
Learning to Train and to Explain a Deep Survival Model with Large-Scale Ovarian Cancer Transcriptomic Data

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
2.1.1. TCGA Gene Expression and Clinical Data
2.1.2. In-House Validation Dataset ICO-OV
2.1.3. External Validation Dataset JGOG-OV
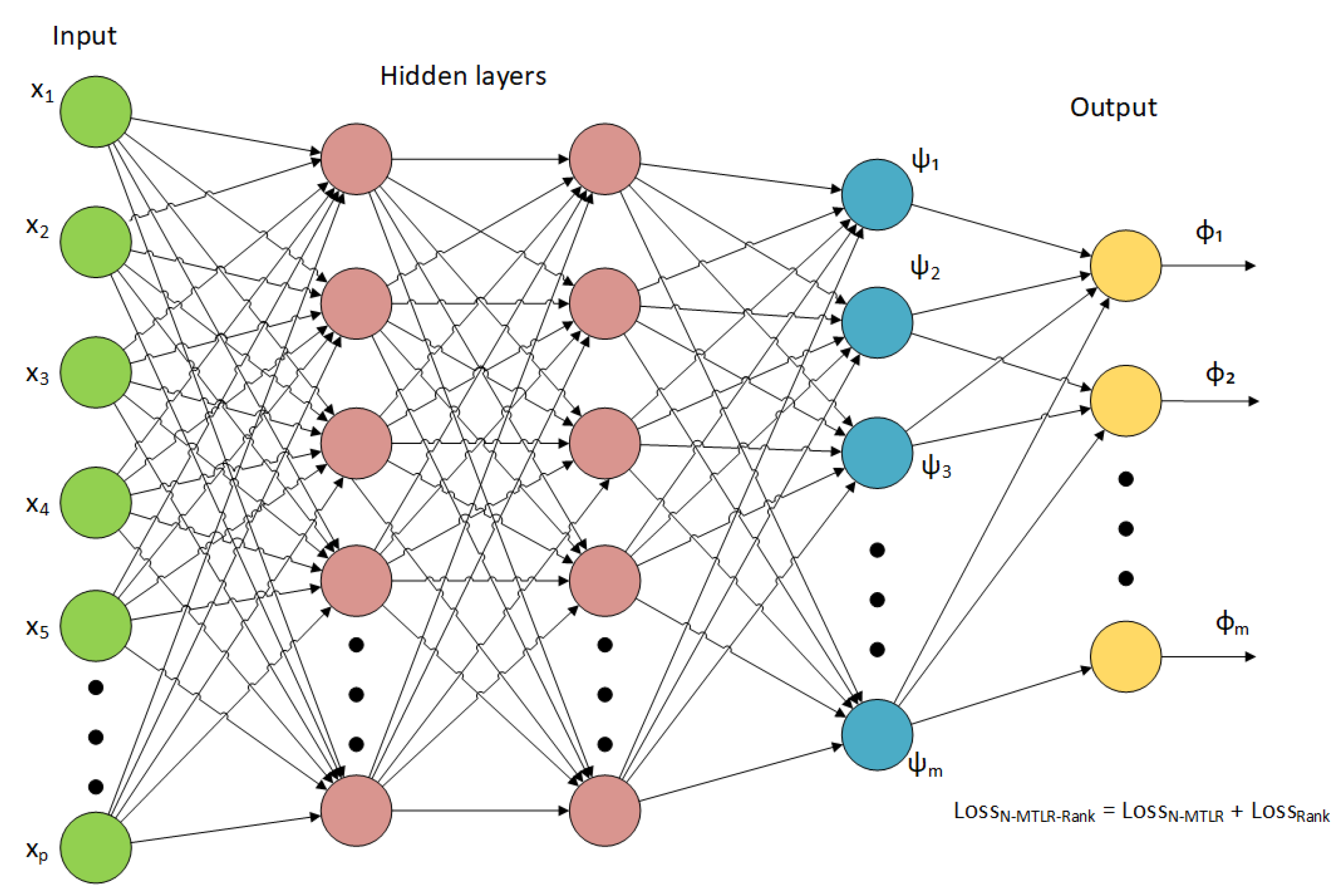
2.2. Proposed Deep Survival Model
2.3. Model Training and Validation
- Number of layers: 1–8.
