
Germline Single-Nucleotide Polymorphism GFI1-36N Causes Alterations in Mitochondrial Metabolism and Leads to Increased ROS-Mediated DNA Damage in a Murine Model of Human Acute Myeloid Leukemia

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Laboratory Animal Strain
2.2. Generation of MLL-AF9 Leukemic Mice
2.3. Collection of Leukemic Cells
2.4. Generation of GFI1-36N-K562 Cells
2.5. Quantification of DNA Double-Strand Breaks (DSBs)
2.6. Seahorse Experiments
2.7. Mitochondrial Membrane Potential (MMP), Mitochondrial Mass (Mito-Mass), and ROS Measurement
2.8. Glucose Consumption and Lactate Secretion
2.9. Mass Spectrometry (MS)
3. Results
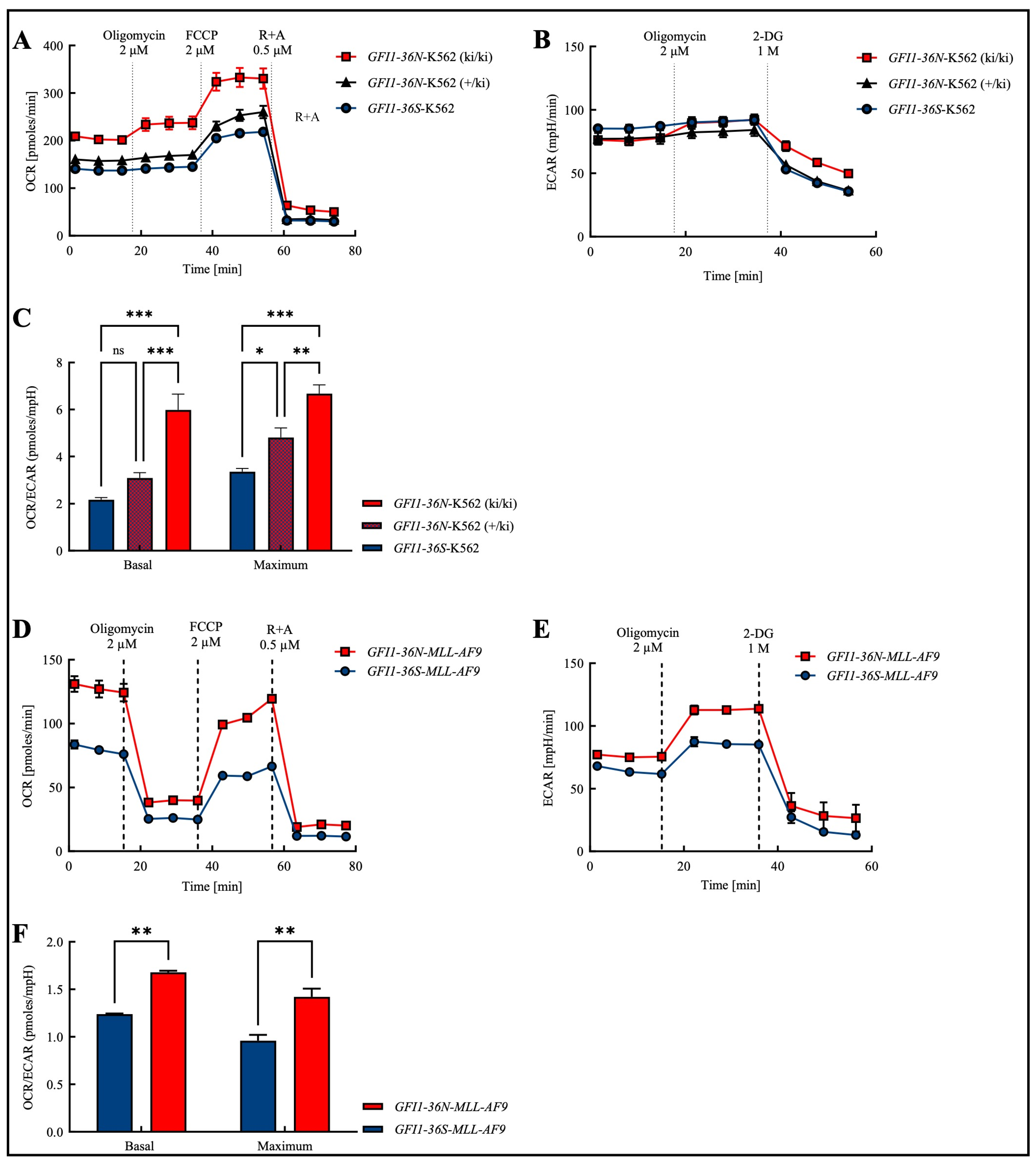
3.1. Altered Mitochondrial Metabolism in GFI1-36N Leukemic Cells
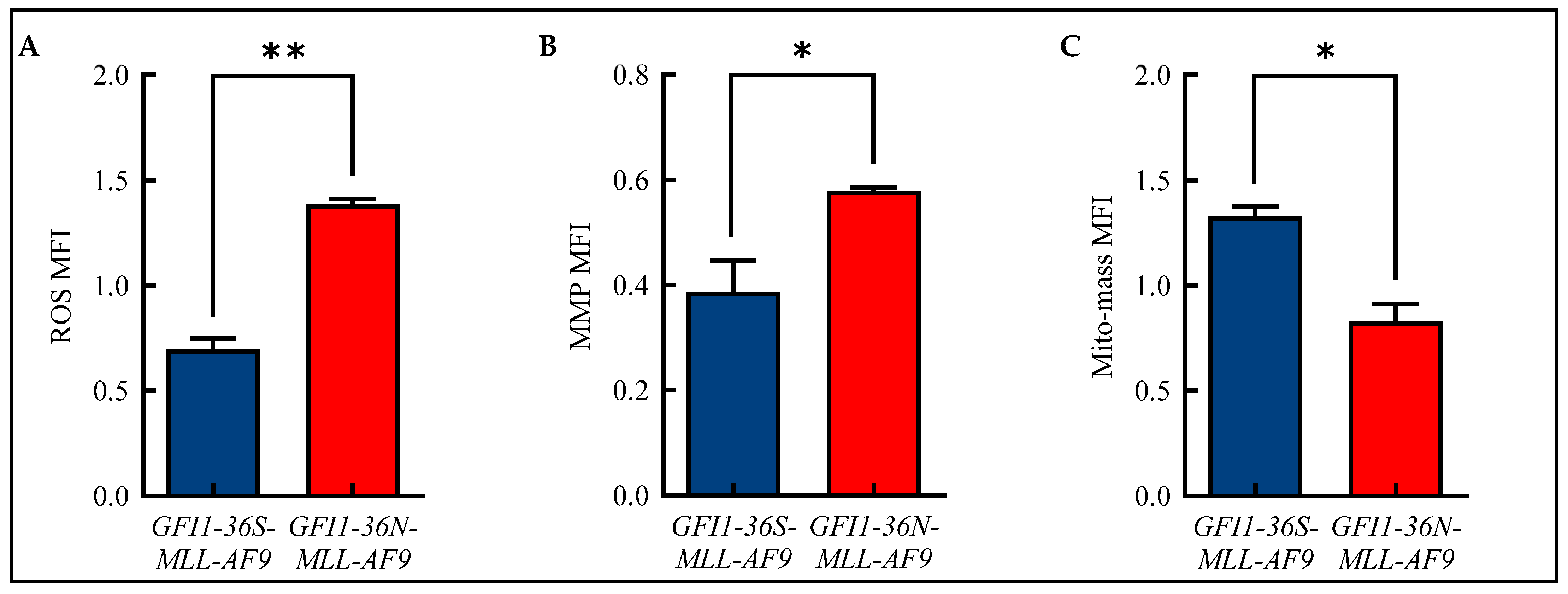
3.2. Increased ROS and MMP Levels in GFI1-36N-MLL-AF9 Cells
3.3. ROS-Induced DNA Damage in GFI1-36N-MLL-AF9 Cells
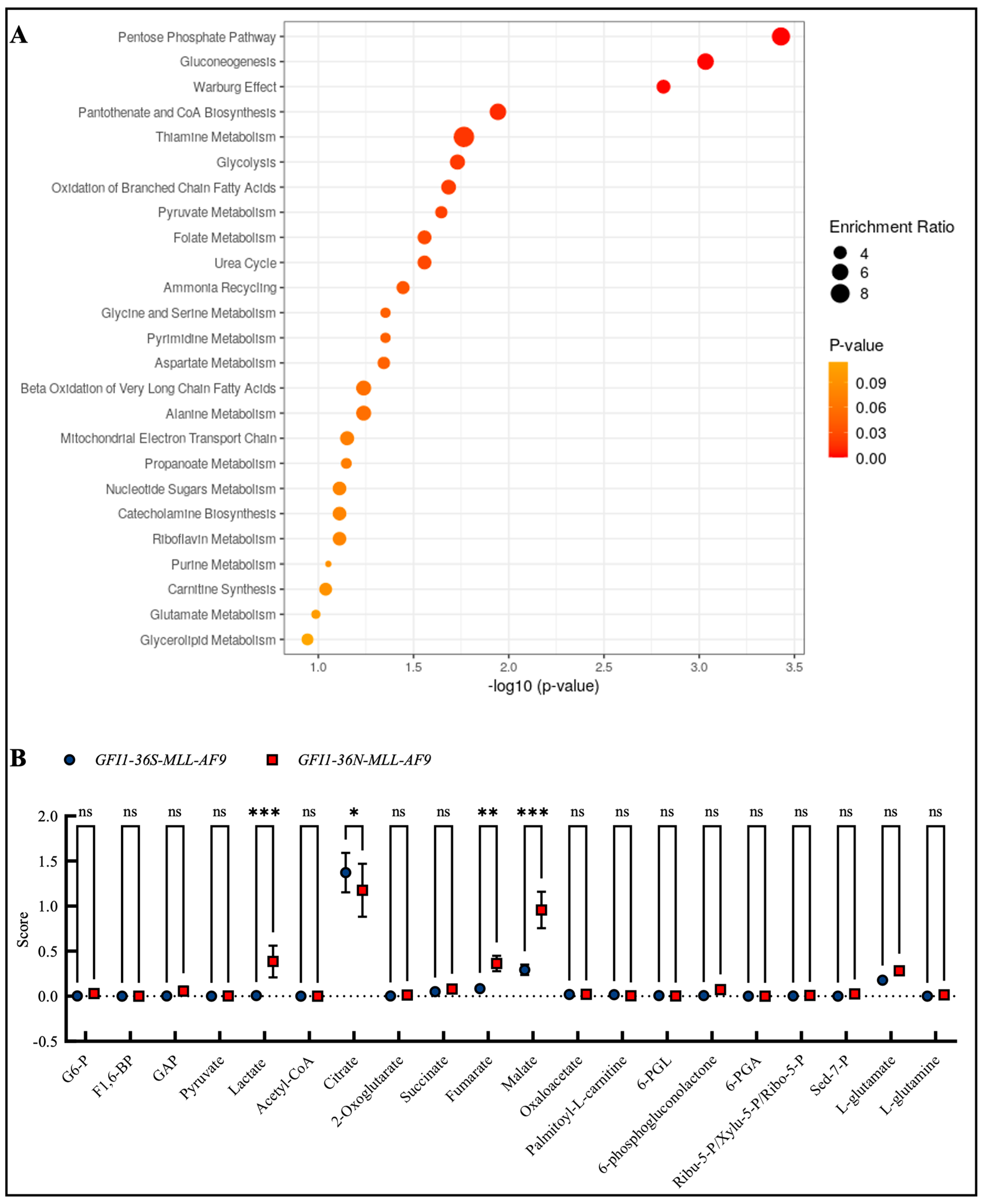
3.4. GFI1-36N-MLL-AF9 Cells Exhibit Pro-Oncogenic Metabolic Changes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vadnais, C.; Chen, R.; Fraszczak, J.; Yu, Z.; Boulais, J.; Pinder, J.; Frank, D.; Khandanpour, C.; Hébert, J.; Dellaire, G.; et al. GFI1 facilitates efficient DNA repair by regulating PRMT1 dependent methylation of MRE11 and 53BP1. Nat. Commun. 2018, 9, 1418. [Google Scholar] [CrossRef] [PubMed]
- Khandanpour, C.; Thiede, C.; Valk, P.J.; Sharif-Askari, E.; Nückel, H.; Lohmann, D.; Horsthemke, B.; Siffert, W.; Neubauer, A.; Grzeschik, K.H.; et al. A variant allele of Growth Factor Independence 1 (GFI1) is associated with acute myeloid leukemia. Blood 2010, 115, 2462–2472. [Google Scholar] [CrossRef]
