
AT2R Activation Improves Wound Healing in a Preclinical Mouse Model
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
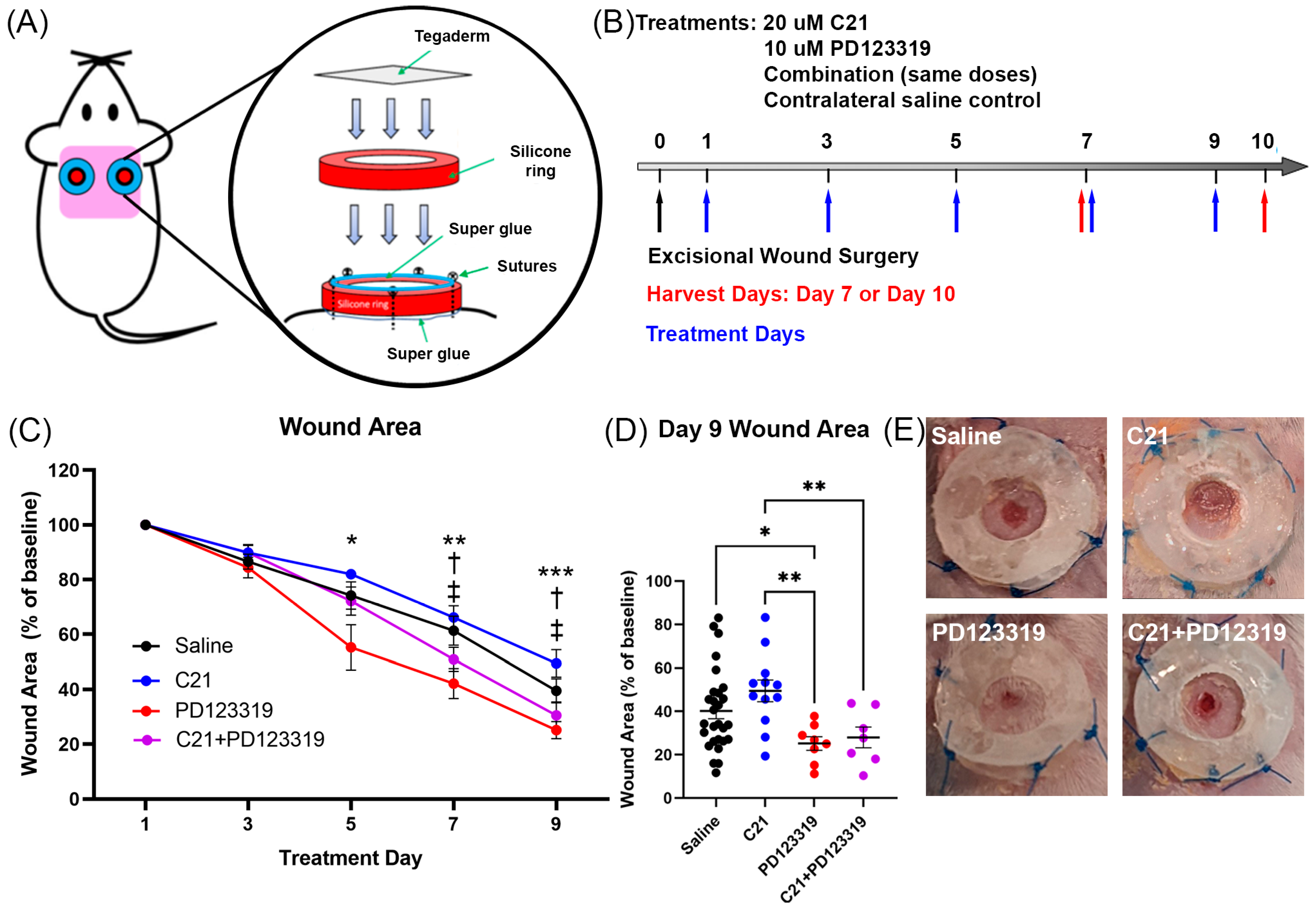
2.1. Mice and Splinted Excisional Wound Model
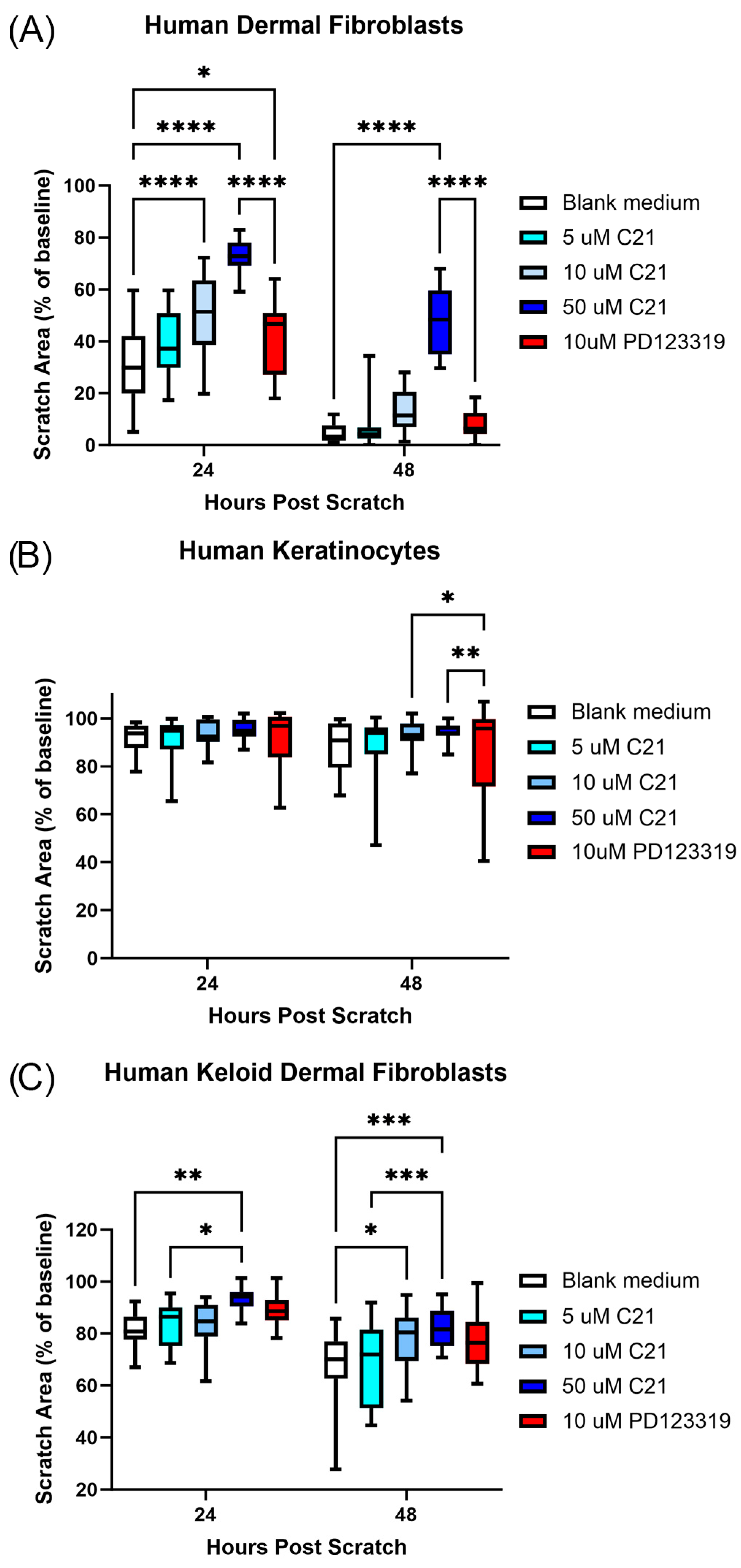
2.2. Scratch Assay
2.3. Histology and Multispectral Multiplex Immunofluorescence Staining
2.4. Collagen and CD31 Imaging Analysis
2.5. Mantra Imaging Analysis
2.6. Quantitative Reverse-Transcription Polymerase Chain Reaction
2.7. Statistical Analysis
3. Results
3.1. AT2R Signal Inhibition Accelerates Wound Closure in a Mouse Excisional Wound Model
3.2. AT2R Activation Reduces Human Skin Cell Migration In Vitro
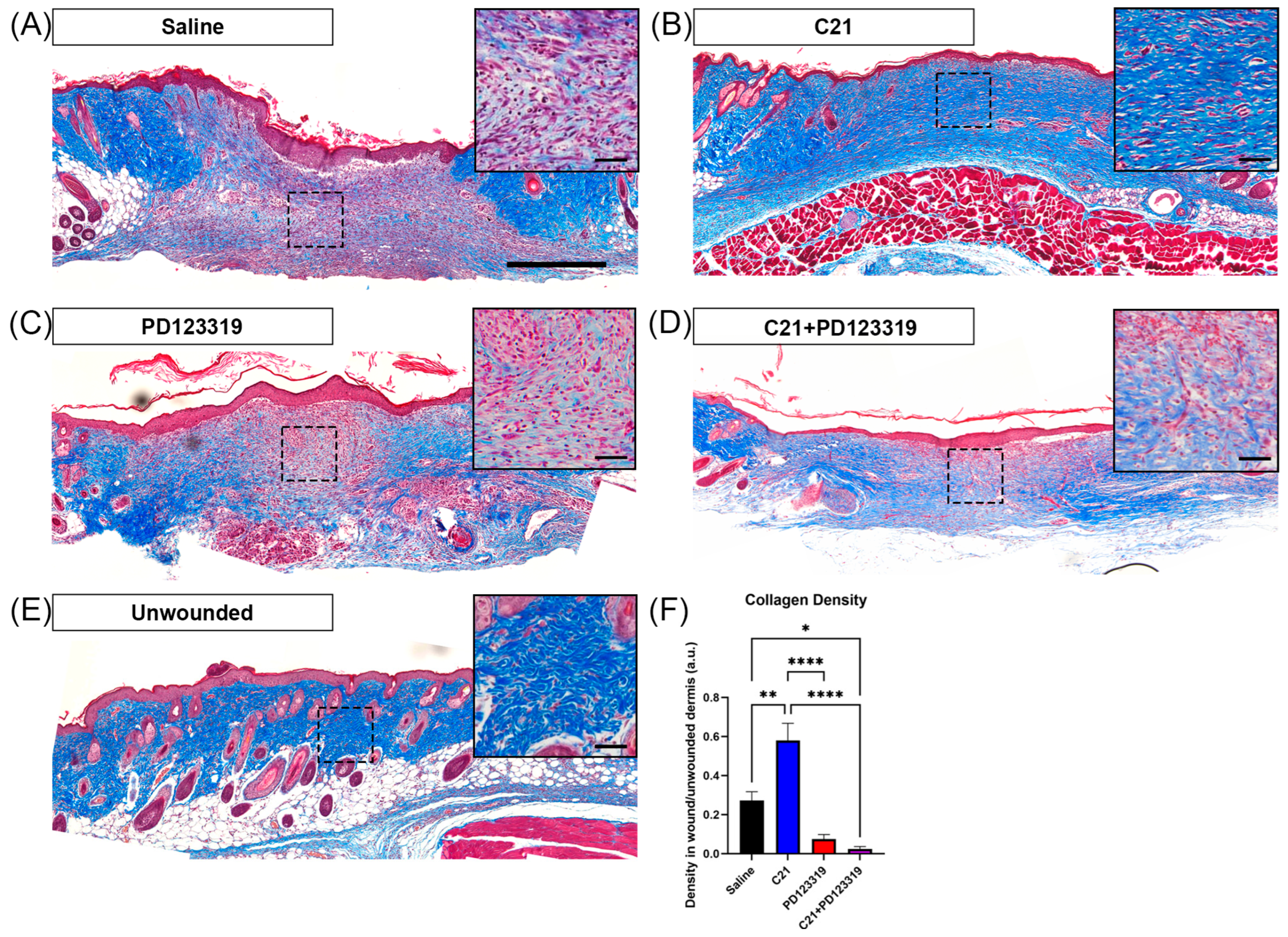
3.3. Increased AT2R Signaling Increases Early Wound Collagen Density and Reduces Cellular Infiltration
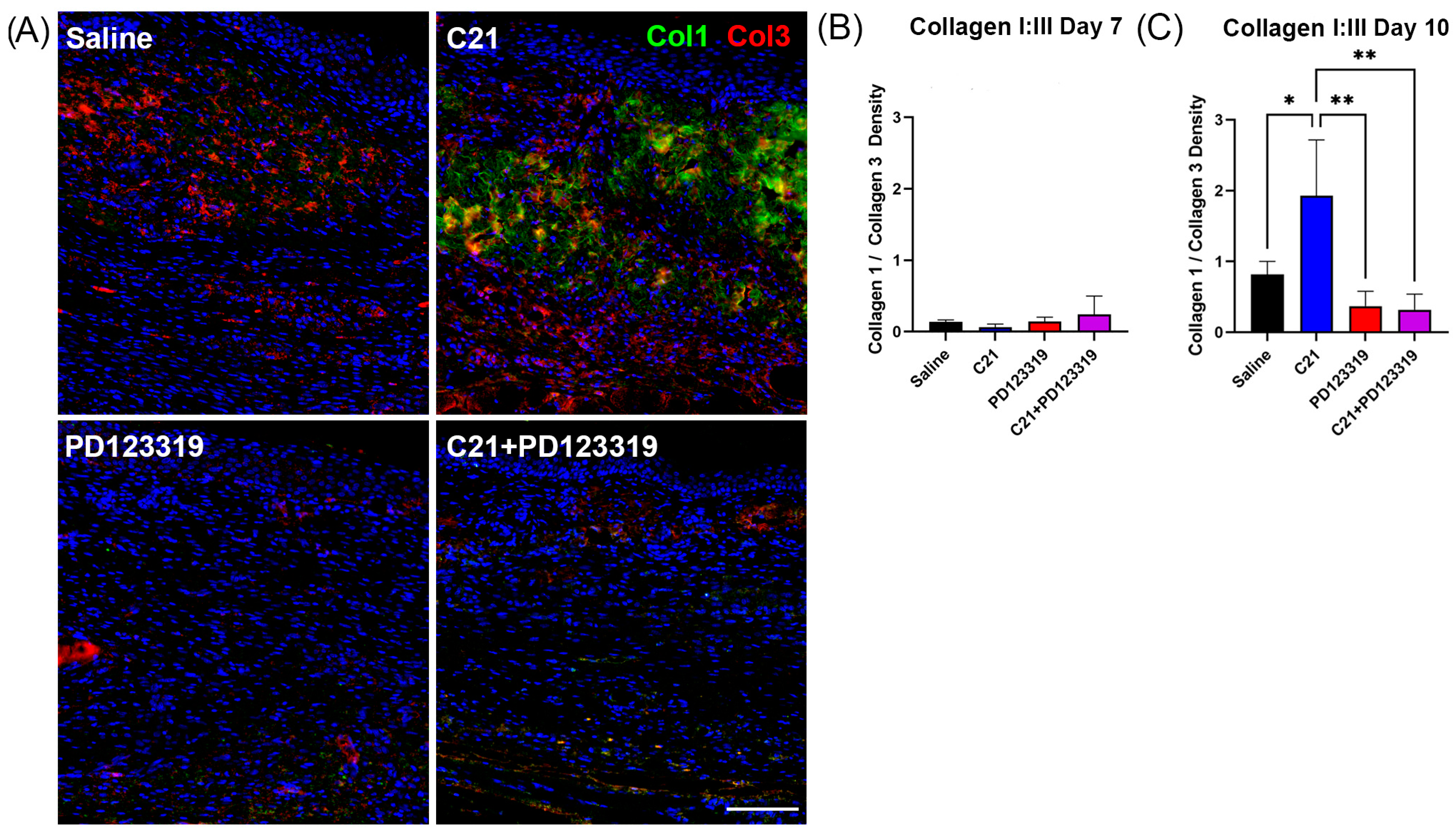
3.4. Collagen I:III Ratio Is Increased with AT2R Signaling by Day 10 Post-Wounding
3.5. AT2R Activation Increases Wound Vascular Density
3.6. AT2R Signal Modulation Alters Wound Remodeling and Immune Cell Gene Transcription
3.7. Increased AT2R Signaling during Wound Healing Promotes an Anti-Inflammatory Immune Cell Microenvironment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Tonnesen, M.G.; Feng, X.; Clark, R.A.F. Angiogenesis in Wound Healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Nunan, R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br. J. Dermatol. 2015, 173, 370–378. [Google Scholar] [CrossRef] [PubMed]
- DesJardins-Park, H.E.; Mascharak, S.; Chinta, M.S.; Wan, D.C.; Longaker, M.T. The Spectrum of Scarring in Craniofacial Wound Repair. Front. Physiol. 2019, 10, 332. [Google Scholar] [CrossRef] [PubMed]
- Monavarian, M.; Kader, S.; Moeinzadeh, S.; Jabbari, E. Regenerative Scar-Free Skin Wound Healing. Tissue Eng. Part B 2019, 25, 294–311. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A.; Roy, S.; McDaniel, J.C. Neutrophils and Wound Repair: Positive Actions and Negative Reactions. Adv. Wound Care 2013, 2, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Oskeritzian, C.A. Mast Cells and Wound Healing. Adv. Wound Care 2012, 1, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Velnar, T.; Bailey, T.; Smrkolj, V. The wound healing process: An overview of the cellular and molecular mechanisms. J. Int. Med. Res. 2009, 37, 1528–1542. [Google Scholar] [CrossRef]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef]
