181, phospho-tau217) measurements, enhancing early detection potential [11,46].
5.6. Challenges and Opportunities
Despite major progress, glycoproteomics faces challenges. Glycan heterogeneity complicates quantification and standardized reference libraries are limited [59]. Moreover, interindividual variability in glycosylation due to genetics, diet, and comorbidities complicates biomarker validation [1,59,60,61].
However, these same features offer opportunities. Disease-specific glycan “fingerprints” could yield highly specific biomarkers and integration with machine learning approaches promises improved classification [3,62,63]. In addition, longitudinal studies are beginning to reveal how glycosylation changes track disease trajectory, paving the way for prognostic applications [62].

Table 1.
Analytical limitations in tau glycoproteomics.
Table 2.
Emerging solutions in tau tau glycoproteomics.
6. Enzymatic Regulators of Glycosylation in Neurodegeneration
The balance of protein glycosylation is tightly regulated by glycosyltransferases and glycosidases, as well as by metabolic flux through the HBP, which generates nucleotide sugars as glycosylation substrates. In Alzheimer’s disease and other tauopathies, dysregulation of these enzymes and pathways alters glycosylation homeostasis, thereby influencing APP metabolism, tau aggregation, receptor signaling, and neuroinflammation [1,50,57].
6.1. Glycosyltransferases in AD and Tauopathies
N-acetylglucosaminyltransferases (GnTs) regulate branching and extension of N-glycans. Upregulation of GnT-III increases bisected N-glycans on tau and APP, promoting pathological processing [16]. Similarly, elevated GnT-V activity has been detected in AD brains, producing β1,6-GlcNAc branching that alters synaptic receptor function [32,64,65].
Sialyltransferases, which attach sialic acid residues, are dysregulated in AD, leading to reduced sialylation of neuronal adhesion molecules such as NCAM and impaired synaptic remodeling [18]. Altered sialylation also contributes to immune dysfunction by modifying microglial receptor interactions with sialylated ligands [36,66].
In tauopathies beyond AD, such as PSP and CBD, distinct changes in glycosyltransferase expression drive unique glycosylation signatures. Comparative analyses reveal elevated fucosyltransferase activity in PSP, whereas CBD brains show increased sialyltransferase expression, contributing to disease-specific tau glycoforms [11,16,67].
6.2. Glycosidases and Tau Pathology
Glycosidases remove glycans from proteins and modulate glycan turnover. In AD, elevated β-N-acetylglucosaminidase (β-N-OGA) activity reduces protective O-GlcNAcylation of tau, facilitating its hyperphosphorylation and aggregation [20,59,68]. Conversely, inhibition of β-N-OGA increases O-GlcNAcylation and reduces neurodegeneration in tauopathy mouse models [69,70].
Neuraminidases (sialidases) regulate the removal of sialic acids. Increased neuraminidase activity has been linked to loss of polysialylation on NCAM in AD hippocampus, impairing synaptic plasticity [18,32,71]. Additionally, altered lysosomal glycosidase activity disrupts degradation of glycoproteins, contributing to protein aggregation and lysosomal storage pathology observed in AD [3]. Abnormal N-glycosylation of tau promotes hyperphosphorylation and aggregation, while reduced O-GlcNAcylation removes a protective brake on phosphorylation. N- and O-glycosylation of APP and secretases regulate Aβ generation, with aberrant patterns favoring amyloidogenic processing [16,71]. Synaptic receptors and adhesion molecules require proper glycosylation for stability and plasticity [72]. Their dysregulation contributes to synaptic failure [73]. Immune receptors (TREM2, CD33, and RAGE) and cytokines are glycosylated, shaping microglial and astrocytic activation, while complement protein glycosylation regulates synapse pruning. Finally, altered activity of glycosyltransferases, glycosidases, and the HBP underlies these shifts. Together, these processes highlight glycosylation as both a pathogenic mechanism and therapeutic target [72,73]. A schematic overview of glycosylation is presented in Figure 1.
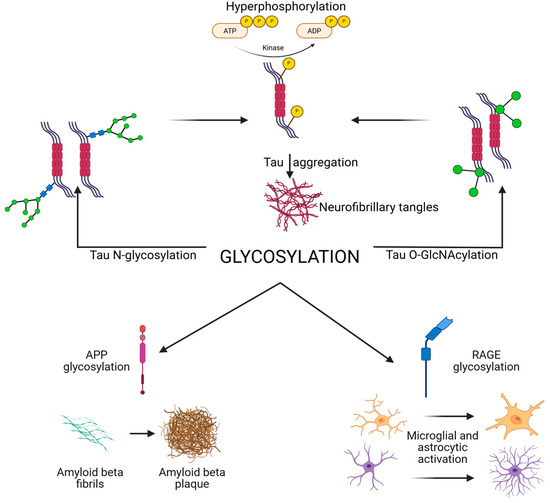
Figure 1.
Schematic overview of glycosylation in AD and tauopathies, illustrating how glycosylation affects tau APP and Aβ, synaptic, and immune receptors, and the enzymatic and metabolic regulators (HBP, OGT, OGA, and GnTs). Created in BioRender. Iordache, M. (2025) https://BioRender.com/fbogzud (accessed on 18 October 2025). APP, amyloid precursor protein; HBP, hexosamine biosynthetic pathway; N-acetylglucosaminyltransferases; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; RAGE, receptor for advanced glycation end products.
6.3. Hexosamine Biosynthetic Pathway and Metabolic Regulation
HBP provides UDP-N-acetylglucosamine (UDP-GlcNAc), the substrate for O-GlcNAcylation [60]. Reduced glucose flux into the HBP, due to impaired brain glucose metabolism in AD, diminishes O-GlcNAcylation levels [60,72,73]. This metabolic defect links systemic insulin resistance and type 2 diabetes to enhanced risk of AD and tauopathies [74,75,76].
Experimental restoration of HBP flux via glucosamine supplementation increases O-GlcNAcylation and reduces tau phosphorylation in animal models, highlighting its therapeutic potential [68]. Similarly, caloric restriction and metabolic interventions that enhance glucose utilization may indirectly restore protective glycosylation patterns [68,77,78].
6.4. Crosstalk with Phosphorylation Pathways
The interplay between glycosylation and phosphorylation is tightly controlled by enzymes. Reduced O-GlcNAc transferase (OGT) activity decreases tau O-GlcNAcylation, thereby exposing phosphorylation sites for kinases such as GSK3β [30,57,79,80]. Conversely, enhancing OGT activity shifts the balance toward protective O-GlcNAcylation [6,56].
This enzymatic tug-of-war suggests that targeting glycosylation enzymes may indirectly modulate tau phosphorylation, providing a dual mechanism for therapeutic intervention [34,68,81].
6.5. Enzymatic Dysregulation as Biomarkers
Aberrant expression of glycosylation enzymes is detectable in CSF and serum, offering biomarker potential [3]. Elevated OGA and reduced OGT levels correlate with higher CSF tau and worse cognitive outcomes [3,29,82,83]. Similarly, altered glycosyltransferase expression patterns in peripheral blood mononuclear cells reflect disease stage and may serve as accessible biomarkers [39,76].
6.6. Clinical Correlations of Tau Glycosylation with Cognitive Decline and Disease Staging
Emerging evidence indicates that aberrant tau glycosylation patterns are not only neuropathological hallmarks but may also correlate with measurable clinical outcomes. In AD, elevated N-glycosylated tau in brain tissue and CSF has been associated with advanced Braak stages and greater neurofibrillary tangle burden, reflecting a molecular shift toward aggregation-prone conformers [29,39]. The Braak staging system assigns cases to six stages based on the presence of Lewy pathology in the medulla oblongata (I), pons (II), mesencephalon (III), amygdala and other limbic structures (IV), association neocortex (V), and first order sensory association neocortex (VI). Conversely, reduced O-GlcNAcylation levels in tau correlate with accelerated cognitive decline and lower Mini-Mental State Examination (MMSE) scores, suggesting a loss of neuroprotective regulation [76]. Longitudinal cohort studies have shown that specific CSF glycoforms of tau—particularly those carrying high-mannose N-glycans or lacking O-GlcNAc at Ser400 and Thr245—predict progression from mild cognitive impairment (MCI) to Alzheimer’s dementia over 2–4 years [76,82]. Furthermore, glyco-profiling in CSF and plasma using lectin-based assays or glycoproteomic MS has demonstrated potential for distinguishing AD from other tauopathies such as PSP and CBD, based on differences in glycan branching and sialylation patterns [82], as shown in Table 3. Collectively, these findings suggest that the glycosylation state of tau serves as a dynamic biochemical correlate of disease stage and may complement phosphorylated tau and Aβ42 as part of a next-generation biomarker panel for early detection and disease monitoring [83].
Table 3.
Disease-specific glycosylation signatures in tauopathies.