- Layer width: 8–2048.
- Learning rate for Adam optimizer [37]: 0.00001–0.1.
- Weight decay [38]: 0–0.9.
- Dropout rate [39]: 0–0.6.
- Discretization scheme: equidistant or Kaplan–Meier quantiles [15].
- Interpolation scheme: constant density interpolation (CDI) or constant hazard interpolation (CHI) [15].
- , ranking loss parameter in (4): 0–1.
- , ranking loss parameter in (4): 0.1–100.
2.4. Model Selection and Interpretation
3. Results
3.1. Training and Comparing Deep Survival Networks
3.2. Validating with ICO-OV Dataset
3.3. Interpreting N-MTLR-Rank with PatternAttribution
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BRCA | Breast cancer gene or Invasive Breast Carcinoma |
| BS | Brier Score |
| CDI | Constant Density Interpolation |
| CESC | Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma |
| CHI | Constant Hazard Interpolation |
| C-index | Concordance Index |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| FF | Fresh Frozen |
| GDC | Genomics Data Commons |
| GSEA | Gene-Set Enrichment Analysis |
| GSVA | Gene-Set Variation Analysis |
| HGSOC | High-Grade Serous Ovarian Carcinoma |
| HRD | Homologous Recombination Deficiency |
| IBS | Integrated Brier Score |
| ICO | Institut de Cancérologie de l’Ouest |
| LSTM | Long Short-Term Memory |
| MSigDB | Molecular Signatures Database |
| MTLR | Multi-Task Logistic Regression |
| N-MTLR | Neural Multi-Task Logistic Regression |
| OV | High-Grade Serous Ovarian Cystadenocarcinoma |
| PARP | Poly(ADP-Ribose) Polymerase |
| PMF | Probability Mass Function |
| TCGA | The Cancer Genome Atlas |
| TCGA-CDR | TCGA Pan-Cancer Clinical Data Resource |
| UCEC | Uterine Corpus Endometrial Carcinoma |
| UCS | Uterine Carcinosarcoma |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomarkers Cancer 2019, 11, 1179299X1986081. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, M.; Scotto, G.; Tuninetti, V.; Giannone, G.; Valabrega, G. The Role of PARP Inhibitors in the Ovarian Cancer Microenvironment: Moving Forward From Synthetic Lethality. Front. Oncol. 2021, 11, 689829. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zhang, F.; Zhong, X.; Wei, J.; Xiao, H.; Tu, R. Immune Subtypes Characterization Identifies Clinical Prognosis, Tumor Microenvironment Infiltration, and Immune Response in Ovarian Cancer. Front. Mol. Biosci. 2022, 9, 801156. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2012, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-nnet: An artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput. Biol. 2018, 14, e1006076. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; He, M. Comprehensive pathway-related genes signature for prognosis and recurrence of ovarian cancer. PeerJ 2020, 8, e10437. [Google Scholar] [CrossRef]
- Faraggi, D.; Simon, R. A neural network model for survival data. Stat. Med. 1995, 14, 73–82. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef]
- Lee, C.; Zame, W.; Yoon, J. DeepHit: A Deep Learning Approach to Survival Analysis with Competing Risks. In Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018; p. 8. [Google Scholar]