- Xue, M.; Jackson, C.J. Extracellular Matrix Reorganization During Wound Healing and Its Impact on Abnormal Scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.C.; Bodnar, R.; Wells, A. Matrix control of scarring. Cell. Mol. Life Sci. 2011, 68, 1871–1881. [Google Scholar] [CrossRef]
- Knight, K.R.; Lepore, D.A.; Horne, R.S.; Ritz, M.; Hurley, J.V.; Kumta, S.; O’Brien, B.M. Collagen content of uninjured skin and scar tissue in foetal and adult sheep. Int. J. Exp. Pathol. 1993, 74, 583–591. [Google Scholar] [PubMed]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.; LeVatte, T.; Boudreau, C.; Midgen, C.; Gratzer, P.; Marshall, J.; Bezuhly, M. Angiotensin II Type I Receptor Blockade Is Associated with Decreased Cutaneous Scar Formation in a Rat Model. Plast. Reconstr. Surg. 2019, 144, 803e–813e. [Google Scholar] [CrossRef] [PubMed]
- Dow, T.; Selman, T.; Williams, J.; Bezuhly, M. The Role of ACE-Inhibitors and ARBs in reducing hypertrophic scarring following bilateral breast reduction. J. Plast. Reconstr. Aesthetic Surg. 2023, 78, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Fatima, N.; Patel, S.N.; Hussain, T. Angiotensin II Type 2 Receptor: A Target for Protection Against Hypertension, Metabolic Dysfunction, and Organ Remodeling. Hypertension 2021, 77, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, J.; Gareau, A.J.; Byun, S.; Paletz, J.L.; Tang, D.; Williams, J.; LeVatte, T.; Bezuhly, M. Effect of Compound 21, a Selective Angiotensin II Type 2 Receptor Agonist, in a Murine Xenograft Model of Dupuytren Disease. Plast. Reconstr. Surg. 2017, 140, 686e–696e. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E. Historical perspective of the renin-angiotensin system. Mol. Biotechnol. 2003, 24, 27–39. [Google Scholar] [CrossRef]
- Kaschina, E.; Grzesiak, A.; Li, J.; Foryst-Ludwig, A.; Timm, M.; Rompe, F.; Sommerfeld, M.; Kemnitz, U.R.; Curato, C.; Namsolleck, P.; et al. Angiotensin II type 2 receptor stimulation: A novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 2008, 118, 2523–2532. [Google Scholar] [CrossRef]
- Bruce, E.; Shenoy, V.; Rathinasabapathy, A.; Espejo, A.; Horowitz, A.; Oswalt, A.; Francis, J.; Nair, A.; Unger, T.; Raizada, M.K.; et al. Selective activation of angiotensin AT2 receptors attenuates progression of pulmonary hypertension and inhibits cardiopulmonary fibrosis. Br. J. Pharmacol. 2015, 172, 2219–2231. [Google Scholar] [CrossRef] [PubMed]