6.7. Therapeutic Implications
Enzymes regulating glycosylation are increasingly viewed as druggable targets. OGA inhibitors, such as thiamet-G, are under preclinical investigation for enhancing tau O-GlcNAcylation [39,57,66,84]. By preserving polysialylation, sialidase inhibitors may protect synaptic plasticity [81]. Small molecules targeting glycosyltransferases are less developed but hold potential for rebalancing glycosylation networks in neurodegeneration [8,81,85,86,87]. The glycosylation processes involved in AD and other tauopathies are summarized in Table 4.
Table 4.
Summary of glycosylation processes in AD and tauopathies.
7. Discussion
Growing evidence demonstrates the role of glycosylation as a central, driver of AD and related tauopathies. Unlike phosphorylation, glycosylation provides a multidimensional regulatory layer that influences protein stability, trafficking, aggregation, and clearance [1,19,89].
7.1. Glycosylation and the Hierarchy of Pathological Events
One of the persistent debates in AD research concerns the sequence of pathological events. To this end, glycosylation research offers new perspectives. Evidence suggests that N-glycosylation of tau occurs early, priming the protein for hyperphosphorylation and aggregation [64,66,90]. In parallel, aberrant glycosylation of APP biases processing toward amyloidogenic cleavage [5,91]. Thus, glycosylation abnormalities may precede both amyloid and tau pathologies, positioning them as an upstream “common denominator”.
7.2. Protective Versus Pathogenic Roles
Not all glycosylation is deleterious. O-GlcNAcylation of tau exemplifies a protective modification by competing with phosphorylation sites, which reduces aggregation and toxicity [16,77,92,93]. Conversely, reduced O-GlcNAcylation in AD brains correlates with worsened pathology [57,77,94]. Similarly, polysialylation of NCAM supports synaptic plasticity, but its reduction contributes to connectivity deficits [64,65,95,96]. The dualistic nature of glycosylation complicates therapeutic strategies. Hence, interventions must distinguish between protective and pathogenic glycosylation events [22,97].
7.3. Crosstalk with Metabolism and Phosphorylation
A recurring theme is the integration of glycosylation with broader cellular networks. Metabolic dysfunction, particularly impaired glucose utilization, reduces flux through the HBP, limiting substrate availability for O-GlcNAcylation [3,98,99]. This explains why AD brains show reduced O-GlcNAc despite increased phosphorylation pressure. Moreover, enzymatic regulators such as OGT and OGA directly shape the phosphorylation landscape of tau by modulating glycosylation at competing sites [20,79,100], as presented in Figure 2.
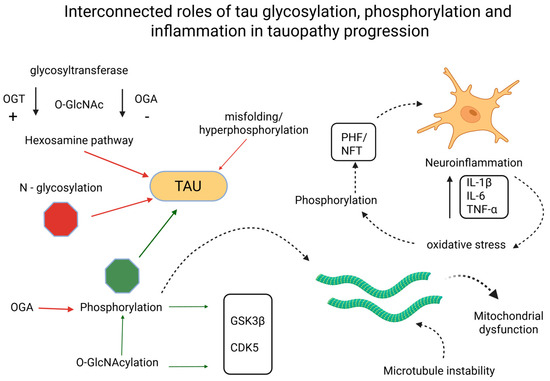
Figure 2.
Interconnected roles of tau glycosylation, phosphorylation, and inflammation in tauopathy progression. Created in BioRender. Iordache, M. (2025) https://BioRender.com/psx2nng (accessed on 18 October 2025). CDK5, cyclin-dependent kinase 5; GSK3β, glycogen synthase kinase 3 beta; IL-1β, interleukin 1β; IL-6, interleukin 6; NFT, neurofibrillary tangle; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; O-GlcNAc, O-linked N-acetylglucosamine; N-glycosylation, asparagine-linked glycosylation; PHF, paired helical filament; TNF-α, tumor necrosis factor α.
7.4. Neuroinflammation as a Glycosylation-Driven Amplifier
Neuroinflammation plays a key role in the neurodegenerative process. Glycosylation critically modulates immune receptor signaling [27]. TREM2 requires N-glycosylation for stability. Mutations impairing this modification reduce microglial Aβ clearance [27,101,102]. Conversely, aberrant glycosylation of CD33 enhances its inhibitory signaling, further dampening microglial phagocytosis [66,103]. Glycosylation of complement proteins amplifies maladaptive synaptic pruning [9,104]. Collectively, these findings highlight glycosylation as a molecular amplifier of neuroinflammatory cascades.
7.5. Biomarker Potential and Translational Challenges
Glycoproteomics offers promising avenues for biomarker discovery. Altered glycosylation of tau, APP, and inflammatory proteins has been consistently detected in CSF and serum [42,105,106,107]. These patterns not only differentiate AD from controls but may also discriminate between tauopathies such as PSP and CBD [67,108]. However, technical barriers remain. Glycan heterogeneity complicates quantification and interindividual variability poses validation challenges [1,109].
7.6. Therapeutic Perspectives
Therapeutic manipulation of glycosylation is a rapidly advancing field. OGA inhibitors that increase protective O-GlcNAcylation of tau are under active investigation [110]. Strategies targeting glycosyltransferases or glycosidases could theoretically rebalance glycosylation networks, though selectivity remains a major obstacle [5,16,111,112]. In addition, targeting glycosylation of immune receptors such as RAGE and TREM2 may offer immunomodulatory benefits [24,27,113].
7.7. Remaining Controversies
Despite major progress, several controversies remain. It is unclear whether glycosylation changes are primary drivers of pathology or secondary adaptations to cellular stress. Some argue that abnormal glycosylation results from metabolic and inflammatory dysfunction rather than initiating pathology [27,114]. Others suggest that glycosylation shifts act as compensatory mechanisms, initially protective but maladaptive when chronic [9,115]. Clarifying these temporal relationships will be essential for therapeutic translation.
Glycosylation has emerged as a pivotal post-translational modification in neurodegeneration, shaping the trajectory of pathology through its multifaceted impact on protein structure, stability, trafficking, and signaling. Across this review, the following themes consistently emerge. Aberrant N-glycosylation primes tau for hyperphosphorylation and aggregation while reduced O-GlcNAcylation removes a critical brake against pathogenic modifications [91,116]. N- and O-glycosylation of APP and its processing enzymes profoundly influence amyloidogenic cleavage, with downstream effects on Aβ burden [5,117]. Glycosylation of receptors and adhesion molecules determines synaptic resilience, while glycosylation of immune receptors (TREM2, CD33, and RAGE) amplifies maladaptive neuroinflammation [19,27,118]. Enzymes such as OGT, OGA, and glycosyltransferases orchestrate glycosylation balance. Their dysregulation, often linked to impaired glucose metabolism and HBP flux, couples systemic metabolic dysfunction to neurodegeneration [3,119]. Glycoproteomic studies reveal distinct glycosylation signatures in brain, CSF, and serum, offering diagnostic and prognostic markers and providing disease-specific fingerprints that differentiate AD from other tauopathies [11,34,120].
Collectively, glycosylation abnormalities are not epiphenomena but core contributors to disease mechanisms, interwoven with phosphorylation, inflammation, and metabolism [44,121,122].
7.8. Glycosylation-Focused Guidelines for Tauopathies: Toward Consensus on Tau Pathology
To help the field converge on comparable and clinically relevant measurements, we propose the following minimum reporting standards and decision rules for glycosylation in tauopathies, expanding beyond prior reviews by integrating site-resolved glycosylation with assay readiness and disease-specific signatures, as illustrated in Figure 3.
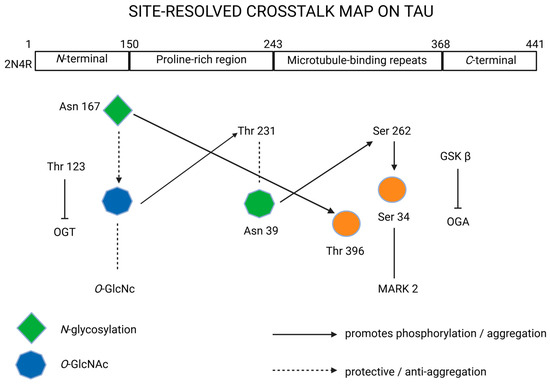
Figure 3.
Site-resolved crosstalk on tau protein. Created in BioRender. Iordache, M. (2025) https://BioRender.com/zq3mkae (accessed on 18 October 2025). 2N4R, tau isoform with 2 N-terminal inserts and 4 microtubule-binding repeats; Asn, asparagine; C-terminal, carboxy-terminal region; GSKβ, glycogen synthase kinase 3-β; MARK2, microtubule affinity-regulating kinase 2; N-glycosylation, N-linked glycosylation; N-terminal, N-terminal region; OGA, O-GlcNAcase; O-GlcNAc, O-linked N-acetylglucosamine; OGT, O-GlcNAc transferase; Ser, serine; Thr, threonine.