- Hönes, J.M.; Botezatu, L.; Helness, A.; Vadnais, C.; Vassen, L.; Robert, F.; Hergenhan, S.M.; Thivakaran, A.; Schütte, J.; Al-Matary, Y.S.; et al. GFI1 as a novel prognostic and therapeutic factor for AML/MDS. Leukemia 2016, 30, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Botezatu, L.; Michel, L.C.; Makishima, H.; Schroeder, T.; Germing, U.; Haas, R.; van der Reijden, B.; Marneth, A.E.; Bergevoet, S.M.; Jansen, J.H.; et al. GFI1(36N) as a therapeutic and prognostic marker for myelodysplastic syndrome. Exp. Hematol. 2016, 44, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, J.; Sun, K.; Frank, D.; Neumann, F.; Hüve, J.; Budde, P.M.; Liu, L.; Xie, X.; Patnana, P.K.; Ahmed, H.M.M.; et al. Presence of the GFI1-36N single nucleotide polymorphism enhances the response of MLL-AF9 leukemic cells to CDK4/6 inhibition. Front. Oncol. 2022, 12, 903691. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Patnana, P.K.; Vorwerk, J.; Mao, L.; Gopal, L.M.; Jung, N.; Hennig, T.; Ruhnke, L.; Frenz, J.M.; Kuppusamy, M.; et al. Germ line variant GFI1-36N affects DNA repair and sensitizes AML cells to DNA damage and repair therapy. Blood 2023, 142, 2175–2191. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular stress responses in radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Fiolka, K.; Hertzano, R.; Vassen, L.; Zeng, H.; Hermesh, O.; Avraham, K.B.; Dührsen, U.; Möröy, T. Gfi1 and Gfi1b act equivalently in haematopoiesis, but have distinct, non-overlapping functions in inner ear development. EMBO Rep. 2006, 7, 326–333. [Google Scholar] [CrossRef]