- Vicore Pharma Holding AB. Interim Report Apr 1–Jun 30, 2021; Vicore Pharma Holding AB: Gothenburg, Sweden, 2021. [Google Scholar]
- Boudreau, C.; LeVatte, T.; Jones, C.; Gareau, A.; Legere, S.; Bezuhly, M. The Selective Angiotensin II Type 2 Receptor Agonist Compound 21 Reduces Abdominal Adhesions in Mice. J. Surg. Res. 2020, 256, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-W.; Cheng, B.; Yu, W.-L.; Sun, R.-X.; Zeng, D.; Wang, J.; Liao, Y.-X.; Fu, X.-B. Angiotensin II regulates phosphoinositide 3 kinase/Akt cascade via a negative crosstalk between AT1 and AT2 receptors in skin fibroblasts of human hypertrophic scars. Life Sci. 2006, 79, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Barsha, G.; Walton, S.L.; Kwok, E.; Mirabito Colafella, K.M.; Pinar, A.A.; Hilliard Krause, L.M.; Gaspari, T.A.; Widdop, R.E.; Samuel, C.S.; Denton, K.M. Relaxin Attenuates Organ Fibrosis via an Angiotensin Type 2 Receptor Mechanism in Aged Hypertensive Female Rats. Kidney360 2021, 2, 1781–1792. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Matsuura, K.; Tanaka, Y.; Honda, T.; Nishida, T.; Inui, M. Signaling mechanism underlying the promotion of keratinocyte migration by angiotensin II. Mol. Pharmacol. 2015, 87, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Brait, V.H.; Arumugam, T.V.; Evans, M.A.; Kim, H.A.; Widdop, R.E.; Drummond, G.R.; Sobey, C.G.; Jones, E.S. Neuroprotective effect of an angiotensin receptor type 2 agonist following cerebral ischemia in vitro and in vivo. Exp. Transl. Stroke Med. 2012, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Sampson, A.K.; Irvine, J.C.; Shihata, W.A.; Dragoljevic, D.; Lumsden, N.; Huet, O.; Barnes, T.; Unger, T.; Steckelings, U.M.; Jennings, G.L.; et al. Compound 21, a selective agonist of angiotensin AT2 receptors, prevents endothelial inflammation and leukocyte adhesion in vitro and in vivo. Br. J. Pharmacol. 2016, 173, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Junge, K.; Klinge, U.; Rosch, R.; Mertens, P.R.; Kirch, J.; Klosterhalfen, B.; Lynen, P.; Schumpelick, V. Decreased collagen type I/III ratio in patients with recurring hernia after implantation of alloplastic prostheses. Langenbeck’s Arch. Surg. 2004, 389, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Wess, T.J. Collagen fibril form and function. Adv. Protein Chem. 2005, 70, 341–374. [Google Scholar] [CrossRef]
