
Utility of Genetic Testing in Patients with Transthyretin Amyloid Cardiomyopathy: A Brief Review

 ,
,
Abstract
:1. Introduction
2. Utility of Family Screening
3. Utility of Genetic Testing in Clinical Characterization and Anticipation of Symptoms
4. Utility of Genetic Testing for the Initiation of a Specific Treatment
5. Utility of Genetic Testing in Prognosis Assessment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and Treatment of Cardiac Amyloidosis. A Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. J. Heart Fail. 2021, 23, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of Transthyretin-Related Hereditary Amyloidosis for Clinicians. Orphanet J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Ando, Y. Recent Advances in Transthyretin Amyloidosis Therapy. Transl. Neurodegener. 2014, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-Type Transthyretin Amyloidosis as a Cause of Heart Failure with Preserved Ejection Fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 2017, 135, 1357–1377. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.-L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile Systemic Amyloidosis Affects 25% of the Very Aged and Associates with Genetic Variation in Alpha2-Macroglobulin and Tau: A Population-Based Autopsy Study. Ann. Med. 2008, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J. Am. Coll. Cardiol. 2016, 68, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- González-López, E.; López-Sainz, Á.; Garcia-Pavia, P. Diagnosis and Treatment of Transthyretin Cardiac Amyloidosis. Progress and Hope. Rev. Esp. Cardiol. (Engl. Ed.) 2017, 70, 991–1004. [Google Scholar] [CrossRef]
- Ohmori, H.; Ando, Y.; Makita, Y.; Onouchi, Y.; Nakajima, T.; Saraiva, M.J.M.; Terazaki, H.; Suhr, O.; Sobue, G.; Nakamura, M.; et al. Common Origin of the Val30Met Mutation Responsible for the Amyloidogenic Transthyretin Type of Familial Amyloidotic Polyneuropathy. J. Med. Genet. 2004, 41, e51. [Google Scholar] [CrossRef]
- Zaros, C.; Genin, E.; Hellman, U.; Saporta, M.A.; Languille, L.; Wadington-Cruz, M.; Suhr, O.; Misrahi, M.; Planté-Bordeneuve, V. On the Origin of the Transthyretin Val30Met Familial Amyloid Polyneuropathy. Ann. Hum. Genet. 2008, 72, 478–484. [Google Scholar] [CrossRef]
- Conceição, I.; González-Duarte, A.; Obici, L.; Schmidt, H.H.-J.; Simoneau, D.; Ong, M.-L.; Amass, L. “Red-Flag” Symptom Clusters in Transthyretin Familial Amyloid Polyneuropathy. J. Peripher. Nerv. Syst. 2016, 21, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Conceição, I.; Damy, T.; Romero, M.; Galán, L.; Attarian, S.; Luigetti, M.; Sadeh, M.; Sarafov, S.; Tournev, I.; Ueda, M. Early Diagnosis of ATTR Amyloidosis through Targeted Follow-up of Identified Carriers of TTR Gene Mutations. Amyloid 2019, 26, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Planté-Bordeneuve, V.; Ferreira, A.; Lalu, T.; Zaros, C.; Lacroix, C.; Adams, D.; Said, G. Diagnostic Pitfalls in Sporadic Transthyretin Familial Amyloid Polyneuropathy (TTR-FAP). Neurology 2007, 69, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Shamloo, A.S.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Europace 2022, 24, 1307–1367. [Google Scholar] [CrossRef]