- Zuber, J.; Radtke, I.; Pardee, T.S.; Zhao, Z.; Rappaport, A.R.; Luo, W.; McCurrach, M.E.; Yang, M.M.; Dolan, M.E.; Kogan, S.C.; et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009, 23, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Loberg, M.A.; Eudy, E.; Schwartz, L.S.; Mujica, K.D.; Trowbridge, J.J. Heritable genetic background alters survival and phenotype of Mll-AF9-induced leukemias. Exp. Hematol. 2020, 89, 61–67.e3. [Google Scholar] [CrossRef] [PubMed]
- Marneth, A.E.; Botezatu, L.; Hönes, J.M.; Israël, J.C.L.; Schütte, J.; Vassen, L.; Lams, R.F.; Bergevoet, S.M.; Groothuis, L.; Mandoli, A.; et al. GFI1 is required for RUNX1/ETO positive acute myeloid leukemia. Haematologica 2018, 103, e395–e399. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.T.; Seo, H.S.; Zhang, T.; Wang, Y.; Jiang, B.; Li, Q.; Buckley, D.L.; Nabet, B.; Roberts, J.M.; Paulk, J.; et al. MELK is not necessary for the proliferation of basal-like breast cancer cells. Elife 2017, 6, e26693. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Zhu, B.; Wang, L.; Chen, C.; Hong, M.; Huang, Y.; Li, H.; Han, H.; Cai, B.; et al. Increasing the efficiency and targeting range of cytidine base editors through fusion of a single-stranded DNA-binding protein domain. Nat. Cell Biol. 2020, 22, 740–750. [Google Scholar] [CrossRef]
- Liu, L.; Patnana, P.K.; Xie, X.; Frank, D.; Nimmagadda, S.C.; Su, M.; Zhang, D.; Koenig, T.; Rosenbauer, F.; Liebmann, M.; et al. GFI1B acts as a metabolic regulator in hematopoiesis and acute myeloid leukemia. Leukemia 2022, 36, 2196–2207. [Google Scholar] [CrossRef]
- Patnana, P.K.; Liu, L.; Frank, D.; Nimmagadda, S.C.; Behrens, M.; Ahmed, H.; Xie, X.; Liebmann, M.; Wei, L.; Gerdemann, A.; et al. Dose-dependent expression of GFI1 alters metabolism in the haematopoietic progenitors and MLL::AF9-induced leukaemic cells. Br. J. Haematol. 2023, 202, 1033–1048. [Google Scholar] [CrossRef]