- Fotso, S. Deep Neural Networks for Survival Analysis Based on a Multi-Task Framework. arXiv 2018, arXiv:1801.05512. [Google Scholar]
- Gensheimer, M.F.; Narasimhan, B. A Scalable Discrete-Time Survival Model for Neural Networks. arXiv 2018, arXiv:1805.00917. [Google Scholar] [CrossRef] [PubMed]
- Kvamme, H.; Borgan, O.; Scheel, I. Time-to-Event Prediction with Neural Networks and Cox Regression. arXiv 2019, arXiv:1907.00825. [Google Scholar]
- Kvamme, H.; Borgan, O. Continuous and Discrete-Time Survival Prediction with Neural Networks. arXiv 2019, arXiv:1910.06724. [Google Scholar] [CrossRef]
- Yousefi, S.; Amrollahi, F.; Amgad, M.; Dong, C.; Lewis, J.E.; Song, C.; Gutman, D.A.; Halani, S.H.; Velazquez Vega, J.E.; Brat, D.J.; et al. Predicting clinical outcomes from large scale cancer genomic profiles with deep survival models. Sci. Rep. 2017, 7, 11707. [Google Scholar] [CrossRef] [PubMed]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. arXiv 2014, arXiv:1312.6114. [Google Scholar]
- Way, G.P.; Greene, C.S. Evaluating deep variational autoencoders trained on pan-cancer gene expression. arXiv 2017, arXiv:1711.04828. [Google Scholar]
- Way, G.P.; Greene, C.S. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. In Proceedings of the Pacific Symposium on Biocomputing, Waimea, HI, USA, 3–7 January 2018; Volume 23, pp. 80–91. [Google Scholar]
- Kim, S.; Kim, K.; Choe, J.; Lee, I.; Kang, J. Improved survival analysis by learning shared genomic information from pan-cancer data. Bioinformatics 2020, 36, i389–i398. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.J.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Networks. arXiv 2014, arXiv:1406.2661. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. Technical Report. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Meng, X.; Wang, X.; Zhang, X.; Zhang, C.; Zhang, Z.; Zhang, K.; Wang, S. A Novel Attention-Mechanism Based Cox Survival Model by Exploiting Pan-Cancer Empirical Genomic Information. Cells 2022, 11, 1421. [Google Scholar] [CrossRef]
- Menand, E.S.; Jrad, N.; Marion, J.M.; Morel, A.; Chauvet, P. Predicting Clinical Outcomes of Ovarian Cancer Patients: Deep Survival Models and Transfer Learning. In Proceedings of the 31st European Safety and Reliability Conference (ESREL 2021), Angers, France, 19–23 September 2021; pp. 371–375. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Menand, E.S.; Jrad, N.; Marion, J.M.; Morel, A.; Chauvet, P. Gene expression RNA-sequencing survival analysis of high-grade serous ovarian carcinoma: A comparative study. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021; pp. 3414–3419. [Google Scholar] [CrossRef]
- Bergstra, J.; Komer, B.; Eliasmith, C.; Yamins, D.; Cox, D.D. Hyperopt: A Python library for model selection and hyperparameter optimization. Comput. Sci. Discov. 2015, 8, 014008. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, S.; Yoshihara, K.; Baba, T.; Shimada, M.; Yoshida, H.; Kajiyama, H.; Oda, K.; Mandai, M.; Okamoto, A.; Enomoto, T.; et al. Prognostic relevance of HRDness gene expression signature in ovarian high-grade serous carcinoma; JGOG3025-TR2 study. Br. J. Cancer 2023, 128, 1095–1104. [Google Scholar] [CrossRef]