- Kittleson, M.M.; Maurer, M.S.; Ambardekar, A.V.; Bullock-Palmer, R.P.; Chang, P.P.; Eisen, H.J.; Nair, A.P.; Nativi-Nicolau, J.; Ruberg, F.L.; American Heart Association Heart Failure and Transplantation Committee of the Council on Clinical Cardiology. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement from the American Heart Association. Circulation 2020, 142, e7–e22. [Google Scholar] [CrossRef]
- Obici, L.; Kuks, J.B.; Buades, J.; Adams, D.; Suhr, O.B.; Coelho, T.; Kyriakides, T. European Network for TTR-FAP (ATTReuNET) Recommendations for Presymptomatic Genetic Testing and Management of Individuals at Risk for Hereditary Transthyretin Amyloidosis. Curr. Opin. Neurol. 2016, 29, S27–S35. [Google Scholar] [CrossRef]
- Manganelli, F.; Fabrizi, G.M.; Luigetti, M.; Mandich, P.; Mazzeo, A.; Pareyson, D. Hereditary Transthyretin Amyloidosis Overview. Neurol. Sci. 2022, 43, 595–604. [Google Scholar] [CrossRef]
- Sekijima, Y. Hereditary Transthyretin Amyloidosis. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Di Stefano, V.; Lupica, A.; Alonge, P.; Pignolo, A.; Augello, S.M.; Gentile, F.; Gagliardo, A.; Giglia, F.; Brinch, D.; Cappello, M.; et al. Genetic Screening for Hereditary Transthyretin Amyloidosis with Polyneuropathy in Western Sicily: Two Years of Experience in a Neurological Clinic. Eur. J. Neurol. 2024, 31, e16065. [Google Scholar] [CrossRef]
- Maestro-Benedicto, A.; Vela, P.; de Frutos, F.; Mora, N.; Pomares, A.; Gonzalez-Vioque, E.; Briceño, A.; Cabrera, E.; Cobo-Marcos, M.; Dominguez, F.; et al. Frequency of Hereditary Transthyretin Amyloidosis among Elderly Patients with Transthyretin Cardiomyopathy. Eur. J. Heart Fail. 2022, 24, 2367–2373. [Google Scholar] [CrossRef]
- Gallego Delgado, M.; Gayán Ordás, J.; Eiros, R.; García Berrocal, B.; Sánchez, P.L.; Villacorta, E. Importance of Genetic Study in Elderly Patients with Transthyretin Cardiac Amyloidosis. Med. Clin. 2023, 161, 382–385. [Google Scholar] [CrossRef]
- García-García, E.; González-Romero, G.M.; Martín-Pérez, E.M.; de Zapata Cornejo, E.D.; Escobar-Aguilar, G.; Cárdenas Bonnet, M.F. Real-World Data and Machine Learning to Predict Cardiac Amyloidosis. Int. J. Environ. Res. Public Health 2021, 18, 908. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, V.; Prinzi, F.; Luigetti, M.; Russo, M.; Tozza, S.; Alonge, P.; Romano, A.; Sciarrone, M.A.; Vitali, F.; Mazzeo, A.; et al. Machine Learning for Early Diagnosis of ATTRv Amyloidosis in Non-Endemic Areas: A Multicenter Study from Italy. Brain Sci. 2023, 13, 805. [Google Scholar] [CrossRef] [PubMed]
