
Peptide-Based Inhibitors of Protein–Protein Interactions (PPIs): A Case Study on the Interaction Between SARS-CoV-2 Spike Protein and Human Angiotensin-Converting Enzyme 2 (hACE2)


Abstract
1. Introduction
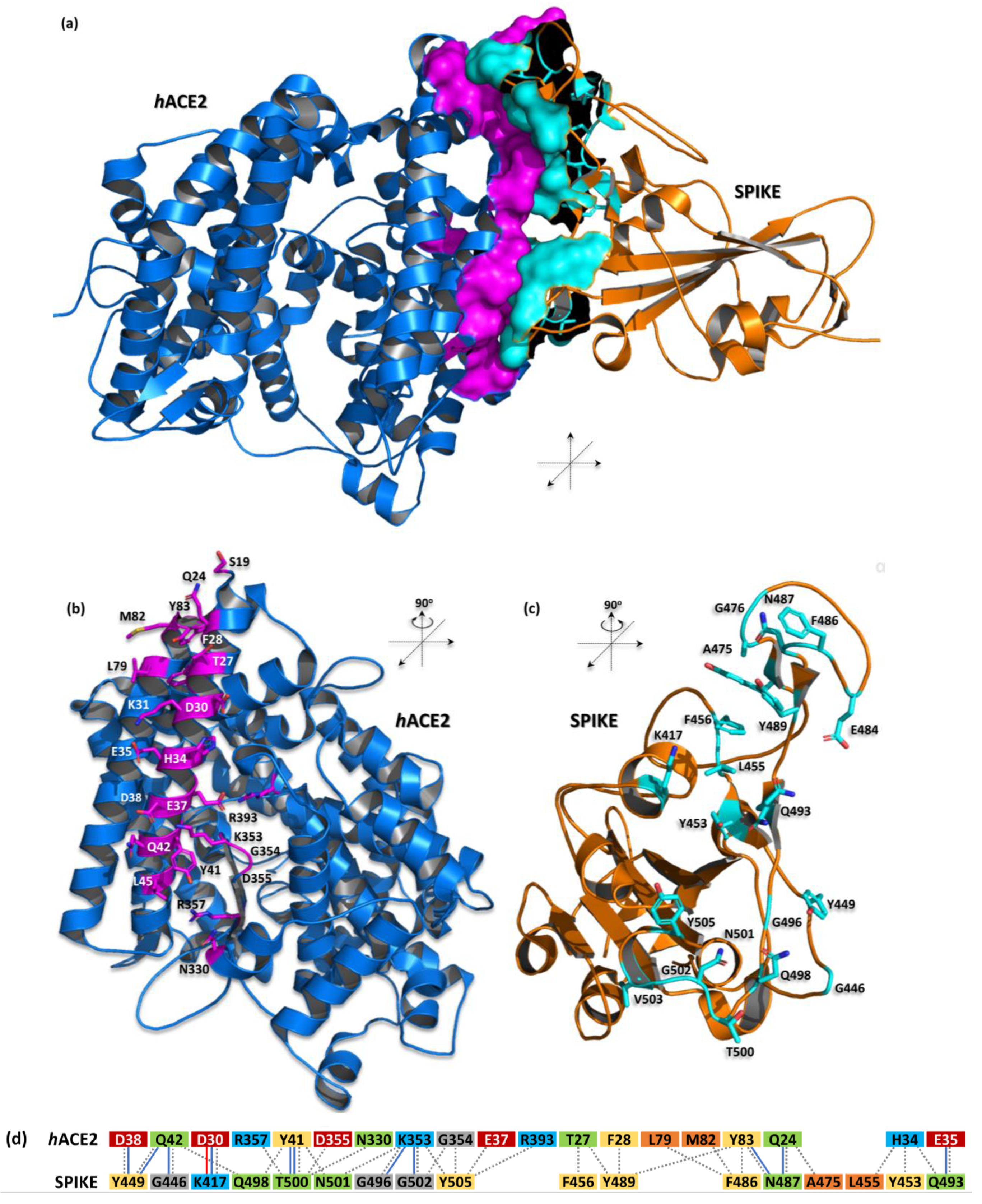
2. Complex Structure of hACE and Spike Proteins
3. Native Peptides
4. Mutated Peptides
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorganic Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Cabri, W.; Cantelmi, P.; Corbisiero, D.; Fantoni, T.; Ferrazzano, L.; Martelli, G.; Mattellone, A.; Tolomelli, A. Therapeutic Peptides Targeting PPI in Clinical Development: Overview, Mechanism of Action and Perspectives. Front. Mol. Biosci. 2021, 8, 697586. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A Global Review on Short Peptides: Frontiers and Perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Guarracino, D.A.; Iannaccone, J.; Cabrera, A.; Kancharla, S. Harnessing the Therapeutic Potential and Biological Activity of Antiviral Peptides. ChemBioChem 2022, 23, e202200415. [Google Scholar] [CrossRef]
- Kahan, R.; Worm, D.J.; de Castro, G.V.; Ng, S.; Barnard, A. Modulators of Protein–Protein Interactions as Antimicrobial Agents. RSC Chem. Biol. 2021, 2, 387–409. [Google Scholar] [CrossRef]
- Poluri, K.M.; Gulati, K.; Tripathi, D.K.; Nagar, N. Protein-Protein Interactions in Host–Pathogen Interactions. In Protein-Protein Interactions: Pathophysiological and Therapeutic Aspects: Volume II; Poluri, K.M., Gulati, K., Tripathi, D.K., Nagar, N., Eds.; Springer Nature: Singapore, 2023; pp. 207–264. ISBN 978-981-9924-23-3. [Google Scholar]
- de Groot, N.S.; Burgas, M.T. Bacteria Use Structural Imperfect Mimicry to Hijack the Host Interactome. PLoS Comput. Biol. 2020, 16, e1008395. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Yao, S.; Ge, H.; Zhu, Y.; Chen, K.; Chen, W.; Zhang, Y.; Zhu, W.; Wang, H.; et al. Discovery of Potential Small Molecular SARS-CoV-2 Entry Blockers Targeting the Spike Protein. Acta Pharmacol. Sin. 2022, 43, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, Y.; Cardoze, S.M.; Obineche, O.W.; Melo, C.; Persaud, A.; Fernández Romero, J.A. Small Molecules Targeting SARS-CoV-2 Spike Glycoprotein Receptor-Binding Domain. ACS Omega 2022, 7, 28779–28789. [Google Scholar] [CrossRef]