- Wang, T.; Gu, Q.; Zhao, J.; Mei, J.; Shao, M.; Pan, Y.; Zhang, J.; Wu, H.; Zhang, Z.; Liu, F. Calcium alginate enhances wound healing by up-regulating the ratio of collagen types I/III in diabetic rats. Int. J. Clin. Exp. Pathol. 2015, 8, 6636–6645. [Google Scholar]
- Fu, M.X.; Knecht, K.J.; Thorpe, S.R.; Baynes, J.W. Role of oxygen in cross-linking and chemical modification of collagen by glucose. Diabetes 1992, 41 (Suppl. S2), 42–48. [Google Scholar] [CrossRef] [PubMed]
- Laverdet, B.; Danigo, A.; Girard, D.; Magy, L.; Demiot, C.; Desmoulière, A. Skin innervation: Important roles during normal and pathological cutaneous repair. Histol. Histopathol. 2015, 30, 875–892. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed]
- Dreieicher, E.; Beck, K.-F.; Lazaroski, S.; Boosen, M.; Tsalastra-Greul, W.; Beck, M.; Fleming, I.; Schaefer, L.; Pfeilschifter, J. Nitric oxide inhibits glomerular TGF-beta signaling via SMOC-1. J. Am. Soc. Nephrol. 2009, 20, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhu, L.; Fu, Z.; Guo, X.; Wang, K.; Hu, X.; Chen, J. Low Molecular Weight Heparin Inhibits Circulating Fibrocytes Differentiation by Modulating Neuronal Nitric Oxide Synthase and TGF-β1/Smad Pathway. Cell. Physiol. Biochem. 2012, 30, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.P.; Most, D.; Efron, D.T.; Tantry, U.; Fischel, M.H.; Barbul, A. The role of iNOS in wound healing. Surgery 2001, 130, 225–229. [Google Scholar] [CrossRef]
- Kolattukudy, P.E.; Niu, J. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ. Res. 2012, 110, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Asano, M.; O’Reilly, A.; Farquhar, A.; Yang, Y.; Amar, S. p53 is an important regulator of CCL2 gene expression. Curr. Mol. Med. 2012, 12, 929–943. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chang, Q.; Lu, F. Oxygen-releasing biomaterials for chronic wounds breathing: From theoretical mechanism to application prospect. Mater. Today Bio 2023, 20, 100687. [Google Scholar] [CrossRef] [PubMed]
- Zaja-Milatovic, S.; Richmond, A. CXC chemokines and their receptors: A case for a significant biological role in cutaneous wound healing. Histol. Histopathol. 2008, 23, 1399–1407. [Google Scholar] [CrossRef]