- Valdrez, K.; Silva, S.; Coelho, T.; Alves, E. Awareness and Motives for Use and Non-Use of Preimplantation Genetic Diagnosis in Familial Amyloid Polyneuropathy Mutation Carriers. Prenat. Diagn. 2014, 34, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y. Transthyretin (ATTR) Amyloidosis: Clinical Spectrum, Molecular Pathogenesis and Disease-Modifying Treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Lemos, C.; Coelho, T.; Alves-Ferreira, M.; Martins-da-Silva, A.; Sequeiros, J.; Mendonça, D.; Sousa, A. Overcoming Artefact: Anticipation in 284 Portuguese Kindreds with Familial Amyloid Polyneuropathy (FAP) ATTRV30M. J. Neurol. Neurosurg. Psychiatry 2014, 85, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Cauquil, C.; Theaudin, M.; Rousseau, A.; Algalarrondo, V.; Slama, M.S. Current and Future Treatment of Amyloid Neuropathies. Expert Rev. Neurother. 2014, 14, 1437–1451. [Google Scholar] [CrossRef] [PubMed]
- Reinés, J.B.; Vera, T.R.; Martín, M.U.; Serra, H.A.; Campins, M.M.C.; Millán, J.M.D.; Lezaun, C.G.; Cruz, M.R. Epidemiology of Transthyretin-Associated Familial Amyloid Polyneuropathy in the Majorcan Area: Son Llàtzer Hospital Descriptive Study. Orphanet J. Rare Dis. 2014, 9, 29. [Google Scholar] [CrossRef]
- Álvarez Rubio, J.; Manovel Sánchez, A.J.; González-Costello, J.; García-Pavía, P.; Limeres Freire, J.; García-Pinilla, J.M.; Zorio Grima, E.; García-Álvarez, A.; Valverde Gómez, M.; Espinosa Castro, M.Á.; et al. Characterization of Hereditary Transthyretin Cardiac Amyloidosis in Spain. Rev. Esp. Cardiol. (Engl. Ed.) 2022, 75, 488–495. [Google Scholar] [CrossRef]
- González-Moreno, J.; Gaya-Barroso, A.; Losada-López, I.; Rodríguez, A.; Bosch-Rovira, T.; Ripoll-Vera, T.; Usón, M.; Figuerola, A.; Descals, C.; Montalà, C.; et al. Val50Met Hereditary Transthyretin Amyloidosis: Not Just a Medical Problem, but a Psychosocial Burden. Orphanet J. Rare Dis. 2021, 16, 266. [Google Scholar] [CrossRef]
- Chandrashekar, P.; Alhuneafat, L.; Mannello, M.; Al-Rashdan, L.; Kim, M.M.; Dungu, J.; Alexander, K.; Masri, A. Prevalence and Outcomes of p.Val142Ile TTR Amyloidosis Cardiomyopathy: A Systematic Review. Circ. Genom. Precis. Med. 2021, 14, e003356. [Google Scholar] [CrossRef]
- de Frutos, F.; Ochoa, J.P.; Gómez-González, C.; Reyes-Leiva, D.; Aróstegui, J.I.; Casasnovas, C.; Barriales-Villa, R.; Sevilla, T.; Gonzalez-Lopez, E.; Ramil, E.; et al. Phenotype and Clinical Outcomes of Glu89Lys Hereditary Transthyretin Amyloidosis: A New Endemic Variant in Spain. Amyloid 2022, 30, 199–207. [Google Scholar] [CrossRef] [PubMed]
- González-Moreno, J.; Losada-López, I.; Cisneros-Barroso, E.; Garcia-Pavia, P.; González-Costello, J.; Muñoz-Beamud, F.; Campistol, J.M.; Fernandez-Torron, R.; Chapman, D.; Amass, L. A Descriptive Analysis of ATTR Amyloidosis in Spain from the Transthyretin Amyloidosis Outcomes Survey. Neurol. Ther. 2021, 10, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, M.; Carpinteiro, A.; Kersting, D.; Jakstaite, A.-M.; Hagenacker, T.; Schlosser, T.-W.; Rischpler, C.; Rassaf, T.; Luedike, P. Rare Variant (p.Ser43Asn) of Familial Transthyretin Amyloidosis Associated with Isolated Cardiac Phenotype: A Case Series with Literature Review. Mol. Genet. Genom. Med. 2021, 9, e1581. [Google Scholar] [CrossRef] [PubMed]