- Plitzko, B.; Loesgen, S. Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism. Bio Protoc. 2018, 8, e2850. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell. 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr. Cancer Drug Targets. 2018, 18, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Rassool, F.V.; Gaymes, T.J.; Omidvar, N.; Brady, N.; Beurlet, S.; Pla, M.; Reboul, M.; Lea, N.; Chomienne, C.; Thomas, N.S.; et al. Reactive oxygen species, DNA damage, and error-prone repair: A model for genomic instability with progression in myeloid leukemia? Cancer Res. 2007, 67, 8762–8771. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Collins, L.B.; Chen, T.H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef] [PubMed]
- Kaweme, N.M.; Zhou, S.; Changwe, G.J.; Zhou, F. The significant role of redox system in myeloid leukemia: From pathogenesis to therapeutic applications. Biomark. Res. 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef]
- Oberley, T.D.; Oberley, L.W. Antioxidant enzyme levels in cancer. Histol. Histopathol. 1997, 12, 525–535. [Google Scholar]
- Sun, S.Y. N-acetylcysteine, reactive oxygen species and beyond. Cancer Biol. Ther. 2010, 9, 109–110. [Google Scholar] [CrossRef]
- Brandt, U. Oxidoreduktasen und reaktive Sauerstoffspezies. In Löffler/Petrides Biochemie und Pathobiochemie; Heinrich, P.C., Müller, M., Graeve, L., Koch, H.G., Eds.; Springer: Heidelberg, Germany, 2022; pp. 329–335. [Google Scholar]