- Takamatsu, S.; Hillman, R.T.; Yoshihara, K.; Baba, T.; Shimada, M.; Yoshida, H.; Kajiyama, H.; Oda, K.; Mandai, M.; Okamoto, A.; et al. Molecular classification of ovarian high-grade serous/endometrioid carcinomas through multi-omics analysis: JGOG3025-TR2 study. Br. J. Cancer 2024, 131, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.N.; Greiner, R.; Lin, H.C.; Baracos, V. Learning Patient-Specific Cancer Survival Distributions as a Sequence of Dependent Regressors. Adv. Neural Inf. Process. Syst. 2011, 24, 10. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2017, arXiv:1412.6980. [Google Scholar]
- Loshchilov, I.; Hutter, F. Decoupled Weight Decay Regularization. arXiv 2019, arXiv:1711.05101. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Nair, V.; Hinton, G.E. Rectified Linear Units Improve Restricted Boltzmann Machines. In Proceedings of the 27th International Conference on Machine Learning (ICML-10), Haifa, Israel, 21–24 June 2010; p. 8. [Google Scholar]
- Klambauer, G.; Unterthiner, T.; Mayr, A.; Hochreiter, S. Self-Normalizing Neural Networks. arXiv 2017, arXiv:1706.02515. [Google Scholar]
- Kindermans, P.J.; Schütt, K.T.; Alber, M.; Müller, K.R.; Erhan, D.; Kim, B.; Dähne, S. Learning how to explain neural networks: PatternNet and PatternAttribution. arXiv 2017, arXiv:1705.05598. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Giunchiglia, E.; Nemchenko, A.; van der Schaar, M. RNN-SURV: A Deep Recurrent Model for Survival Analysis. In Artificial Neural Networks and Machine Learning—ICANN 2018; Kůrková, V., Manolopoulos, Y., Hammer, B., Iliadis, L., Maglogiannis, I., Eds.; Series Title: Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2018; Volume 11141, pp. 23–32. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Langdon, S.P.; Herrington, C.S.; Hollis, R.L.; Gourley, C. Estrogen Signaling and Its Potential as a Target for Therapy in Ovarian Cancer. Cancers 2020, 12, 1647. [Google Scholar] [CrossRef]
- van der Ploeg, P.; Uittenboogaard, A.; Thijs, A.M.; Westgeest, H.M.; Boere, I.A.; Lambrechts, S.; van de Stolpe, A.; Bekkers, R.L.; Piek, J.M. The effectiveness of monotherapy with PI3K/AKT/mTOR pathway inhibitors in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2021, 163, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Garsed, D.W.; Pandey, A.; Fereday, S.; Kennedy, C.J.; Takahashi, K.; Alsop, K.; Hamilton, P.T.; Hendley, J.; Chiew, Y.E.; Traficante, N.; et al. The genomic and immune landscape of long-term survivors of high-grade serous ovarian cancer. Nat. Genet. 2022, 54, 1853–1864. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | TCGA-OV | ICO-OV | JGOG-OV |
|---|---|---|---|
| Age at pathologic diagnosis | |||
| Count | 372 | 12 | 274 |
| Mean (SD) | 59.60 (11.38) | 63.17 (12.64) | 61.22 (11.41) |
| Median (IQR) | 59.00 (17.00) | 67.50 (20.50) | 61.50 (15.00) |
| Q1, Q3 | 51.00, 68 | 50.50, 71 | 54.00, 69 |
| Min, Max | 30.00, 87 | 46.00, 86 | 28.00, 89 |
| Missing | 0 | 0 | 0 |
| Clinical stage | |||
| Count (%) | 372 | 12 | 274 |
| Stage IB | 1 (8.33%) | ||
| Stage IC | 1 (0.27%) | ||
| Stage II | 1 (8.33%) | ||
| Stage IIA | 3 (0.81%) | 7 (2.55%) | |