- Swiecicki, P.L.; Zhen, D.B.; Mauermann, M.L.; Kyle, R.A.; Zeldenrust, S.R.; Grogan, M.; Dispenzieri, A.; Gertz, M.A. Hereditary ATTR Amyloidosis: A Single-Institution Experience with 266 Patients. Amyloid 2015, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Puffer, R.C.; Spinner, R.J.; Bi, H.; Sharma, R.; Wang, Y.; Theis, J.D.; McPhail, E.D.; Poterucha, J.J.; Niu, Z.; Klein, C.J. Fatal TTR Amyloidosis with Neuropathy from Domino Liver p.Val71Ala Transplant. Neurol. Genet. 2019, 5, e351. [Google Scholar] [CrossRef]
- Chaves, M.; Bettini, M.; Marciano, S.; Sáez, S.; Cristiano, E.; Rugiero, M. Presentations of transthyretin associated familial amyloid polyneuropathy in Argentina. Medicina 2016, 76, 105–108. [Google Scholar]
- Nakase, T.; Yamashita, T.; Matsuo, Y.; Nomura, T.; Sasada, K.; Masuda, T.; Misumi, Y.; Takamatsu, K.; Oda, S.; Furukawa, Y.; et al. Hereditary ATTR Amyloidosis with Cardiomyopathy Caused by the Novel Variant Transthyretin Y114S (p.Y134S). Intern. Med. 2019, 58, 2695–2698. [Google Scholar] [CrossRef]
- Klaassen, S.H.C.; Lemmink, H.H.; Bijzet, J.; Glaudemans, A.W.J.M.; Bos, R.; Plattel, W.; van den Berg, M.P.; Slart, R.H.J.A.; Nienhuis, H.L.A.; van Veldhuisen, D.J.; et al. Late Onset Cardiomyopathy as Presenting Sign of ATTR A45G Amyloidosis Caused by a Novel TTR Mutation (p.A65G). Cardiovasc. Pathol. 2017, 29, 19–22. [Google Scholar] [CrossRef]
- Damy, T.; Costes, B.; Hagège, A.A.; Donal, E.; Eicher, J.-C.; Slama, M.; Guellich, A.; Rappeneau, S.; Gueffet, J.-P.; Logeart, D.; et al. Prevalence and Clinical Phenotype of Hereditary Transthyretin Amyloid Cardiomyopathy in Patients with Increased Left Ventricular Wall Thickness. Eur. Heart J. 2016, 37, 1826–1834. [Google Scholar] [CrossRef]
- Rodrigues, P.; Dias Frias, A.; Gouveia, P.; Trêpa, M.; Fontes Oliveira, M.; Costa, R.; Reis, H.; Amorim, I.; Palma, P.; Cyrne Carvalho, H.; et al. Radionuclide Imaging in the Diagnosis of Transthyretin Cardiac Amyloidosis: Different Sensitivity in Early-Onset V30M Mutation? JACC Cardiovasc. Imaging 2021, 14, 1072–1074. [Google Scholar] [CrossRef]
- Musumeci, M.B.; Cappelli, F.; Russo, D.; Tini, G.; Canepa, M.; Milandri, A.; Bonfiglioli, R.; Di Bella, G.; My, F.; Luigetti, M.; et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc. Imaging 2020, 13, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Gagliardi, C.; Milandri, A. Analogies and Disparities among Scintigraphic Bone Tracers in the Diagnosis of Cardiac and Non-Cardiac ATTR Amyloidosis. J. Nucl. Cardiol. 2019, 26, 1638–1641. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Elliott, P.; Damy, T.; Nativi-Nicolau, J.; Berk, J.L.; Velazquez, E.J.; Boman, K.; Gundapaneni, B.; Patterson, T.A.; Schwartz, J.H.; et al. Efficacy of Tafamidis in Patients with Hereditary and Wild-Type Transthyretin Amyloid Cardiomyopathy: Further Analyses From ATTR-ACT. JACC Heart Fail. 2021, 9, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Mathew, V.; Wang, A.K. Inotersen: New Promise for the Treatment of Hereditary Transthyretin Amyloidosis. Drug Des. Dev. Ther. 2019, 13, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- González-Duarte, A.; Berk, J.L.; Quan, D.; Mauermann, M.L.; Schmidt, H.H.; Polydefkis, M.; Waddington-Cruz, M.; Ueda, M.; Conceição, I.M.; Kristen, A.V.; et al. Analysis of Autonomic Outcomes in APOLLO, a Phase III Trial of the RNAi Therapeutic Patisiran in Patients with Hereditary Transthyretin-Mediated Amyloidosis. J. Neurol. 2020, 267, 703–712. [Google Scholar] [CrossRef]
- Di Stefano, V.; Fava, A.; Gentile, L.; Guaraldi, P.; Leonardi, L.; Poli, L.; Tagliapietra, M.; Vastola, M.; Fanara, S.; Ferrero, B.; et al. Italian Real-Life Experience of Patients with Hereditary Transthyretin Amyloidosis Treated with Patisiran. Pharmacogenomics Pers. Med. 2022, 15, 499–514. [Google Scholar] [CrossRef]
- Habtemariam, B.A.; Karsten, V.; Attarwala, H.; Goel, V.; Melch, M.; Clausen, V.A.; Garg, P.; Vaishnaw, A.K.; Sweetser, M.T.; Robbie, G.J.; et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-Acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin. Pharmacol. Ther. 2021, 109, 372–382. [Google Scholar] [CrossRef]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Planté-Bordeneuve, V.; Berk, J.L.; González-Duarte, A.; Gillmore, J.D.; Low, S.-C.; Sekijima, Y.; et al. Efficacy and Safety of Vutrisiran for Patients with Hereditary Transthyretin-Mediated Amyloidosis with Polyneuropathy: A Randomized Clinical Trial. Amyloid 2023, 30, 18–26. [Google Scholar] [CrossRef]
- Obici, L.; Ajroud-Driss, S.; Lin, K.-P.; Berk, J.L.; Gillmore, J.D.; Kale, P.; Koike, H.; Danese, D.; Aldinc, E.; Chen, C.; et al. Impact of Vutrisiran on Quality of Life and Physical Function in Patients with Hereditary Transthyretin-Mediated Amyloidosis with Polyneuropathy. Neurol. Ther. 2023, 12, 1759–1775. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Rosenthal, J.L.; Grodin, J.L.; Maurer, M.S.; Grogan, M.; Cheng, R.K. ATTR Amyloidosis: Current and Emerging Management Strategies: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncology 2021, 3, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Dasgupta, N.R.; Rissing, S.M.; Smith, J.; Feigenbaum, H. Safety and Efficacy of a TTR Specific Antisense Oligonucleotide in Patients with Transthyretin Amyloid Cardiomyopathy. Amyloid 2017, 24, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Bakalakos, A.; Rubino, M.; Verrillo, F.; Diana, G.; De Michele, G.; Altobelli, I.; Lioncino, M.; Perna, A.; Falco, L.; et al. Targeted Therapies in Pediatric and Adult Patients With Hypertrophic Heart Disease: From Molecular Pathophysiology to Personalized Medicine. Circ. Heart Fail. 2023, 16, e010687. [Google Scholar] [CrossRef]