Table 5 presents recommended reporting standards, disease signatures, validation rules, and translation framework for tau glycoproteomics.
Table 5.
Recommended reporting standards, disease signatures, validation rules, and translation framework for tau glycoproteomics.
8. Future Directions
While significant progress has been made, several research priorities and therapeutic opportunities remain. Longitudinal human studies are needed to determine whether glycosylation changes precede, accompany, or follow amyloid and tau pathology [9,123]. This temporal clarity will inform whether glycosylation can serve as a true early biomarker or therapeutic entry point.
High-resolution glycoproteomics using advanced MS, lectin arrays, and nanoparticle probes should be applied to large, well-characterized cohorts [3]. Integration with imaging and fluid biomarkers will improve diagnostic specificity and enable personalized medicine [3,42,124]. Clinical translation of OGA inhibitors and other glycosylation-modulating compounds should be pursued cautiously with attention to the dualistic nature of glycosylation, protective vs. pathogenic [5]. Parallel approaches may include sialidase inhibitors to preserve polysialylation, or small-molecule modulators of glycosyltransferases [5,24,125].
Glycosylation does not act isolated. Integrating glycosylation with phosphoproteomics, metabolomics, and transcriptomics will reveal multi-layered disease networks [1,126,127]. Such approaches can identify convergent pathways and potential drug targets. Comparative studies across PSP, CBD, and FTLD will refine understanding of disease-specific glycosylation signatures and highlight both shared and divergent mechanisms [11,128,129,130]. This comparative lens will sharpen differential diagnosis and inform tailored interventions. Translation will require standardization of glycoproteomic assays, establishment of reference databases, and validation in multicenter cohorts [13,131,132,133]. Importantly, therapeutic interventions targeting glycosylation must demonstrate CNS specificity while minimizing systemic off-target effects [16,17,19,134,135,136,137,138,139,140,141,142].
Taken together, these findings position glycosylation at the crossroads of molecular pathology, systemic metabolism, and neuroimmune signaling in AD and related tauopathies. While the evidence underscores both pathogenic and protective aspects of glycosylation, it also highlights unresolved questions regarding timing, causality, and therapeutic feasibility [135,143]. The convergence of biochemical, glycoproteomic, and translational studies emphasizes that glycosylation is not a peripheral phenomenon but a central determinant of disease trajectory [133,144]. This integrative perspective sets the stage for the concluding remarks, which synthesize the mechanistic insights, biomarker potential, and therapeutic avenues into a broader framework for future research and clinical translation [11,105].
9. Conclusions
Glycosylation represents a critical but underappreciated frontier in neurodegeneration research. By modulating protein folding, phosphorylation, aggregation, immune activation, and synaptic function, it acts as both a pathogenic driver and protective modifier. Aberrant N-glycosylation of tau and APP promotes aggregation and amyloidogenic processing while reduced O-GlcNAcylation and polysialylation remove protective mechanisms. In parallel, altered glycosylation of receptors and inflammatory mediators amplifies neuroinflammation and accelerates disease progression.
These insights highlight glycosylation as both a biomarker source and a therapeutic target at the convergence of technological advances. Progress in glycoproteomics now allows detection of disease-specific glycan signatures in CSF and serum, supporting early diagnosis and differential classification among tauopathies. Therapeutic strategies aimed at rebalancing glycosylation, such as OGA inhibitors or glycosyltransferase modulators, are promising but require careful consideration of specificity and systemic effects.
Overall, by integrating glycosylation research with broader molecular and clinical frameworks of neurodegeneration, a new path can be opened toward tangible opportunities for biomarker development, earlier detection, improved prognostic tools, and innovative therapies.
Author Contributions
Conceptualization, A.-C.B., M.P.I., M.C. and A.B.; methodology, A.B., E.R., M.B. and C.T.; software, A.-C.B., M.P.I. and M.C.; validation, E.R., M.B. and C.T.; formal analysis, M.P.I., A.B., E.R., M.B. and C.T.; investigation, A.-C.B., M.P.I., M.C. and A.B.; resources, A.-C.B., M.P.I., M.C., A.B., E.R., M.B. and C.T.; data curation, E.R., M.B. and C.T.; writing—original draft preparation, A.-C.B. and M.P.I.; writing—review and editing, M.C. and A.B.; visualization, M.P.I., M.C. and A.B.; supervision, E.R., M.B. and C.T.; project administration, E.R., M.B. and C.T.; funding acquisition, A.-C.B., M.P.I., M.C., A.B., E.R., M.B. and C.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
All authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gao, G.; Li, C.; Fan, W.; Zhang, M.; Li, X.; Chen, W.; Li, W.; Liang, R.; Li, Z.; Zhu, X. Brilliant Glycans and Glycosylation: Seq and Ye Shall Find. Int. J. Biol. Macromol. 2021, 189, 279–291. [Google Scholar] [CrossRef]
- Kim, Y.; Park, H.; Kim, Y.; Kim, S.-H.; Lee, J.H.; Yang, H.; Kim, S.J.; Li, C.M.; Lee, H.; Na, D.-H.; et al. Pathogenic Role of RAGE in Tau Transmission and Memory Deficits. Biol. Psychiatry 2023, 93, 829–841. [Google Scholar] [CrossRef]
- Yi, L.; Fu, M.; Shao, Y.; Tang, K.; Yan, Y.; Ding, C.-F. Bifunctional Super-Hydrophilic Mesoporous Nanocomposite: A Novel Nanoprobe for Investigation of Glycosylation and Phosphorylation in Alzheimer’s Disease. J. Chromatogr. A 2022, 1676, 463236. [Google Scholar] [CrossRef]
- Maria, C.; Rauter, A.P. Nucleoside Analogues: N-Glycosylation Methodologies, Synthesis of Antiviral and Antitumor Drugs and Potential against Drug-Resistant Bacteria and Alzheimer’s Disease. Carbohydr. Res. 2023, 532, 108889. [Google Scholar] [CrossRef] [PubMed]
- Tachida, Y.; Iijima, J.; Takahashi, K.; Suzuki, H.; Kizuka, Y.; Yamaguchi, Y.; Tanaka, K.; Nakano, M.; Takakura, D.; Kawasaki, N.; et al. O-GalNAc Glycosylation Determines Intracellular Trafficking of APP and Aβ Production. J. Biol. Chem. 2023, 299, 104905. [Google Scholar] [CrossRef]
- Li, W.; Li, H.-L.; Wang, J.-Z.; Liu, R.; Wang, X. Abnormal Protein Post-Translational Modifications Induces Aggregation and Abnormal Deposition of Protein, Mediating Neurodegenerative Diseases. Cell Biosci. 2024, 14, 22. [Google Scholar] [CrossRef]
- Tsay, H.-J.; Huang, Y.-C.; Chen, Y.-J.; Lee, Y.-H.; Hsu, S.-M.; Tsai, K.-C.; Yang, C.-N.; Huang, F.-L.; Shie, F.-S.; Lee, L.-C.; et al. Identifying N-Linked Glycan Moiety and Motifs in the Cysteine-Rich Domain Critical for N-Glycosylation and Intracellular Trafficking of SR-AI and MARCO. J. Biomed. Sci. 2016, 23, 27. [Google Scholar] [CrossRef]