- Bermudez, D.M.; Xu, J.; Herdrich, B.J.; Radu, A.; Mitchell, M.E.; Liechty, K.W. Inhibition of stromal cell-derived factor-1α further impairs diabetic wound healing. J. Vasc. Surg. 2011, 53, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T.; Nakajima, T.; Tachibana, K.; Iizasa, H.; Bleul, C.C.; Yoshie, O.; Matsushima, K.; Yoshida, N.; Springer, T.A.; Kishimoto, T. Molecular cloning and characterization of a murine pre-B-cell growth-stimulating factor/stromal cell-derived factor 1 receptor, a murine homolog of the human immunodeficiency virus 1 entry coreceptor fusin. Proc. Natl. Acad. Sci. USA 1996, 93, 14726–14729. [Google Scholar] [CrossRef]
- Wulff, B.C.; Parent, A.E.; Meleski, M.A.; Dipietro, L.A.; Schrementi, M.E.; Wilgus, T.A. Mast Cells Contribute to Scar Formation During Fetal Wound Healing. J. Investig. Dermatol. Symp. Proc. 2012, 132, 458–465. [Google Scholar] [CrossRef]
- Marshall, J.S.; Bienenstock, J. The role of mast cells in inflammatory reactions of the airways, skin and intestine. Curr. Opin. Immunol. 1994, 6, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Phipps, K.D.; Gebremeskel, S.; Gillis, J.; Hong, P.; Johnston, B.; Bezuhly, M. Alternatively activated M2 macrophages improve autologous Fat Graft survival in a mouse model through induction of angiogenesis. Plast. Reconstr. Surg. 2015, 135, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Faghih, M.; Hosseini, S.M.; Smith, B.; Ansari, A.M.; Lay, F.; Ahmed, A.K.; Inagami, T.; Marti, G.P.; Harmon, J.W.; Walston, J.D.; et al. Knockout of Angiotensin AT2 receptors accelerates healing but impairs quality. Aging 2015, 7, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, P.; Braye, F.; Dayan, G. Re-epithelialization of adult skin wounds: Cellular mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 344–365. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.E.; Bergeron, A.; Thurlow, P.; Selim, M.A.; Bowers, E.V.; Kuang, A.; Levinson, H. Angiotensin-II mediates nonmuscle myosin II activation and expression and contributes to human keloid disease progression. Mol. Med. 2011, 17, 1196–1203. [Google Scholar] [CrossRef]
- Lo, J.F.; Brennan, M.; Merchant, Z.; Chen, L.; Guo, S.; Eddington, D.T.; DiPietro, L.A. Microfluidic Wound Bandage: Localized Oxygen Modulation of Collagen Maturation. Wound Repair Regen. 2013, 21, 226–234. [Google Scholar] [CrossRef]