| Stage IIB | 3 (0.81%) | 19 (6.93%) | |
| Stage IIC | 15 (4.03%) | ||
| Stage IIIA | 7 (1.88%) | 2 (16.67%) | 19 (6.93%) |
| Stage IIIB | 13 (3.49%) | 32 (11.68%) | |
| Stage IIIC | 270 (72.58%) | 7 (58.33%) | 138 (50.36%) |
| Stage IV | 57 (15.32%) | 1 (8.33%) | 53 (19.34%) |
| Missing | 3 (0.81%) | 0 | 6 (2.19%) |
| Histological grade | |||
| Count (%) | 372 | 12 | 274 |
| G1 | 1 (0.27%) | ||
| G2 | 42 (11.29%) | ||
| G3 | 319 (85.75%) | 12 (100%) | 274 (100%) |
| G4 | 1 (0.27%) | ||
| GB | 2 (0.54%) | ||
| GX | 5 (1.34%) | ||
| Missing | 2 (0.54%) | 0 | 0 |
| OS | |||
| Count (%) | 372 | 12 | 274 |
| 0 | 143 (38.44%) | 7 (58.33%) | 210 (76.64%) |
| 1 | 229 (61.56%) | 5 (41.67%) | 64 (23.36%) |
| Missing | 0 | 0 | 0 |
| OS.time | |||
| Count | 372 | 12 | 274 |
| Mean (SD) | 1187.17 (943.74) | 2132.75 (1609.92) | 1021.62 (321.42) |
| Median (IQR) | 1024.00 (1141.75) | 1677.50 (2786.50) | 1072.00 (297.00) |
| Q1, Q3 | 517.25, 1659 | 954.00, 3740.5 | 932.25, 1229.25 |
| Min, Max | 8.00, 5481 | 8.00, 4646 | 56.00, 1639 |
| Missing | 0 | 0 | 0 |
| Collection | TCGA-OV, 1 y. | TCGA-OV, 5 y. |
|---|---|---|
| HALLMARK_ALLOGRAFT_REJECTION | 0.54% | 25.81% |
| HALLMARK_E2F_TARGETS | - | 13.98% |
| HALLMARK_ESTROGEN_RESPONSE_EARLY | 0.81% | 6.72% |
| HALLMARK_ESTROGEN_RESPONSE_LATE | - | 2.42% |
| HALLMARK_G2M_CHECKPOINT | - | 8.33% |
| HALLMARK_HYPOXIA | 0.27% | 1.88% |
| HALLMARK_IL2_STAT5_SIGNALING | 2.96% | 20.97% |
| HALLMARK_MTORC1_SIGNALING | 0.54% | 5.91% |
| HALLMARK_MYC_TARGETS_V1 | - | 3.23% |
| HALLMARK_PANCREAS_BETA_CELLS | 1.34% | 1.61% |
| HALLMARK_UNFOLDED_PROTEIN_RESPONSE | - | 0.54% |
| HALLMARK_WNT_BETA_CATENIN_SIGNALING | - | 0.27% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spirina Menand, E.; De Vries-Brilland, M.; Tessier, L.; Dauvé, J.; Campone, M.; Verrièle, V.; Jrad, N.; Marion, J.-M.; Chauvet, P.; Passot, C.; et al. Learning to Train and to Explain a Deep Survival Model with Large-Scale Ovarian Cancer Transcriptomic Data. Biomedicines 2024, 12, 2881. https://doi.org/10.3390/biomedicines12122881
Spirina Menand E, De Vries-Brilland M, Tessier L, Dauvé J, Campone M, Verrièle V, Jrad N, Marion J-M, Chauvet P, Passot C, et al. Learning to Train and to Explain a Deep Survival Model with Large-Scale Ovarian Cancer Transcriptomic Data. Biomedicines. 2024; 12(12):2881. https://doi.org/10.3390/biomedicines12122881
Chicago/Turabian StyleSpirina Menand, Elena, Manon De Vries-Brilland, Leslie Tessier, Jonathan Dauvé, Mario Campone, Véronique Verrièle, Nisrine Jrad, Jean-Marie Marion, Pierre Chauvet, Christophe Passot, and et al. 2024. "Learning to Train and to Explain a Deep Survival Model with Large-Scale Ovarian Cancer Transcriptomic Data" Biomedicines 12, no. 12: 2881. https://doi.org/10.3390/biomedicines12122881
APA StyleSpirina Menand, E., De Vries-Brilland, M., Tessier, L., Dauvé, J., Campone, M., Verrièle, V., Jrad, N., Marion, J.-M., Chauvet, P., Passot, C., & Morel, A. (2024). Learning to Train and to Explain a Deep Survival Model with Large-Scale Ovarian Cancer Transcriptomic Data. Biomedicines, 12(12), 2881. https://doi.org/10.3390/biomedicines12122881