- Lau, E.Y.; Negrete, O.A.; Bennett, W.F.D.; Bennion, B.J.; Borucki, M.; Bourguet, F.; Epstein, A.; Franco, M.; Harmon, B.; He, S.; et al. Discovery of Small-Molecule Inhibitors of SARS-CoV-2 Proteins Using a Computational and Experimental Pipeline. Front. Mol. Biosci. 2021, 8, 678701. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; pp. 1–23. ISBN 978-1-4939-2438-7. [Google Scholar]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus Infections and Immune Responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular Interaction and Inhibition of SARS-CoV-2 Binding to the ACE2 Receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the Novel Coronavirus from the Ongoing Wuhan Outbreak and Modeling of Its Spike Protein for Risk of Human Transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bachiller, M.I.; Brzozowska, I.; Odolczyk, N.; Zielenkiewicz, U.; Zielenkiewicz, P.; Rademann, J. Mapping Protein–Protein Interactions of the Resistance-Related Bacterial Zeta Toxin–Epsilon Antitoxin Complex (Ε2ζ2) with High Affinity Peptide Ligands Using Fluorescence Polarization. Toxins 2016, 8, 222. [Google Scholar] [CrossRef]
- Odolczyk, N.; Marzec, E.; Winiewska-Szajewska, M.; Poznański, J.; Zielenkiewicz, P. Native Structure-Based Peptides as Potential Protein-Protein Interaction Inhibitors of SARS-CoV-2 Spike Protein and Human ACE2 Receptor. Molecules 2021, 26, 2157. [Google Scholar] [CrossRef]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef]
- Rezaei Araghi, R.; Keating, A.E. Designing Helical Peptide Inhibitors of Protein–Protein Interactions. Curr. Opin. Struct. Biol. 2016, 39, 27–38. [Google Scholar] [CrossRef]
- Ho, T.-Y.; Wu, S.-L.; Chen, J.-C.; Wei, Y.-C.; Cheng, S.-E.; Chang, Y.-H.; Liu, H.-J.; Hsiang, C.-Y. Design and Biological Activities of Novel Inhibitory Peptides for SARS-CoV Spike Protein and Angiotensin-Converting Enzyme 2 Interaction. Antivir. Res. 2006, 69, 70–76. [Google Scholar] [CrossRef]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef]
- Ling, R.; Dai, Y.; Huang, B.; Huang, W.; Yu, J.; Lu, X.; Jiang, Y. In Silico Design of Antiviral Peptides Targeting the Spike Protein of SARS-CoV-2. Peptides 2020, 130, 170328. [Google Scholar] [CrossRef]
- Larue, R.C.; Xing, E.; Kenney, A.D.; Zhang, Y.; Tuazon, J.A.; Li, J.; Yount, J.S.; Li, P.-K.; Sharma, A. Rationally Designed ACE2-Derived Peptides Inhibit SARS-CoV-2. Bioconjugate Chem. 2021, 32, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemBioChem 2020, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Pei, P.; Qin, H.; Chen, J.; Wang, F.; He, C.; He, S.; Hong, B.; Liu, K.; Qiao, R.; Fan, H.; et al. Computational Design of Ultrashort Peptide Inhibitors of the Receptor-Binding Domain of the SARS-CoV-2 S Protein. Brief. Bioinform. 2021, 22, bbab243. [Google Scholar] [CrossRef]
- Abouhajar, F.; Chaudhuri, R.; Valiulis, S.N.; Stuart, D.D.; Malinick, A.S.; Xue, M.; Cheng, Q. Label-Free Analysis of Binding and Inhibition of SARS-CoV-19 Spike Proteins to ACE2 Receptor with ACE2-Derived Peptides by Surface Plasmon Resonance. ACS Appl. Bio Mater. 2022, 6, 182–190. [Google Scholar] [CrossRef]