- Carter, M.J.; Frykberg, R.G.; Oropallo, A.; Sen, C.K.; Armstrong, D.G.; Nair, H.K.R.; Serena, T.E. Efficacy of Topical Wound Oxygen Therapy in Healing Chronic Diabetic Foot Ulcers: Systematic Review and Meta-Analysis. Adv. Wound Care 2023, 12, 177–186. [Google Scholar] [CrossRef]
- Hajhosseini, B.; Kuehlmann, B.A.; Bonham, C.A.; Kamperman, K.J.; Gurtner, G.C. Hyperbaric Oxygen Therapy: Descriptive Review of the Technology and Current Application in Chronic Wounds. Plast. Reconstr. Surg. Glob. Open 2020, 8, e3136. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.; Saville, C.R.; Murray, P.J.; Cruickshank, S.M.; Hardman, M.J. Local arginase 1 activity is required for cutaneous wound healing. J. Investig. Dermatol. 2013, 133, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qian, L.; Pi, L.; Meng, X. A therapeutic role of exosomal lncRNA H19 from adipose mesenchymal stem cells in cutaneous wound healing by triggering macrophage M2 polarization. Cytokine 2023, 165, 156175. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Wu, Y.-P.; Yang, C.-C.; Yi, Z.; Zeng, N.; Xu, Y.; Zeng, H.; Deng, P.; Zhang, Q.; Wu, M. Knockout of E2F1 enhances the polarization of M2 phenotype macrophages to accelerate the wound healing process. Kaohsiung J. Med. Sci. 2020, 36, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Lyu, L.; Cai, Y.; Zhang, G.; Jing, Z.; Liang, J.; Zhang, R.; Dang, X.; Zhang, C. Exosomes derived from M2 macrophages induce angiogenesis to promote wound healing. Front. Mol. Biosci. 2022, 9, 1008802. [Google Scholar] [CrossRef]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [PubMed]
- Assis, D.V.; Campos, A.C.P.; Paschoa, A.F.N.; Santos, T.F.; Fonoff, E.T.; Pagano, R.L. Systemic and Peripheral Mechanisms of Cortical Stimulation-Induced Analgesia and Refractoriness in a Rat Model of Neuropathic Pain. Int. J. Mol. Sci. 2023, 24, 7796. [Google Scholar] [CrossRef]
- Cioato, S.G.; Fernandes Medeiros, L.; Costa Lopes, B.; De Souza, A.; Richardt Medeiros, H.; Fagundes Assumpcao, J.A.; Caumo, W.; Roesler, R.; Torres, I.L.S. Antinociceptive and neurochemical effects of a single dose of IB-MECA in chronic pain rat models. Purinergic Signal. 2020, 16, 573–584. [Google Scholar] [CrossRef]
- Xie, Y.; Fang, F.; Su, P.; Xiao, J.; Zheng, H.; Zhuang, Y. Quantification of axonal ingrowth and functional recovery in a myocutaneous flap model in rats with strong clinical implications. Wound Repair Regen. 2020, 28, 823–833. [Google Scholar] [CrossRef]