- Akasaka-Manya, K.; Manya, H. The Role of APP O-Glycosylation in Alzheimer’s Disease. Biomolecules 2020, 10, 1569. [Google Scholar] [CrossRef]
- Wang, A.C.; Jensen, E.H.; Rexach, J.E.; Vinters, H.V.; Hsieh-Wilson, L.C. Loss of O-GlcNAc Glycosylation in Forebrain Excitatory Neurons Induces Neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 15120–15125. [Google Scholar] [CrossRef]
- Mashal, Y.; Abdelhady, H.; Iyer, A.K. Comparison of Tau and Amyloid-β Targeted Immunotherapy Nanoparticles for Alzheimer’s Disease. Biomolecules 2022, 12, 1001. [Google Scholar] [CrossRef]
- Fastenau, C.; Bunce, M.; Keating, M.; Wickline, J.; Hopp, S.C.; Bieniek, K.F. Distinct Patterns of Plaque and Microglia Glycosylation in Alzheimer’s Disease. Brain Pathol. 2024, 34, e13267. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, Y.; Liu, X.; Ren, X.; Zhang, W.; Dong, M.; Ge, Y.; Yu, Y.; Ye, M. Development of Complementary Enrichment Strategies for Analysis of N-Linked Intact Glycopeptides and Potential Site-Specific Glycoforms in Alzheimer’s Disease. Talanta 2026, 297, 128595. [Google Scholar] [CrossRef]
- Gupta, R.; Sahu, M.; Srivastava, D.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Post-Translational Modifications: Regulators of Neurodegenerative Proteinopathies. Ageing Res. Rev. 2021, 68, 101336. [Google Scholar] [CrossRef]
- Krawczuk, D.; Kulczyńska-Przybik, A.; Mroczko, B. The Potential Regulators of Amyloidogenic Pathway of APP Processing in Alzheimer’s Disease. Biomedicines 2025, 13, 1513. [Google Scholar] [CrossRef]
- Kizuka, Y.; Kitazume, S.; Taniguchi, N. N-Glycan and Alzheimer’s Disease. Biochim. Biophys. Acta BBA—Gen. Subj. 2017, 1861, 2447–2454. [Google Scholar] [CrossRef]
- Suzuki, S.; Itoh, M. Synergistic Effects of Mutation and Glycosylation on Disease Progression. Front. Mol. Biosci. 2025, 12, 1550815. [Google Scholar] [CrossRef]
- Liang, C.; Yuan, Z.; Yang, S.; Zhu, Y.; Chen, Z.; Can, D.; Lei, A.; Li, H.; Leng, L.; Zhang, J. Mannose Promotes β-Amyloid Pathology by Regulating BACE1 Glycosylation in Alzheimer’s Disease. Adv. Sci. 2025, 12, 2409105. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Ishihara, S.; Nobuhara, M.; Higashide, H.; Funamoto, S. Glycosylation Status of Nicastrin Influences Catalytic Activity and Substrate Preference of γ-Secretase. Biochem. Biophys. Res. Commun. 2018, 502, 98–103. [Google Scholar] [CrossRef]
- Cathrine, R.; Lukose, B.; Rani, P. G82S RAGE Polymorphism Influences Amyloid-RAGE Interactions Relevant in Alzheimer’s Disease Pathology. PLoS ONE 2020, 15, e0225487. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, M.; Sun, J.; Guo, A.; Fernando, R.L.; Chen, Y.; Peng, P.; Zhao, G.; Deng, Y. DPP-4 Inhibitor Improves Learning and Memory Deficits and AD-like Neurodegeneration by Modulating the GLP-1 Signaling. Neuropharmacology 2019, 157, 107668. [Google Scholar] [CrossRef]
- Aljadaan, A.M.; AlSaadi, A.M.; Shaikh, I.A.; Whitby, A.; Ray, A.; Kim, D.-H.; Carter, W.G. Characterization of the Anticholinesterase and Antioxidant Properties of Phytochemicals from Moringa Oleifera as a Potential Treatment for Alzheimer’s Disease. Biomedicines 2025, 13, 2148. [Google Scholar] [CrossRef]
- Ma, L.; Allen, M.; Sakae, N.; Ertekin-Taner, N.; Graff-Radford, N.R.; Dickson, D.W.; Younkin, S.G.; Sevlever, D. Expression and Processing Analyses of Wild Type and p.R47H TREM2 Variant in Alzheimer’s Disease Brains. Mol. Neurodegener. 2016, 11, 72. [Google Scholar] [CrossRef]
- Li, J.Q.; Wang, L.H.; Zhan, Q.W.; Liu, Y.L.; Zhang, Q.; Li, J.F.; Fan, F.F. Mapping Quantitative Trait Loci for Five Forage Quality Traits in a Sorghum-Sudangrass Hybrid. Genet. Mol. Res. 2015, 14, 13266–13273. [Google Scholar] [CrossRef]
- Tolstova, A.P.; Adzhubei, A.A.; Mitkevich, V.A.; Petrushanko, I.Y.; Makarov, A.A. Docking and Molecular Dynamics-Based Identification of Interaction between Various Beta-Amyloid Isoforms and RAGE Receptor. Int. J. Mol. Sci. 2022, 23, 11816. [Google Scholar] [CrossRef]
- Mocanu, A.-I.; Mocanu, H.; Moldovan, C.; Soare, I.; Niculet, E.; Tatu, A.L.; Vasile, C.I.; Diculencu, D.; Postolache, P.A.; Nechifor, A. Some Manifestations of Tuberculosis in Otorhinolaryngology—Case Series and a Short Review of Related Data from South-Eastern Europe. Infect. Drug Resist. 2022, 15, 2753–2762. [Google Scholar] [CrossRef]
- Park, J.; Ji, I.J.; An, H.J.; Kang, M.; Kang, S.; Kim, D.; Yoon, S. Disease-Associated Mutations of TREM2 Alter the Processing of N-Linked Oligosaccharides in the Golgi Apparatus. Traffic 2015, 16, 510–518. [Google Scholar] [CrossRef]
- Shirotani, K.; Hatta, D.; Wakita, N.; Watanabe, K.; Iwata, N. The Role of TREM2 N-Glycans in Trafficking to the Cell Surface and Signal Transduction of TREM2. J. Biochem. 2022, 172, 347–353. [Google Scholar] [CrossRef]
- Tang, X.; Schindler, R.L.; Di Lucente, J.; Oloumi, A.; Tena, J.; Harvey, D.; Lebrilla, C.B.; Zivkovic, A.M.; Jin, L.-W.; Maezawa, I. Unique N-Glycosylation Signatures in Human iPSC Derived Microglia Activated by Aβ Oligomer and Lipopolysaccharide. Sci. Rep. 2025, 15, 12348. [Google Scholar] [CrossRef]
- Gizaw, S.T.; Ohashi, T.; Tanaka, M.; Hinou, H.; Nishimura, S.-I. Glycoblotting Method Allows for Rapid and Efficient Glycome Profiling of Human Alzheimer’s Disease Brain, Serum and Cerebrospinal Fluid towards Potential Biomarker Discovery. Biochim. Biophys. Acta BBA—Gen. Subj. 2016, 1860, 1716–1727. [Google Scholar] [CrossRef]
- Kumar, A.; Karuppagounder, S.S.; Chen, Y.; Corona, C.; Kawaguchi, R.; Cheng, Y.; Balkaya, M.; Sagdullaev, B.T.; Wen, Z.; Stuart, C.; et al. 2-Deoxyglucose Drives Plasticity via an Adaptive ER Stress-ATF4 Pathway and Elicits Stroke Recovery and Alzheimer’s Resilience. Neuron 2023, 111, 2831–2846.e10. [Google Scholar] [CrossRef]
- Iordache, M.P.; Buliman, A.; Costea-Firan, C.; Gligore, T.C.I.; Cazacu, I.S.; Stoian, M.; Teoibaș-Şerban, D.; Blendea, C.-D.; Protosevici, M.G.-I.; Tanase, C.; et al. Immunological and Inflammatory Biomarkers in the Prognosis, Prevention, and Treatment of Ischemic Stroke: A Review of a Decade of Advancement. Int. J. Mol. Sci. 2025, 26, 7928. [Google Scholar] [CrossRef]
- Ramos-Martinez, I.; Martínez-Loustalot, P.; Lozano, L.; Issad, T.; Limón, D.; Díaz, A.; Perez-Torres, A.; Guevara, J.; Zenteno, E. Neuroinflammation Induced by Amyloid Β25–35 Modifies Mucin-Type O-Glycosylation in the Rat’s Hippocampus. Neuropeptides 2018, 67, 56–62. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, Q.; Yu, Q.; Johnson, J.; Shipman, R.; Zhong, X.; Huang, J.; Asthana, S.; Carlsson, C.; Okonkwo, O.; et al. In-Depth Site-Specific Analysis of N-Glycoproteome in Human Cerebrospinal Fluid and Glycosylation Landscape Changes in Alzheimer’s Disease. Mol. Cell. Proteom. 2021, 20, 100081. [Google Scholar] [CrossRef]
- Kobeissy, F.; Kobaisi, A.; Peng, W.; Barsa, C.; Goli, M.; Sibahi, A.; El Hayek, S.; Abdelhady, S.; Ali Haidar, M.; Sabra, M.; et al. Glycomic and Glycoproteomic Techniques in Neurodegenerative Disorders and Neurotrauma: Towards Personalized Markers. Cells 2022, 11, 581. [Google Scholar] [CrossRef]