- Russo, M.; Gentile, L.; Di Stefano, V.; Di Bella, G.; Minutoli, F.; Toscano, A.; Brighina, F.; Vita, G.; Mazzeo, A. Use of Drugs for ATTRv Amyloidosis in the Real World: How Therapy Is Changing Survival in a Non-Endemic Area. Brain Sci. 2021, 11, 545. [Google Scholar] [CrossRef]
- Falcão de Campos, C.; Conceição, I. Updated Evaluation of the Safety, Efficacy and Tolerability of Tafamidis in the Treatment of Hereditary Transthyretin Amyloid Polyneuropathy. Drug Healthc. Patient Saf. 2023, 15, 51–62. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Damy, T.; Fontana, M.; Hutchinson, M.; Lachmann, H.J.; Martinez-Naharro, A.; Quarta, C.C.; Rezk, T.; Whelan, C.J.; Gonzalez-Lopez, E.; et al. A New Staging System for Cardiac Transthyretin Amyloidosis. Eur. Heart J. 2018, 39, 2799–2806. [Google Scholar] [CrossRef]
- Obi, C.A.; Mostertz, W.C.; Griffin, J.M.; Judge, D.P. ATTR Epidemiology, Genetics, and Prognostic Factors. Methodist Debakey Cardiovasc. J. 2022, 18, 17–26. [Google Scholar] [CrossRef]
- Bézard, M.; Kharoubi, M.; Galat, A.; Poullot, E.; Guendouz, S.; Fanen, P.; Funalot, B.; Moktefi, A.; Lefaucheur, J.-P.; Abulizi, M.; et al. Natural History and Impact of Treatment with Tafamidis on Major Cardiovascular Outcome-Free Survival Time in a Cohort of Patients with Transthyretin Amyloidosis. Eur. J. Heart Fail. 2021, 23, 264–274. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Maurer, M.S.; Judge, D.P.; Zeldenrust, S.; Skinner, M.; Kim, A.Y.; Falk, R.H.; Cheung, K.N.; Patel, A.R.; Pano, A.; et al. Prospective Evaluation of the Morbidity and Mortality of Wild-Type and V122I Mutant Transthyretin Amyloid Cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS). Am. Heart J. 2012, 164, 222–228. [Google Scholar] [CrossRef] [PubMed]

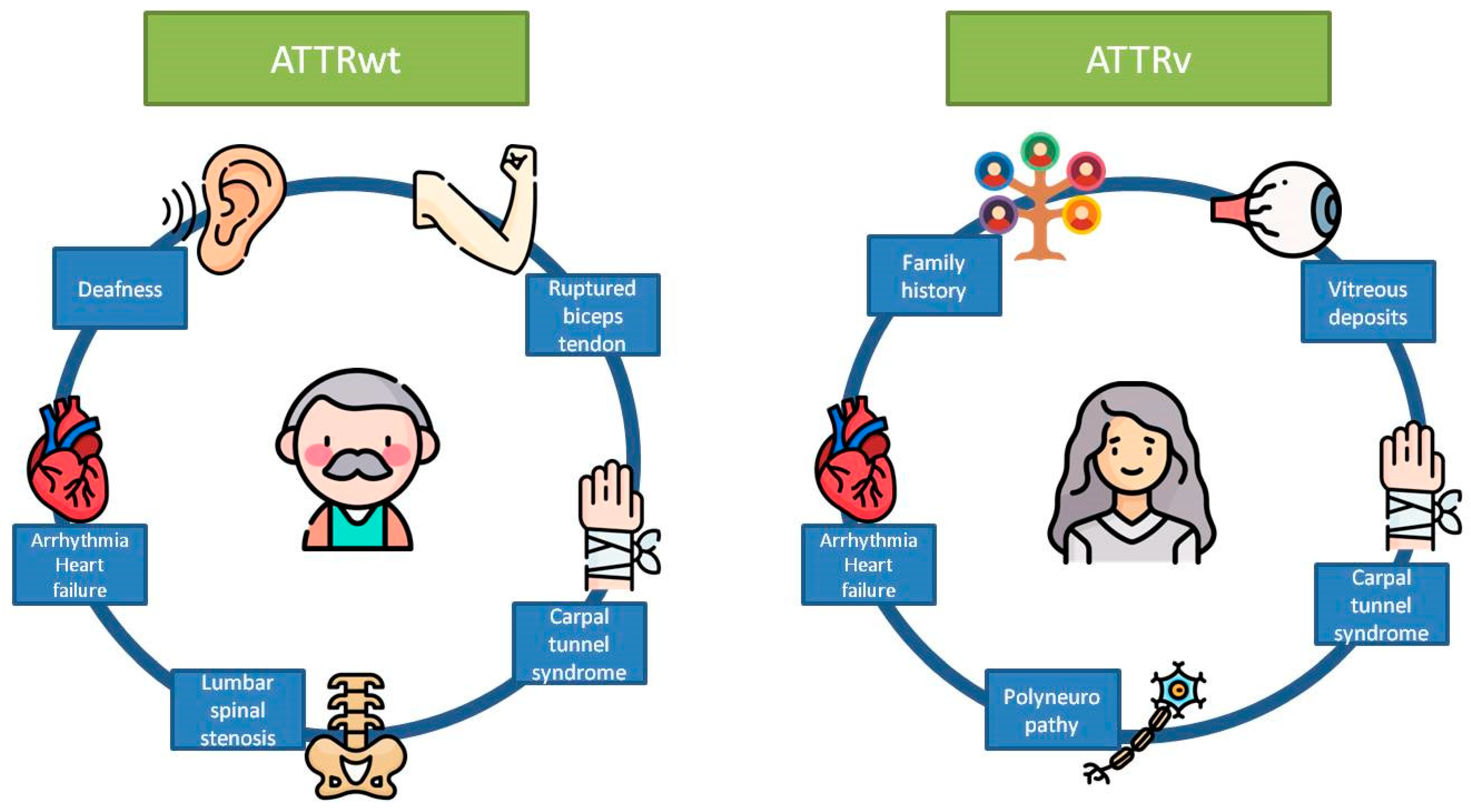

{kind=link}
{kind=link}
| Localization | Type | Red Flag | Amyloidosis Where It Is Most Frequently Found |
|---|---|---|---|
| Extracardiac | Clinical | Ruptured biceps tendon | ATTRwt |
| Carpal tunnel syndrome | ATTRwt and ATTRv | ||
| Deafness | ATTRwt | ||
| Lumbar spinal stenosis | ATTRwt | ||
| Polyneuropathy | ATTRv | ||
| Vitreous deposits | ATTRv | ||
| Family history | ATTRv | ||
| Cardiac | Clinical | Heart failure | ATTRwt and ATTRv |
| Atrial fibrillation | ATTRwt and ATTRv | ||
| ECG | Pseudoinfarct pattern | ATTRwt and ATTRv | |
| Low QRS voltage | ATTRwt and ATTRv | ||
| Laboratory | Disproportionally elevated NT-proBNP and troponin | ATTRwt and ATTRv | |
| Echocardiogram | Granular sparkling of myocardium | ATTRwt and ATTRv | |
| Increased right ventricular wall thickness | ATTRwt and ATTRv | ||
| Increased valve thickness | ATTRwt and ATTRv | ||
| Pericardial effusion | ATTRwt and ATTRv | ||
| Reduced longitudinal strain with apical sparing pattern | ATTRwt and ATTRv | ||
| Cardiac magnetic resonance | Abnormal gadolinium kinetics | ATTRwt and ATTRv | |