- Chitsike, L.; Krstenansky, J.; Duerksen-Hughes, P.J. ACE2 : S1 RBD Interaction-Targeted Peptides and Small Molecules as Potential COVID-19 Therapeutics. Adv. Pharmacol. Pharm. Sci. 2021, 2021, 1828792. [Google Scholar] [CrossRef]
- Karoyan, P.; Vieillard, V.; Gómez-Morales, L.; Odile, E.; Guihot, A.; Luyt, C.-E.; Denis, A.; Grondin, P.; Lequin, O. Human ACE2 Peptide-Mimics Block SARS-CoV-2 Pulmonary Cells Infection. Commun. Biol. 2021, 4, 197. [Google Scholar] [CrossRef]
- Kolaskar, A.S.; Tongaonkar, P.C. A Semi-Empirical Method for Prediction of Antigenic Determinants on Protein Antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering Human ACE2 to Optimize Binding to the Spike Protein of SARS Coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef]
- Chopra, A.; Shukri, A.H.; Adhikary, H.; Lukinović, V.; Hoekstra, M.; Cowpland, M.; Biggar, K.K. A Peptide Array Pipeline for the Development of Spike-ACE2 Interaction Inhibitors. Peptides 2022, 158, 170898. [Google Scholar] [CrossRef]
- Rajpoot, S.; Ohishi, T.; Kumar, A.; Pan, Q.; Banerjee, S.; Zhang, K.Y.J.; Baig, M.S. A Novel Therapeutic Peptide Blocks SARS-CoV-2 Spike Protein Binding with Host Cell ACE2 Receptor. Drugs R D 2021, 21, 273–283. [Google Scholar] [CrossRef]
- Odolczyk, N.; Klim, J.; Podsiadła-Białoskórska, M.; Winiewska-Szajewska, M.; Szolajska, E.; Zielenkiewicz, U.; Poznański, J.; Zielenkiewicz, P. Improvement of Native Structure-Based Peptides as Efficient Inhibitors of Protein-Protein Interactions of SARS-CoV-2 Spike Protein and Human ACE2. Front. Mol. Biosci. 2022, 9, 983014. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A. Clinical Trials of Repurposed Antivirals for SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Spinner, C.D.; Gottlieb, R.L.; Criner, G.J.; Arribas López, J.R.; Cattelan, A.M.; Soriano Viladomiu, A.; Ogbuagu, O.; Malhotra, P.; Mullane, K.M.; Castagna, A.; et al. Effect of Remdesivir vs Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Ahmad, K.; Saeed, M.; Alharbi, A.M.; Barreto, G.E.; Ashraf, G.M.; Choi, I. Peptide Based Therapeutics and Their Use for the Treatment of Neurodegenerative and Other Diseases. Biomed. Pharmacother. 2018, 103, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Heydari, H.; Golmohammadi, R.; Mirnejad, R.; Tebyanian, H.; Fasihi-Ramandi, M.; Moosazadeh Moghaddam, M. Antiviral Peptides against Coronaviridae Family: A Review. Peptides 2021, 139, 170526. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, R.; Sharma, S.; Thakur, A.; Liou, J.-P. Beyond the Vaccines: A Glance at the Small Molecule and Peptide-Based Anti-COVID19 Arsenal. J. Biomed. Sci. 2022, 29, 65. [Google Scholar] [CrossRef]