- Schloss, J.V. Is Dolichol Pathway Dysfunction a Significant Factor in Alzheimer’s Disease? Inflammopharmacology 2025, 33, 4651–4658. [Google Scholar] [CrossRef]
- Huang, C.-W.; Rust, N.; Wu, H.-F.; Hart, G. Altered O-GlcNAcylation and Mitochondrial Dysfunction, a Molecular Link between Brain Glucose Dysregulation and Sporadic Alzheimer’s Disease. Neural Regen. Res. 2023, 18, 779. [Google Scholar] [CrossRef]
- Medina, M.; Hernández, F.; Avila, J. New Features about Tau Function and Dysfunction. Biomolecules 2016, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, C.; Li, L.; Chin, L.-S. Differential Analysis of N-Glycopeptide Abundance and N-Glycosylation Site Occupancy for Studying Protein N-Glycosylation Dysregulation in Human Disease. BIO-Protoc. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.; Ibrahim, M.; Alkorbi, N.; Alkuwari, S.; Pedersen, S.; Rathore, H.A. Epoxide Hydrolase Inhibitors for the Treatment of Alzheimer’s Disease and Other Neurological Disorders: A Comprehensive Review. Biomedicines 2025, 13, 2073. [Google Scholar] [CrossRef] [PubMed]
- Demirev, A.V.; Song, H.-L.; Cho, M.-H.; Cho, K.; Peak, J.-J.; Yoo, H.J.; Kim, D.-H.; Yoon, S.-Y. V232M Substitution Restricts a Distinct O-Glycosylation of PLD3 and Its Neuroprotective Function. Neurobiol. Dis. 2019, 129, 182–194. [Google Scholar] [CrossRef]
- Fang, P.; Yu, X.; Ding, M.; Qifei, C.; Jiang, H.; Shi, Q.; Zhao, W.; Zheng, W.; Li, Y.; Ling, Z.; et al. Ultradeep N-Glycoproteome Atlas of Mouse Reveals Spatiotemporal Signatures of Brain Aging and Neurodegenerative Diseases. Nat. Commun. 2025, 16, 5568. [Google Scholar] [CrossRef] [PubMed]
- Gaunitz, S.; Tjernberg, L.O.; Schedin-Weiss, S. What Can N-Glycomics and N-Glycoproteomics of Cerebrospinal Fluid Tell Us about Alzheimer Disease? Biomolecules 2021, 11, 858. [Google Scholar] [CrossRef]
- Rajabally, Y.A. Chronic Inflammatory Demyelinating Polyneuropathy with Positive Anti-myelin Associated Glycoprotein Antibodies: Back to Clinical Basics. Eur. J. Neurol. 2023, 30, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Frenkel-Pinter, M.; Stempler, S.; Tal-Mazaki, S.; Losev, Y.; Singh-Anand, A.; Escobar-Álvarez, D.; Lezmy, J.; Gazit, E.; Ruppin, E.; Segal, D. Altered Protein Glycosylation Predicts Alzheimer’s Disease and Modulates Its Pathology in Disease Model Drosophila. Neurobiol. Aging 2017, 56, 159–171. [Google Scholar] [CrossRef]
- Rusu, E.; Necula, L.G.; Neagu, A.I.; Alecu, M.; Stan, C.; Albulescu, R.; Tanase, C.P. Current Status of Stem Cell Therapy: Opportunities and Limitations. Turk. J. Biol. 2016, 40, 955–967. [Google Scholar] [CrossRef]
- Ahmad, W.; Shabbiri, K.; Ahmad, I. Prediction of Human Tau 3D Structure, and Interplay between O-β-GlcNAc and Phosphorylation Modifications in Alzheimer’s Disease: C. Elegans as a Suitable Model to Study These Interactions in Vivo. Biochem. Biophys. Res. Commun. 2020, 528, 466–472. [Google Scholar] [CrossRef]
- Meuret, C.J.; Hu, Y.; Smadi, S.; Bantugan, M.A.; Xian, H.; Martinez, A.E.; Krauss, R.M.; Ma, Q.-L.; Nedelkov, D.; Yassine, H.N. An Association of CSF Apolipoprotein E Glycosylation and Amyloid-Beta 42 in Individuals Who Carry the APOE4 Allele. Alzheimers Res. Ther. 2023, 15, 96. [Google Scholar] [CrossRef]
- Popa, M.-L.; Popa, A.; Tanase, C.; Gheorghisan-Galateanu, A.-A. Acanthosis Nigricans: To Be or Not to Be Afraid (Review). Oncol. Lett. 2019, 17, 4133–4138. [Google Scholar] [CrossRef]
- Tena, J.; Tang, X.; Zhou, Q.; Harvey, D.; Barajas-Mendoza, M.; Jin, L.; Maezawa, I.; Zivkovic, A.M.; Lebrilla, C.B. Glycosylation Alterations in Serum of Alzheimer’s Disease Patients Show Widespread Changes in N-glycosylation of Proteins Related to Immune Function, Inflammation, and Lipoprotein Metabolism. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2022, 14, e12309. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-Translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2021, 11, 595532. [Google Scholar] [CrossRef]
- Ercan, E.; Eid, S.; Weber, C.; Kowalski, A.; Bichmann, M.; Behrendt, A.; Matthes, F.; Krauss, S.; Reinhardt, P.; Fulle, S.; et al. A Validated Antibody Panel for the Characterization of Tau Post-Translational Modifications. Mol. Neurodegener. 2017, 12, 87. [Google Scholar] [CrossRef]
- Thummel, R.; Li, L.; Tanase, C.; Sarras, M.P.; Godwin, A.R. Differences in Expression Pattern and Function between Zebrafish Hoxc13 Orthologs: Recruitment of Hoxc13b into an Early Embryonic Role. Dev. Biol. 2004, 274, 318–333. [Google Scholar] [CrossRef]
- Okła, E.; Hołota, M.; Michlewska, S.; Zawadzki, S.; Miłowska, K.; Sánchez-Nieves, J.; Gómez, R.; De La Mata, F.J.; Bryszewska, M.; Ionov, M. Crossing Barriers: PEGylated Gold Nanoparticles as Promising Delivery Vehicles for siRNA Delivery in Alzheimer’s Disease. Biomedicines 2025, 13, 2108. [Google Scholar] [CrossRef]
- Park, E.; He, C.; Abbasi, A.Z.; Tian, M.; Huang, S.; Wang, L.; Georgiou, J.; Collingridge, G.L.; Fraser, P.E.; Henderson, J.T.; et al. Brain Microenvironment-Remodeling Nanomedicine Improves Cerebral Glucose Metabolism, Mitochondrial Activity and Synaptic Function in a Mouse Model of Alzheimer’s Disease. Biomaterials 2025, 318, 123142. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xu, K.; Xu, Z.; De Las Rivas, M.; Wang, C.; Li, X.; Lu, J.; Zhou, Y.; Delso, I.; Merino, P.; et al. The Small Molecule Luteolin Inhibits N-Acetyl-α-Galactosaminyltransferases and Reduces Mucin-Type O-Glycosylation of Amyloid Precursor Protein. J. Biol. Chem. 2017, 292, 21304–21319. [Google Scholar] [CrossRef] [PubMed]
- Bukke, V.N.; Villani, R.; Archana, M.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Ferraro, L.; Serviddio, G.; Cassano, T. The Glucose Metabolic Pathway as A Potential Target for Therapeutics: Crucial Role of Glycosylation in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7739. [Google Scholar] [CrossRef]
- Hoshi, K.; Ito, H.; Abe, E.; Fuwa, T.J.; Kanno, M.; Murakami, Y.; Abe, M.; Murakami, T.; Yoshihara, A.; Ugawa, Y.; et al. Transferrin Biosynthesized in the Brain Is a Novel Biomarker for Alzheimer’s Disease. Metabolites 2021, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Constantinoiu, S.; Cochior, D. Severe Acute Pancreatitis—Determinant Factors and Current Therapeutic Conduct. Chirurgia 2018, 113, 385. [Google Scholar] [CrossRef]
- Pan, K.; Chen, S.; Wang, Y.; Yao, W.; Gao, X. MicroRNA-23b Attenuates Tau Pathology and Inhibits Oxidative Stress by Targeting GnT-III in Alzheimer’s Disease. Neuropharmacology 2021, 196, 108671. [Google Scholar] [CrossRef]
- Rosewood, T.J.; Nho, K.; Risacher, S.L.; Liu, S.; Gao, S.; Shen, L.; Foroud, T.; Saykin, A.J.; for the Alzheimer’s Disease Neuroimaging Initiative. Pathway Enrichment in Genome-wide Analysis of Longitudinal Alzheimer’s Disease Biomarker Endophenotypes. Alzheimer’s Dement. 2024, 20, 8639–8650. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, Y.; Huang, H.; Xue, Y.; Chen, S.; Gao, X. DHEC Mesylate Attenuates Pathologies and Aberrant Bisecting N-Glycosylation in Alzheimer’s Disease Models. Neuropharmacology 2024, 248, 109863. [Google Scholar] [CrossRef]