| Elevated native T1 values | ATTRwt and ATTRv | ||
| Increased extracellular volume | ATTRwt and ATTRv |
| Variant | Prevalence | Average Age of Onset in Years | Main Organs Involved | Prognosis | Reference |
|---|---|---|---|---|---|
| p.Val50Met | Unknown 1:106 in Japan | <50 | Neurologic (early onset) and neurologic/mixed (late onset) | Median of 28 years in early onset and 10 years in late onset | [29,30] |
| p.Val142Ile | 0.3–1.6% in general population. Among African descent 3–3.5%. | 7–8th decade of life | Cardiac | Median survival 3–5 years | [31] |
| p.Glu109Lys | 50 patients described | 40 | Cardiac and neurologic predominant. Ophthalmologic in advanced. | Median life expectancy 61.6 years | [32] |
| p.Glu109Gln | Unknown | 60 | Neurologic and mixed | Unknown | [33] |
| p.Ser43Asn | Very rare. Unknown. | 40–50 | Cardiac | Before the age of 55 | [34] |
| p.Thr60Ala | Unknown | 63 | Cardiac | Median survival 56.8 months | [35] |
| p.Val71Ala | Unknown | 30–40 | Cardiac and neurologic | Unknown | [36] |
| p.Tyr114Cys | Unknown | 52 | Neurologic | Unknown | [37] |
| p.Tyr134Ser | Unknown | 65 | Mixed | Survival time 10.6 years | [38] |
| p.Ala65Gly | Unknown | 56–78 | Late onset cardiomyopathy | Unknown | [39] |
| p.Val50Met | No p.Val50Met | |
|---|---|---|
| Clinical evaluation | -NIS -BP supine/orthostatic -BMI | -BP supine/orthostatic -BMI |
| Neurophysiology | -NCS -Sudorimetry -HRDB -QST | -Not necessary |
| Biomarkers | -NT-proBNP -Troponin (only if late onset) -Blood sample | -NT-proBNP -Troponin -Blood sample |
| Cardiac evaluation | -Scintigraphy 99mTc-DPD (only if late onset) -Echo -ECG | -Scintigraphy 99mTc-DPD (only if late onset) -Echo -ECG -MRI |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merino-Merino, A.-M.; Labrador-Gomez, J.; Sanchez-Corral, E.; Delgado-Lopez, P.-D.; Perez-Rivera, J.-A. Utility of Genetic Testing in Patients with Transthyretin Amyloid Cardiomyopathy: A Brief Review. Biomedicines 2024, 12, 25. https://doi.org/10.3390/biomedicines12010025
Merino-Merino A-M, Labrador-Gomez J, Sanchez-Corral E, Delgado-Lopez P-D, Perez-Rivera J-A. Utility of Genetic Testing in Patients with Transthyretin Amyloid Cardiomyopathy: A Brief Review. Biomedicines. 2024; 12(1):25. https://doi.org/10.3390/biomedicines12010025
Chicago/Turabian StyleMerino-Merino, Ana-Maria, Jorge Labrador-Gomez, Ester Sanchez-Corral, Pedro-David Delgado-Lopez, and Jose-Angel Perez-Rivera. 2024. "Utility of Genetic Testing in Patients with Transthyretin Amyloid Cardiomyopathy: A Brief Review" Biomedicines 12, no. 1: 25. https://doi.org/10.3390/biomedicines12010025
APA StyleMerino-Merino, A.-M., Labrador-Gomez, J., Sanchez-Corral, E., Delgado-Lopez, P.-D., & Perez-Rivera, J.-A. (2024). Utility of Genetic Testing in Patients with Transthyretin Amyloid Cardiomyopathy: A Brief Review. Biomedicines, 12(1), 25. https://doi.org/10.3390/biomedicines12010025