- Dahal, A.; Sonju, J.J.; Kousoulas, K.G.; Jois, S.D. Peptides and Peptidomimetics as Therapeutic Agents for COVID-19. Pept. Sci. 2022, 114, e24245. [Google Scholar] [CrossRef]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and Peptide-Based Inhibitors of SARS-CoV-2 Entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Panchal, D.; Kataria, J.; Patel, K.; Crowe, K.; Pai, V.; Azizogli, A.-R.; Kadian, N.; Sanyal, S.; Roy, A.; Dodd-o, J.; et al. Peptide-Based Inhibitors for SARS-CoV-2 and SARS-CoV. Adv. Ther. 2021, 4, 2100104. [Google Scholar] [CrossRef]
- Shah, J.N.; Guo, G.-Q.; Krishnan, A.; Ramesh, M.; Katari, N.K.; Shahbaaz, M.; Abdellattif, M.H.; Singh, S.K.; Dua, K. Peptides-Based Therapeutics: Emerging Potential Therapeutic Agents for COVID-19. Therapies 2022, 77, 319–328. [Google Scholar] [CrossRef]
- Maas, M.N.; Hintzen, J.C.J.; Löffler, P.M.G.; Mecinović, J. Targeting SARS-CoV-2 Spike Protein by Stapled hACE2 Peptides. Chem. Commun. 2021, 57, 3283–3286. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dawber, R.S.; Zhang, P.; Walko, M.; Wilson, A.J.; Wang, X. Peptide-Based Inhibitors of Protein–Protein Interactions: Biophysical, Structural and Cellular Consequences of Introducing a Constraint. Chem. Sci. 2021, 12, 5977–5993. [Google Scholar] [CrossRef] [PubMed]
- Lucana, M.C.; Arruga, Y.; Petrachi, E.; Roig, A.; Lucchi, R.; Oller-Salvia, B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics 2021, 13, 2065. [Google Scholar] [CrossRef] [PubMed]
- Rossino, G.; Marchese, E.; Galli, G.; Verde, F.; Finizio, M.; Serra, M.; Linciano, P.; Collina, S. Peptides as Therapeutic Agents: Challenges and Opportunities in the Green Transition Era. Molecules 2023, 28, 7165. [Google Scholar] [CrossRef]
- Lee, M.F.; Poh, C.L. Strategies to Improve the Physicochemical Properties of Peptide-Based Drugs. Pharm. Res. 2023, 40, 617–632. [Google Scholar] [CrossRef]
- de Vries, R.D.; Schmitz, K.S.; Bovier, F.T.; Predella, C.; Khao, J.; Noack, D.; Haagmans, B.L.; Herfst, S.; Stearns, K.N.; Drew-Bear, J.; et al. Intranasal Fusion Inhibitory Lipopeptide Prevents Direct-Contact SARS-CoV-2 Transmission in Ferrets. Science 2021, 371, 1379–1382. [Google Scholar] [CrossRef]
- Oliveira, E.H.; Monteleone-Cassiano, A.C.; Tavares, L.; Santos, J.C.; Lima, T.M.; Gomes, G.F.; Tanaka, P.P.; Monteiro, C.J.; Munuera, M.; Batah, S.S.; et al. A Mimetic Peptide of ACE2 Protects against SARS-CoV-2 Infection and Decreases Pulmonary Inflammation Related to COVID-19. Antivir. Res. 2024, 229, 105968. [Google Scholar] [CrossRef]
- Fan, H.; He, S.; Han, P.; Hong, B.; Liu, K.; Li, M.; Wang, S.; Tong, Y. Cepharanthine: A Promising Old Drug against SARS-CoV-2. Adv. Biol. 2022, 6, 2200148. [Google Scholar] [CrossRef]