- Finke, J.M.; Ayres, K.R.; Brisbin, R.P.; Hill, H.A.; Wing, E.E.; Banks, W.A. Antibody Blood-Brain Barrier Efflux Is Modulated by Glycan Modification. Biochim. Biophys. Acta BBA—Gen. Subj. 2017, 1861, 2228–2239. [Google Scholar] [CrossRef]
- Blendea, C.-D.; Khan, M.T.; Stoian, M.; Gligore, T.C.I.; Cuculici, Ș.; Stanciu, I.L.; Protosevici, M.G.-I.; Iordache, M.; Buliman, A.; Costea-Firan, C.; et al. Advances in Minimally Invasive Treatments for Prostate Cancer: A Review of the Role of Ultrasound Therapy and Laser Therapy. Balneo PRM Res. J. 2025, 16, 827. [Google Scholar] [CrossRef]
- Gaunitz, S.; Tjernberg, L.O.; Schedin-Weiss, S. The N-glycan Profile in Cortex and Hippocampus Is Altered in Alzheimer Disease. J. Neurochem. 2021, 159, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Nyarko, J.N.K.; Quartey, M.O.; Heistad, R.M.; Pennington, P.R.; Poon, L.J.; Knudsen, K.J.; Allonby, O.; El Zawily, A.M.; Freywald, A.; Rauw, G.; et al. Glycosylation States of Pre- and Post-Synaptic Markers of 5-HT Neurons Differ with Sex and 5-HTTLPR Genotype in Cortical Autopsy Samples. Front. Neurosci. 2018, 12, 545. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, Y.; Huang, H.; Cao, Y.; Pan, K.; Zhou, Y.; He, J.; Yao, W.; Chen, S.; Gao, X. Targeting Aberrant Glycosylation to Modulate Microglial Response and Improve Cognition in Models of Alzheimer’s Disease. Pharmacol. Res. 2024, 202, 107133. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.; Marcus, J.; Pearson, M.; Song, L.; Smith, K.; Terracina, G.; Lee, J.; Hong, K.-L.K.; Lu, S.X.; et al. MK-8719, a Novel and Selective O-GlcNAcase Inhibitor That Reduces the Formation of Pathological Tau and Ameliorates Neurodegeneration in a Mouse Model of Tauopathy. J. Pharmacol. Exp. Ther. 2020, 374, 252–263. [Google Scholar] [CrossRef]
- Adnan, M.; Siddiqui, A.J.; Bardakci, F.; Surti, M.; Badraoui, R.; Patel, M. NEU1-Mediated Extracellular Vesicle Glycosylation in Alzheimer’s Disease: Mechanistic Insights into Intercellular Communication and Therapeutic Targeting. Pharmaceuticals 2025, 18, 921. [Google Scholar] [CrossRef]
- Du, L.; Su, Z.; Wang, S.; Meng, Y.; Xiao, F.; Xu, D.; Li, X.; Qian, X.; Lee, S.B.; Lee, J.; et al. EGFR-Induced and c-Src-Mediated CD47 Phosphorylation Inhibits TRIM21-Dependent Polyubiquitylation and Degradation of CD47 to Promote Tumor Immune Evasion. Adv. Sci. 2023, 10, 2206380. [Google Scholar] [CrossRef]
- Barbu, L.A.; Vasile, L.; Cercelaru, L.; Șurlin, V.; Mogoantă, S.-Ș.; Mogoș, G.F.R.; Țenea Cojan, T.S.; Mărgăritescu, N.-D.; Iordache, M.P.; Buliman, A. Aggressiveness in Well-Differentiated Small Intestinal Neuroendocrine Tumors: A Rare Case and Narrative Literature Review. J. Clin. Med. 2025, 14, 5821. [Google Scholar] [CrossRef]
- Tanase, C.; Enciu, A.M.; Codrici, E.; Popescu, I.D.; Dudau, M.; Dobri, A.M.; Pop, S.; Mihai, S.; Gheorghișan-Gălățeanu, A.-A.; Hinescu, M.E. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? Int. J. Mol. Sci. 2022, 23, 604. [Google Scholar] [CrossRef]
- Dong, L.; Shen, S.; Xu, Y.; Wang, L.; Feng, R.; Zhang, J.; Lu, H. Computational Studies on the Potency and Selectivity of PUGNAc Derivatives Against GH3, GH20, and GH84 β-N-Acetyl-D-Hexosaminidases. Front. Chem. 2019, 7, 235. [Google Scholar] [CrossRef]
- Krüger, L.; Biskup, K.; Schipke, C.G.; Kochnowsky, B.; Schneider, L.-S.; Peters, O.; Blanchard, V. The Cerebrospinal Fluid Free-Glycans Hex1 and HexNAc1Hex1Neu5Ac1 as Potential Biomarkers of Alzheimer’s Disease. Biomolecules 2024, 14, 512. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.Z.; Gaunitz, S.; Kirsebom, B.-E.; Lundin, B.; Hellström, M.; Jejcic, A.; Sköldunger, A.; Wimo, A.; Winblad, B.; Fladby, T.; et al. Blood N-Glycomics Reveals Individuals at Risk for Cognitive Decline and Alzheimer’s Disease. eBioMedicine 2025, 113, 105598. [Google Scholar] [CrossRef]
- Zhou, R.Z.; Vetrano, D.L.; Grande, G.; Duell, F.; Jönsson, L.; Laukka, E.J.; Fredolini, C.; Winblad, B.; Tjernberg, L.; Schedin-Weiss, S. A Glycan Epitope Correlates with Tau in Serum and Predicts Progression to Alzheimer’s Disease in Combination with APOE4 Allele Status. Alzheimer’s Dement. 2023, 19, 3244–3249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.Z.; Duell, F.; Axenhus, M.; Jönsson, L.; Winblad, B.; Tjernberg, L.O.; Schedin-Weiss, S. A Glycan Biomarker Predicts Cognitive Decline in Amyloid- and Tau-Negative Patients. Brain Commun. 2024, 6, fcae371. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kwon, O.-H.; Chung, S. O-GlcNAcylation of Amyloid-β Precursor Protein at Threonine 576 Residue Regulates Trafficking and Processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef]
- Moldovan, C.; Cochior, D.; Gorecki, G.; Rusu, E.; Ungureanu, F.-D. Clinical and Surgical Algorithm for Managing Iatrogenic Bile Duct Injuries during Laparoscopic Cholecystectomy: A Multicenter Study. Exp. Ther. Med. 2021, 22, 1385. [Google Scholar] [CrossRef]
- Fabiano, M.; Oikawa, N.; Kerksiek, A.; Furukawa, J.; Yagi, H.; Kato, K.; Schweizer, U.; Annaert, W.; Kang, J.; Shen, J.; et al. Presenilin Deficiency Results in Cellular Cholesterol Accumulation by Impairment of Protein Glycosylation and NPC1 Function. Int. J. Mol. Sci. 2024, 25, 5417. [Google Scholar] [CrossRef]
- Ohkawa, Y.; Kizuka, Y.; Takata, M.; Nakano, M.; Ito, E.; Mishra, S.; Akatsuka, H.; Harada, Y.; Taniguchi, N. Peptide Sequence Mapping around Bisecting GlcNAc-Bearing N-Glycans in Mouse Brain. Int. J. Mol. Sci. 2021, 22, 8579. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef]
- Reyes, C.D.G.; Hakim, M.A.; Atashi, M.; Goli, M.; Gautam, S.; Wang, J.; Bennett, A.I.; Zhu, J.; Lubman, D.M.; Mechref, Y. LC-MS/MS Isomeric Profiling of N-Glycans Derived from Low-Abundant Serum Glycoproteins in Mild Cognitive Impairment Patients. Biomolecules 2022, 12, 1657. [Google Scholar] [CrossRef]
- Shao, Y.; Yi, L.; Fu, M.; Feng, Q.; Mao, X.; Mao, H.; Yan, Y.; Ding, C.-F. Anti-Nonspecific Hydrophilic Hydrogel for Efficient Capture of N-Glycopeptides from Alzheimer’s Disease Patient’s Serum. Talanta 2023, 253, 124068. [Google Scholar] [CrossRef]
- Yang, K.; Yang, Z.; Chen, X.; Li, W. The Significance of Sialylation on the Pathogenesis of Alzheimer’s Disease. Brain Res. Bull. 2021, 173, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Caramello, A.; Fancy, N.; Tournerie, C.; Eklund, M.; Chau, V.; Adair, E.; Papageorgopoulou, M.; Willumsen, N.; Jackson, J.S.; Hardy, J.; et al. Intracellular Accumulation of Amyloid-ß Is a Marker of Selective Neuronal Vulnerability in Alzheimer’s Disease. Nat. Commun. 2025, 16, 5189. [Google Scholar] [CrossRef]