- Lu, J.; Wu, L.; Jiang, T.; Wang, Y.; Zhao, H.; Gao, Q.; Pan, Y.; Tian, Y.; Zhang, Y. Angiotensin AT2 receptor stimulation inhibits activation of NADPH oxidase and ameliorates oxidative stress in rotenone model of Parkinson’s disease in CATH.a cells. Neurotoxicol. Teratol. 2015, 47, 16–24. [Google Scholar] [CrossRef]
- Pulakat, L.; Sumners, C. Angiotensin Type 2 Receptors: Painful, or Not? Front. Pharmacol. 2020, 11, 571994. [Google Scholar] [CrossRef] [PubMed]
- Bessaguet, F.; Danigo, A.; Bouchenaki, H.; Duchesne, M.; Magy, L.; Richard, L.; Sturtz, F.; Desmoulière, A.; Demiot, C. Neuroprotective effect of angiotensin II type 2 receptor stimulation in vincristine-induced mechanical allodynia. Pain 2018, 159, 2538–2546. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Muralidharan, A. Targeting angiotensin II type 2 receptor pathways to treat neuropathic pain and inflammatory pain. Expert Opin. Ther. Targets 2015, 19, 25–35. [Google Scholar] [CrossRef]
- Bhat, S.A.; Sood, A.; Shukla, R.; Hanif, K. AT2R Activation Prevents Microglia Pro-inflammatory Activation in a NOX-Dependent Manner: Inhibition of PKC Activation and p47(phox) Phosphorylation by PP2A. Mol. Neurobiol. 2019, 56, 3005–3023. [Google Scholar] [CrossRef]
- Georgieva, D.; Georgiev, V. The role of angiotensin II and of its receptor subtypes in the acetic acid-induced abdominal constriction test. Pharmacol. Biochem. Behav. 1999, 62, 229–232. [Google Scholar] [CrossRef]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef]
- Cutolo, M.; Campitiello, R.; Gotelli, E.; Soldano, S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Front. Immunol. 2022, 13, 867260. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef]
- Matavelli, L.C.; Huang, J.; Siragy, H.M. Angiotensin AT2 Receptor Stimulation Inhibits Early Renal Inflammation in Renovascular Hypertension. Hypertension 2011, 57, 308–313. [Google Scholar] [CrossRef]
- Studer, R.; Jaffurs, D.; Stefanovic-Racic, M.; Robbins, P.D.; Evans, C.H. Nitric oxide in osteoarthritis. Osteoarthr. Cartil. 1999, 7, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.-C.; Lai, C.-S.; Chang, C.-H.; Yen, J.-H.; Huang, S.-W.; Feng, C.-H.; Chen, Y.-W.; Li, Z.-Y. Nitric oxide: Is it the culprit for the continued expansion of keloids? Eur. J. Pharmacol. 