- Hijikata, A.; Shionyu-Mitsuyama, C.; Nakae, S.; Shionyu, M.; Ota, M.; Kanaya, S.; Hirokawa, T.; Nakajima, S.; Watashi, K.; Shirai, T. Evaluating Cepharanthine Analogues as Natural Drugs against SARS-CoV-2. FEBS Open Bio 2022, 12, 285–294. [Google Scholar] [CrossRef]
- Williams, B.A.; Jones, C.H.; Welch, V.; True, J.M. Outlook of Pandemic Preparedness in a Post-COVID-19 World. npj Vaccines 2023, 8, 178. [Google Scholar] [CrossRef]

{kind=link}
| Feature 1 | hACE2 | Spike |
|---|---|---|
| No. of interface residues | 19 | 17 |
| Interface area | 825 | 863 |
| No. of H-bonds | 11 | |
| No. of salt bridges | 1 | |
| No. nonbonded contacts | 106 | |
| hACE2: | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| First Author | Peptide Name | S | T | I | E | E | Q | A | K | T | F | L | D | K | F | N | H | E | A | E | D | L | F | Y | Q | S | S | L | A |
| Larue [33] | SAP1 | ||||||||||||||||||||||||||||
| SAP2 | |||||||||||||||||||||||||||||
| SAP5 | |||||||||||||||||||||||||||||
| SAP6 | |||||||||||||||||||||||||||||
| Pei [35] | SI4α | ||||||||||||||||||||||||||||
| SI5α | |||||||||||||||||||||||||||||
| Abouhajar [36] | [30,31,32,33,34,35,36,37,38,39,40,41,42] | ||||||||||||||||||||||||||||
| [22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44] | |||||||||||||||||||||||||||||
| Chitsike [37] | Pep1 | ||||||||||||||||||||||||||||
| hACE2: | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| First Author | Peptide Name | S | T | I | E | E | Q | A | K | T | F | L | D | K | F | N | H | E | A | E | D | L | F | Y | Q | S | S | L | A | |||
| Karoyan [38] | P1 | (1) | ||||||||||||||||||||||||||||||
| P2 | A | L | L | L | L | L | (1) | |||||||||||||||||||||||||
| P3 | A | L | L | L | L | L | A | (1) | ||||||||||||||||||||||||
| P4 | (2) | A | L | L | L | L | L | A | (1) | |||||||||||||||||||||||
| P5 | A | L | L | L | P | L | L | A | (1) | |||||||||||||||||||||||
| P6 | A | L | L | L | L | L | L | (1) | ||||||||||||||||||||||||
| P7 | A | L | Y | L | L | L | L | (1) | ||||||||||||||||||||||||
| P8 | A | L | L | M | L | L | A | (1) | ||||||||||||||||||||||||
| P9 | A | L | Y | M | L | L | (1) | |||||||||||||||||||||||||
| P10 | A | L | Y | M | L | L | A | (1) | ||||||||||||||||||||||||
| Chitsike [37] | Pep2 | (3) | ||||||||||||||||||||||||||||||
| Pep3 | V | Y | R | D | ||||||||||||||||||||||||||||
| Pep4 | P | E | L | L | L | L | E | |||||||||||||||||||||||||
| Pep5 | P | E | L | L | CD | L | L | E | ||||||||||||||||||||||||
| Chopra [41] | BP19 | (4) | I | |||||||||||||||||||||||||||||
| Rajpoot [42] | 13AApi | N | F | K | ||||||||||||||||||||||||||||