- Didonna, A.; Benetti, F. 1 Department of Neurology, University of California San Francisco, San Francisco, CA 94158, USA Post-Translational Modifications in Neurodegeneration. AIMS Biophys. 2015, 3, 27–49. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Gill, D.; Rannikmäe, K.; Traylor, M.; Anderson, C.D.; MEGASTROKE Consortium of the International Stroke Genetics Consortium (ISGC); Lee, J.-M.; Kamatani, Y.; Hopewell, J.C.; Worrall, B.B.; et al. Genetically Determined Levels of Circulating Cytokines and Risk of Stroke: Role of Monocyte Chemoattractant Protein-1. Circulation 2019, 139, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Jin, Z.; Yu, T.; Guo, C.; He, Y.; Kan, Y.; Yan, L.; Wu, L. Effect of Glycosylation on the Enzymatic Degradation of D-Amino Acid-Containing Peptides. Molecules 2025, 30, 441. [Google Scholar] [CrossRef]
- Tang, X.; Tena, J.; Di Lucente, J.; Maezawa, I.; Harvey, D.J.; Jin, L.-W.; Lebrilla, C.B.; Zivkovic, A.M. Transcriptomic and Glycomic Analyses Highlight Pathway-Specific Glycosylation Alterations Unique to Alzheimer’s Disease. Sci. Rep. 2023, 13, 7816. [Google Scholar] [CrossRef]
- Alkuhlani, A.; Gad, W.; Roushdy, M.; Salem, A.-B.M. PUStackNGly: Positive-Unlabeled and Stacking Learning for N-Linked Glycosylation Site Prediction. IEEE Access 2022, 10, 12702–12713. [Google Scholar] [CrossRef]
- Alkuhlani, A.; Gad, W.; Roushdy, M.; Voskoglou, M.G.; Salem, A.M. PTG-PLM: Predicting Post-Translational Glycosylation and Glycation Sites Using Protein Language Models and Deep Learning. Axioms 2022, 11, 469. [Google Scholar] [CrossRef]
- Lebart, M.-C.; Trousse, F.; Valette, G.; Torrent, J.; Denus, M.; Mestre-Frances, N.; Marcilhac, A. Reg-1α, a New Substrate of Calpain-2 Depending on Its Glycosylation Status. Int. J. Mol. Sci. 2022, 23, 8591. [Google Scholar] [CrossRef]
- Liang, N.; Nho, K.; Newman, J.W.; Arnold, M.; Huynh, K.; Meikle, P.J.; Borkowski, K.; Kaddurah-Daouk, R.; the Alzheimer’s Disease Metabolomics Consortium; Kueider-Paisley, A.; et al. Peripheral Inflammation Is Associated with Brain Atrophy and Cognitive Decline Linked to Mild Cognitive Impairment and Alzheimer’s Disease. Sci. Rep. 2024, 14, 17423. [Google Scholar] [CrossRef] [PubMed]
- Losev, Y.; Paul, A.; Frenkel-Pinter, M.; Abu-Hussein, M.; Khalaila, I.; Gazit, E.; Segal, D. Novel Model of Secreted Human Tau Protein Reveals the Impact of the Abnormal N-Glycosylation of Tau on Its Aggregation Propensity. Sci. Rep. 2019, 9, 2254. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.A.; Grant, O.C.; Woods, R.J.; Rebeck, G.W. O-Glycosylation on Cerebrospinal Fluid and Plasma Apolipoprotein E Differs in the Lipid-Binding Domain. Glycobiology 2020, 30, 74–85. [Google Scholar] [CrossRef]
- Hu, Y.; Meuret, C.; Go, S.; Yassine, H.N.; Nedelkov, D. Simple and Fast Assay for Apolipoprotein E Phenotyping and Glycotyping: Discovering Isoform-Specific Glycosylation in Plasma and Cerebrospinal Fluid. J. Alzheimer’s Dis. 2020, 76, 883–893. [Google Scholar] [CrossRef]
- Zhang, Y.; Lan, J.; Zhao, D.; Ruan, C.; Zhou, J.; Tan, H.; Bao, Y. Netrin-1 Upregulates GPX4 and Prevents Ferroptosis after Traumatic Brain Injury via the UNC5B/Nrf2 Signaling Pathway. CNS Neurosci. Ther. 2023, 29, 216–227. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, F.; Fan, J.; Li, X.; Liu, Y.; Chen, W.; Sun, H.; Ma, T.; Wang, Q.; Maihaiti, Y.; et al. Identification and Immune Characteristics of Molecular Subtypes Related to Protein Glycosylation in Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 968190. [Google Scholar] [CrossRef]
- Kronimus, Y.; Albus, A.; Hasenberg, M.; Walkenfort, B.; Seifert, M.; Budeus, B.; Gronewold, J.; Hermann, D.M.; Ross, J.A.; Lochnit, G.; et al. Fc N-glycosylation of Autoreactive Aβ Antibodies as a Blood-based Biomarker for Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 5563–5572. [Google Scholar] [CrossRef] [PubMed]
- Lawler, P.E.; Bollinger, J.G.; Schindler, S.E.; Hodge, C.R.; Iglesias, N.J.; Krishnan, V.; Coulton, J.B.; Li, Y.; Holtzman, D.M.; Bateman, R.J. Apolipoprotein E O-Glycosylation Is Associated with Amyloid Plaques and APOE Genotype. Anal. Biochem. 2023, 672, 115156. [Google Scholar] [CrossRef]
- Ephrame, S.J.; Cork, G.K.; Marshall, V.; Johnston, M.A.; Shawa, J.; Alghusen, I.; Qiang, A.; Denson, A.R.; Carman, M.S.; Fedosyuk, H.; et al. O-GlcNAcylation Regulates Extracellular Signal-Regulated Kinase (ERK) Activation in Alzheimer’s Disease. Front. Aging Neurosci. 2023, 15, 1155630. [Google Scholar] [CrossRef]
- Pinho, T.S.; Correia, S.C.; Perry, G.; Ambrósio, A.F.; Moreira, P.I. Diminished O-GlcNAcylation in Alzheimer’s Disease Is Strongly Correlated with Mitochondrial Anomalies. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2019, 1865, 2048–2059. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Shmueli, M.D.; Raz, C.; Yanku, M.; Zilberzwige, S.; Gazit, E.; Segal, D. Interplay between Protein Glycosylation Pathways in Alzheimer’s Disease. Sci. Adv. 2017, 3, e1601576. [Google Scholar] [CrossRef]
- Boix, C.P.; Lopez-Font, I.; Cuchillo-Ibañez, I.; Sáez-Valero, J. Amyloid Precursor Protein Glycosylation Is Altered in the Brain of Patients with Alzheimer’s Disease. Alzheimer’s Res. Ther. 2020, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Haukedal, H.; Freude, K.K. Implications of Glycosylation in Alzheimer’s Disease. Front. Neurosci. 2021, 14, 625348. [Google Scholar] [CrossRef] [PubMed]
- Helmfors, L.; Boman, A.; Civitelli, L.; Nath, S.; Sandin, L.; Janefjord, C.; McCann, H.; Zetterberg, H.; Blennow, K.; Halliday, G.; et al. Protective Properties of Lysozyme on β-Amyloid Pathology: Implications for Alzheimer Disease. Neurobiol. Dis. 2015, 83, 122–133. [Google Scholar] [CrossRef]
- Vela Navarro, N.; De Nadai Mundim, G.; Cudic, M. Implications of Mucin-Type O-Glycosylation in Alzheimer’s Disease. Molecules 2025, 30, 1895. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhang, Q.; Xu, S.; Yu, Y. The Alteration and Role of Glycoconjugates in Alzheimer’s Disease. Front. Aging Neurosci. 2024, 16, 1398641. [Google Scholar] [CrossRef]
- Hong, X.; Huang, L.; Lei, F.; Li, T.; Luo, Y.; Zeng, M.; Wang, Z. The Role and Pathogenesis of Tau Protein in Alzheimer’s Disease. Biomolecules 2025, 15, 824. [Google Scholar] [CrossRef]
- Nedelkov, D.; Tsokolas, Z.E.; Rodrigues, M.S.; Sible, I.; Han, S.D.; Kerman, B.E.; Renteln, M.; Mack, W.J.; Pascoal, T.A.; Yassine, H.N.; et al. Increased Cerebrospinal Fluid and Plasma apoE Glycosylation Is Associated with Reduced Levels of Alzheimer’s Disease Biomarkers. Alzheimer’s Res. Ther. 2025, 17, 151. [Google Scholar] [CrossRef]
- Olejnik, B.; Ferens-Sieczkowska, M. Lectins and Neurodegeneration: A Glycobiologist’s Perspective. Adv. Clin. Exp. Med. 2025, 34, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Pająk, B.; Kania, E.; Orzechowski, A. Killing Me Softly: Connotations to Unfolded Protein Response and Oxidative Stress in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2016, 2016, 1805304. [Google Scholar] [CrossRef] [PubMed]
- Pimenova, A.A.; Goate, A.M. Novel Presenilin 1 and 2 Double Knock-out Cell Line for in Vitro Validation of PSEN1 and PSEN2 Mutations. Neurobiol. Dis. 2020, 138, 104785. [Google Scholar] [CrossRef]
- Qiu, G.; Cao, L.; Chen, Y.-J. Novel Heterozygous Mutation in Alpha-2-Macroglobulin (A2M) Suppressing the Binding of Amyloid-β (Aβ). Front. Neurol. 2023, 13, 1090900. [Google Scholar] [CrossRef]