- Sentellas, S.; Morales-Ibanez, O.; Zanuy, M.; Albertí, J.J. GSSG/GSH ratios in cryopreserved rat and human hepatocytes as a biomarker for drug induced oxidative stress. Toxicol. In Vitro 2014, 28, 1006–1015. [Google Scholar] [CrossRef]
- Zappe, K.; Pointner, A.; Switzeny, O.J.; Magnet, U.; Tomeva, E.; Heller, J.; Mare, G.; Wagner, K.H.; Knasmueller, S.; Haslberger, A.G. Counteraction of oxidative stress by vitamin E affects epigenetic regulation by increasing global methylation and gene expression of MLH1 and DNMT1 dose dependently in Caco-2 cells. Oxid. Med. Cell Longev. 2018, 2018, 3734250. [Google Scholar] [CrossRef]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; Weijzen, S.; de Vries-Smits, A.M.; Saarloos, I.; de Ruiter, N.D.; Bos, J.L.; Burgering, B.M. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Lebiedzinska, M.; Karkucinska-Wieckowska, A.; Wojtala, A.; Suski, J.M.; Szabadkai, G.; Wilczynski, G.; Wlodarczyk, J.; Diogo, C.V.; Oliveira, P.J.; Tauber, J.; et al. Disrupted ATP synthase activity and mitochondrial hyperpolarisation-dependent oxidative stress is associated with p66Shc phosphorylation in fibroblasts of NARP patients. Int. J. Biochem. Cell Biol. 2013, 45, 141–150. [Google Scholar] [CrossRef]
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023, 19, 401–414. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Botezatu, L.; Michel, L.C.; Helness, A.; Vadnais, C.; Makishima, H.; Hönes, J.M.; Robert, F.; Vassen, L.; Thivakaran, A.; Al-Matary, Y.; et al. Epigenetic therapy as a novel approach for GFI136N-associated murine/human AML. Exp. Hematol. 2016, 44, 713–726.e14. [Google Scholar] [CrossRef]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updat. 2018, 38, 1–11. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vorwerk, J.; Liu, L.; Stadler, T.H.; Frank, D.; Ahmed, H.M.M.; Patnana, P.K.; Kebenko, M.; Dazert, E.; Opalka, B.; von Bubnoff, N.; et al. Germline Single-Nucleotide Polymorphism GFI1-36N Causes Alterations in Mitochondrial Metabolism and Leads to Increased ROS-Mediated DNA Damage in a Murine Model of Human Acute Myeloid Leukemia. Biomedicines 2025, 13, 107. https://doi.org/10.3390/biomedicines13010107
Vorwerk J, Liu L, Stadler TH, Frank D, Ahmed HMM, Patnana PK, Kebenko M, Dazert E, Opalka B, von Bubnoff N, et al. Germline Single-Nucleotide Polymorphism GFI1-36N Causes Alterations in Mitochondrial Metabolism and Leads to Increased ROS-Mediated DNA Damage in a Murine Model of Human Acute Myeloid Leukemia. Biomedicines. 2025; 13(1):107. https://doi.org/10.3390/biomedicines13010107
Chicago/Turabian StyleVorwerk, Jan, Longlong Liu, Theresa Helene Stadler, Daria Frank, Helal Mohammed Mohammed Ahmed, Pradeep Kumar Patnana, Maxim Kebenko, Eva Dazert, Bertram Opalka, Nikolas von Bubnoff, and et al. 2025. "Germline Single-Nucleotide Polymorphism GFI1-36N Causes Alterations in Mitochondrial Metabolism and Leads to Increased ROS-Mediated DNA Damage in a Murine Model of Human Acute Myeloid Leukemia" Biomedicines 13, no. 1: 107. https://doi.org/10.3390/biomedicines13010107
APA StyleVorwerk, J., Liu, L., Stadler, T. H., Frank, D., Ahmed, H. M. M., Patnana, P. K., Kebenko, M., Dazert, E., Opalka, B., von Bubnoff, N., & Khandanpour, C. (2025). Germline Single-Nucleotide Polymorphism GFI1-36N Causes Alterations in Mitochondrial Metabolism and Leads to Increased ROS-Mediated DNA Damage in a Murine Model of Human Acute Myeloid Leukemia. Biomedicines, 13(1), 107. https://doi.org/10.3390/biomedicines13010107