2019, 854, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Ager, E.I.; Neo, J.; Christophi, C. Regulation of colorectal cancer cell epithelial to mesenchymal transition by the renin angiotensin system. J. Gastroenterol. Hepatol. 2016, 31, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Schwentker, A.; Billiar, T.R. Nitric oxide and wound repair. Surg. Clin. N. Am. 2003, 83, 521–530. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Supplier | Location |
|---|---|---|
| Arg1 | Bio-Rad Laboratories | Hercules, CA, USA |
| Ccl2 | Bio-Rad Laboratories | Hercules, CA, USA |
| Col1a1 | Invitrogen (Thermo Fisher) | Waltham, MA, USA |
| Col3a1 | Invitrogen (Thermo Fisher) | Waltham, MA, USA |
| Col6a1 | Bio-Rad Laboratories | Hercules, CA, USA |
| Cxcl1 | Qiagen | Hilden, Germany |
| Cxcl12 | Qiagen | Hilden, Germany |
| Il10 | Invitrogen (Thermo Fisher) | Waltham, MA, USA |
| Il1β | Invitrogen (Thermo Fisher) | Waltham, MA, USA |
| Il6 | Bio-Rad Laboratories | Hercules, CA, USA |
| iNos | Invitrogen (Thermo Fisher) | Waltham, MA, USA |
| Ms4a2 | Qiagen | Hilden, Germany |
| Ngf | Bio-Rad Laboratories | Hercules, CA, USA |
| Pparγ | Bio-Rad Laboratories | Hercules, CA, USA |
| Tgfβ1 | Qiagen | Hilden, Germany |
| Tpsb2 | Qiagen | Hilden, Germany |
| Vegfa | Invitrogen (Thermo Fisher) | Waltham, MA, USA |
| Hprt | Integrated DNA Technologies | Coralville, IA, USA |
| Tbp | Integrated DNA Technologies | Coralville, IA, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, J.M.; Leong, E.K.; Osborne, N.D.; Marshall, J.S.; Bezuhly, M. AT2R Activation Improves Wound Healing in a Preclinical Mouse Model. Biomedicines 2024, 12, 1238. https://doi.org/10.3390/biomedicines12061238
Harrison JM, Leong EK, Osborne ND, Marshall JS, Bezuhly M. AT2R Activation Improves Wound Healing in a Preclinical Mouse Model. Biomedicines. 2024; 12(6):1238. https://doi.org/10.3390/biomedicines12061238
Chicago/Turabian StyleHarrison, Julia M., Edwin K. Leong, Natasha D. Osborne, Jean S. Marshall, and Michael Bezuhly. 2024. "AT2R Activation Improves Wound Healing in a Preclinical Mouse Model" Biomedicines 12, no. 6: 1238. https://doi.org/10.3390/biomedicines12061238
APA StyleHarrison, J. M., Leong, E. K., Osborne, N. D., Marshall, J. S., & Bezuhly, M. (2024). AT2R Activation Improves Wound Healing in a Preclinical Mouse Model. Biomedicines, 12(6), 1238. https://doi.org/10.3390/biomedicines12061238