- Piscitelli, E.; Abeni, E.; Balbino, C.; Angeli, E.; Cocola, C.; Pelucchi, P.; Palizban, M.; Diaspro, A.; Götte, M.; Zucchi, I.; et al. Glycosylation Regulation by TMEM230 in Aging and Autoimmunity. Int. J. Mol. Sci. 2025, 26, 2412. [Google Scholar] [CrossRef]
- Ercan-Herbst, E.; Ehrig, J.; Schöndorf, D.C.; Behrendt, A.; Klaus, B.; Gomez Ramos, B.; Prat Oriol, N.; Weber, C.; Ehrnhoefer, D.E. A Post-Translational Modification Signature Defines Changes in Soluble Tau Correlating with Oligomerization in Early Stage Alzheimer’s Disease Brain. Acta Neuropathol. Commun. 2019, 7, 192. [Google Scholar] [CrossRef] [PubMed]
- Nagae, M.; Yamaguchi, Y.; Taniguchi, N.; Kizuka, Y. 3D Structure and Function of Glycosyltransferases Involved in N-Glycan Maturation. Int. J. Mol. Sci. 2020, 21, 437. [Google Scholar] [CrossRef]
- Ramazi, S.; Dadzadi, M.; Darvazi, M.; Seddigh, N.; Allahverdi, A. Protein Modification in Neurodegenerative Diseases. MedComm 2024, 5, e674. [Google Scholar] [CrossRef]
- Saha, A.; Fernández-Tejada, A. Chemical Biology Tools to Interrogate the Roles of O-GlcNAc in Immunity. Front. Immunol. 2023, 13, 1089824. [Google Scholar] [CrossRef]
- Shi, J.; Ku, X.; Zou, X.; Hou, J.; Yan, W.; Zhang, Y. Comprehensive Analysis of O-Glycosylation of Amyloid Precursor Protein (APP) Using Targeted and Multi-Fragmentation MS Strategy. Biochim. Biophys. Acta BBA—Gen. Subj. 2021, 1865, 129954. [Google Scholar] [CrossRef]
- Singh, Y.; Regmi, D.; Ormaza, D.; Ayyalasomayajula, R.; Vela, N.; Mundim, G.; Du, D.; Minond, D.; Cudic, M. Mucin-Type O-Glycosylation Proximal to β-Secretase Cleavage Site Affects APP Processing and Aggregation Fate. Front. Chem. 2022, 10, 859822. [Google Scholar] [CrossRef] [PubMed]
- Suttapitugsakul, S.; Stavenhagen, K.; Donskaya, S.; Bennett, D.A.; Mealer, R.G.; Seyfried, N.T.; Cummings, R.D. Glycoproteomics Landscape of Asymptomatic and Symptomatic Human Alzheimer’s Disease Brain. Mol. Cell. Proteom. 2022, 21, 100433. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Ohkawa, Y.; Maeda, K.; Kanto, N.; Johnson, E.L.; Harada, Y. N-Glycan Branching Enzymes Involved in Cancer, Alzheimer’s Disease and COPD and Future Perspectives. Biochem. Biophys. Res. Commun. 2022, 633, 68–71. [Google Scholar] [CrossRef]
- Tena, J.; Maezawa, I.; Barboza, M.; Wong, M.; Zhu, C.; Alvarez, M.R.; Jin, L.-W.; Zivkovic, A.M.; Lebrilla, C.B. Regio-Specific N-Glycome and N-Glycoproteome Map of the Elderly Human Brain with and Without Alzheimer’s Disease. Mol. Cell. Proteom. 2022, 21, 100427. [Google Scholar] [CrossRef]
- Urano, Y.; Takahachi, M.; Higashiura, R.; Fujiwara, H.; Funamoto, S.; Imai, S.; Futai, E.; Okuda, M.; Sugimoto, H.; Noguchi, N. Curcumin Derivative GT863 Inhibits Amyloid-Beta Production via Inhibition of Protein N-Glycosylation. Cells 2020, 9, 349. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, X.; Zeng, J.; Yuan, J.; Wang, Z.; Zhou, W.; Zhang, Y. LW-AFC Effects on N-Glycan Profile in Senescence-Accelerated Mouse Prone 8 Strain, a Mouse Model of Alzheimer’s Disease. Aging Dis. 2017, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gopal, S.; Pocock, R.; Xiao, Z. Glycan Mimetics from Natural Products: New Therapeutic Opportunities for Neurodegenerative Disease. Molecules 2019, 24, 4604. [Google Scholar] [CrossRef]
- Wani, W.Y.; Chatham, J.C.; Darley-Usmar, V.; McMahon, L.L.; Zhang, J. O-GlcNAcylation and Neurodegeneration. Brain Res. Bull. 2017, 133, 80–87. [Google Scholar] [CrossRef]
- Weber, P.; Bojarová, P.; Brouzdová, J.; Křen, V.; Kulik, N.; Stütz, A.E.; Thonhofer, M.; Wrodnigg, T.M. Diaminocyclopentane—l-Lysine Adducts: Potent and Selective Inhibitors of Human O-GlcNAcase. Bioorganic Chem. 2024, 148, 107452. [Google Scholar] [CrossRef]
- Wheatley, E.G.; Albarran, E.; White, C.W.; Bieri, G.; Sanchez-Diaz, C.; Pratt, K.; Snethlage, C.E.; Ding, J.B.; Villeda, S.A. Neuronal O-GlcNAcylation Improves Cognitive Function in the Aged Mouse Brain. Curr. Biol. 2019, 29, 3359–3369.e4. [Google Scholar] [CrossRef]
- Wu, F.; Li, W.; Lu, H.; Li, L. Recent Advances in Mass Spectrometry-Based Studies of Post-Translational Modifications in Alzheimer’s Disease. Mol. Cell. Proteom. 2025, 24, 101003. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Wen, R.; Yang, N.; Zhang, T.-N.; Liu, C.-F. Roles of Protein Post-Translational Modifications in Glucose and Lipid Metabolism: Mechanisms and Perspectives. Mol. Med. 2023, 29, 93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, T.; Zhao, H.; Ren, S. Glycosylation in Aging and Neurodegenerative Diseases. Acta Biochim. Biophys. Sin. 2024, 56, 1208–1220. [Google Scholar] [CrossRef]
- Reddy, V.P.; Aryal, P.; Darkwah, E.K. Advanced Glycation End Products in Health and Disease. Microorganisms 2022, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lang, M. New Insight into Protein Glycosylation in the Development of Alzheimer’s Disease. Cell Death Discov. 2023, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Moll, T.; Shaw, P.J.; Cooper-Knock, J. Disrupted Glycosylation of Lipids and Proteins Is a Cause of Neurodegeneration. Brain 2020, 143, 1332–1340. [Google Scholar] [CrossRef]
- Pradeep, P.; Kang, H.; Lee, B. Glycosylation and Behavioral Symptoms in Neurological Disorders. Transl. Psychiatry 2023, 13, 154. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Chin, L.-S.; Pan, S.; Li, L. Human Brain Glycoform Coregulation Network and Glycan Modification Alterations in Alzheimer’s Disease. Sci. Adv. 2024, 10, eadk6911. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Chin, L.-S.; Li, L. Integrative Glycoproteomics Reveals Protein N-Glycosylation Aberrations and Glycoproteomic Network Alterations in Alzheimer’s Disease. Sci. Adv. 2020, 6, eabc5802. [Google Scholar] [CrossRef]
- Schreiner, T.G.; Menéndez-González, M.; Schreiner, O.D.; Ciobanu, R.C. Intrathecal Therapies for Neurodegenerative Diseases: A Review of Current Approaches and the Urgent Need for Advanced Delivery Systems. Biomedicines 2025, 13, 2167. [Google Scholar] [CrossRef]
- Yang, Y.; Gu, Y.; Wan, B.; Ren, X.; Guo, L.-H. Label-Free Electrochemical Biosensing of Small-Molecule Inhibition on O-GlcNAc Glycosylation. Biosens. Bioelectron. 2017, 95, 94–99. [Google Scholar] [CrossRef]
- Lin, T.; Van Husen, L.S.; Yu, Y.; Tjernberg, L.O.; Schedin-Weiss, S. Lack of N-Glycosylation Increases Amyloidogenic Processing of the Amyloid Precursor Protein. Glycobiology 2022, 32, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Buliman, A.; Calin, M.A.; Iordache, M.P. Targeting Anxiety with Light: Mechanistic and Clinical Insights into Photobiomodulation Therapy: A Mini Narrative Review. Balneo PRM Res. J. 2025, 16, 846. [Google Scholar] [CrossRef]
- Adam, R.; Moldovan, C.; Tudorache, S.; Hârșovescu, T.; Orban, C.; Pogărășteanu, M.; Rusu, E. Patellar Resurfacing in Total Knee Arthroplasty, a Never-Ending Controversy; Case Report and Literature Review. Diagnostics 2023, 13, 383. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).