| Odolczyk [43] | pep1c | G | ||||||||||||||||||||||||||||||
| pep1d | G | |||||||||||||||||||||||||||||||
| pep1e | G | |||||||||||||||||||||||||||||||
| J3 | Y | G | ||||||||||||||||||||||||||||||
| Peptide Name | Modified * | Verification Method | IC50 ** | Binding Affinity (KD) ** | Ref. |
|---|---|---|---|---|---|
| SAP1 | LA (IC50); affinity precipitation assays (KD) | 2.39 ± 0.20 mM | 0.53 ± 0.01 mM | [33] | |
| SAP2 | 3.72 ± 0.37 mM | 10.7 ± 4.2 mM | |||
| SAP4 | >7.5 mM | -n/d | |||
| SAP5 | >7.5 mM | n/d | |||
| SI5α | ELISA | (EC50) 1.59 μM | n/d | [35] | |
| [30,31,32,33,34,35,36,37,38,39,40,41,42] | SPR | 0.65 μg/mL | n/d | [36] | |
| [22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44] | 2.00 μg/mL | n/d | |||
| P8 | ELISA (IC50); BLI (KD) | 46 nM | 24 ± 11 nM | [38] | |
| P9 | 53 nM | 0.09 ± 0.08 nM | |||
| P10 | 42 nM | 0.03 ± 0.01 nM | |||
| Pep1 | AlphaScreen™ assay | 80 μM | n/d | [37] | |
| Pep2 | Y | 113 μM | n/d | ||
| Pep3 | Y | 72 μM | n/d | ||
| Pep4 | Y | 42 μM | n/d | ||
| Pep5 | Y | 363 μM | n/d | ||
| BP19 | Y | ELISA | 2.08 ± 0.38 μM | n/d | [41] |
| 13AApi | Y | ELISA | >100 μM | n/d | [42] |
| pep1c | Y | ELISA (IC50); MST (KD) | n/d | 280 ± 60 nM | [27] |
| pep1d | Y | 3.3 mM | 210 ± 50 nM | ||
| pep1e | Y | n/d | 1900 ± 400 nM | ||
| J3 | Y | ELISA (IC50); MST (KD) | n/d | ~50 nM | [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakhmetullina, A.; Zielenkiewicz, P.; Odolczyk, N. Peptide-Based Inhibitors of Protein–Protein Interactions (PPIs): A Case Study on the Interaction Between SARS-CoV-2 Spike Protein and Human Angiotensin-Converting Enzyme 2 (hACE2). Biomedicines 2024, 12, 2361. https://doi.org/10.3390/biomedicines12102361
Rakhmetullina A, Zielenkiewicz P, Odolczyk N. Peptide-Based Inhibitors of Protein–Protein Interactions (PPIs): A Case Study on the Interaction Between SARS-CoV-2 Spike Protein and Human Angiotensin-Converting Enzyme 2 (hACE2). Biomedicines. 2024; 12(10):2361. https://doi.org/10.3390/biomedicines12102361
Chicago/Turabian StyleRakhmetullina, Aizhan, Piotr Zielenkiewicz, and Norbert Odolczyk. 2024. "Peptide-Based Inhibitors of Protein–Protein Interactions (PPIs): A Case Study on the Interaction Between SARS-CoV-2 Spike Protein and Human Angiotensin-Converting Enzyme 2 (hACE2)" Biomedicines 12, no. 10: 2361. https://doi.org/10.3390/biomedicines12102361
APA StyleRakhmetullina, A., Zielenkiewicz, P., & Odolczyk, N. (2024). Peptide-Based Inhibitors of Protein–Protein Interactions (PPIs): A Case Study on the Interaction Between SARS-CoV-2 Spike Protein and Human Angiotensin-Converting Enzyme 2 (hACE2). Biomedicines, 12(10), 2361. https://doi.org/10.3390/biomedicines12102361