
The Role of NAD+ in Metabolic Regulation of Adipose Tissue: Implications for Obesity-Induced Insulin Resistance


Abstract
1. Introduction
2. Impairment of the NAD+/SIRT Pathway in the Obese Adipose Organ
2.1. Animal Studies of NAD+/SIRT Pathway
2.2. In Vitro Studies on 3T3-L1 Adipocytes
2.3. Studies in Human Biology
2.4. The Role of NAD+ in Regulating Brown Adipose Tissue
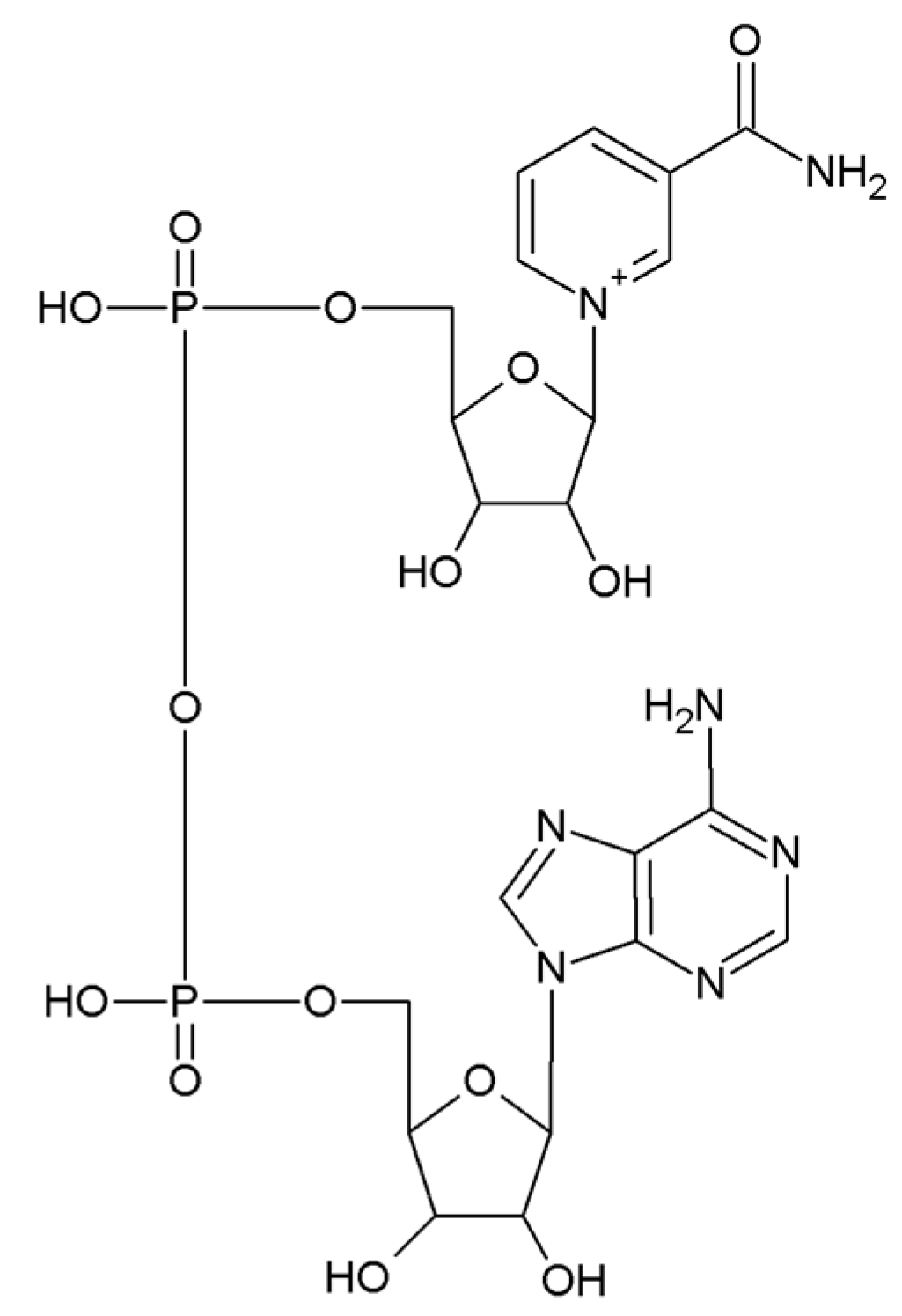
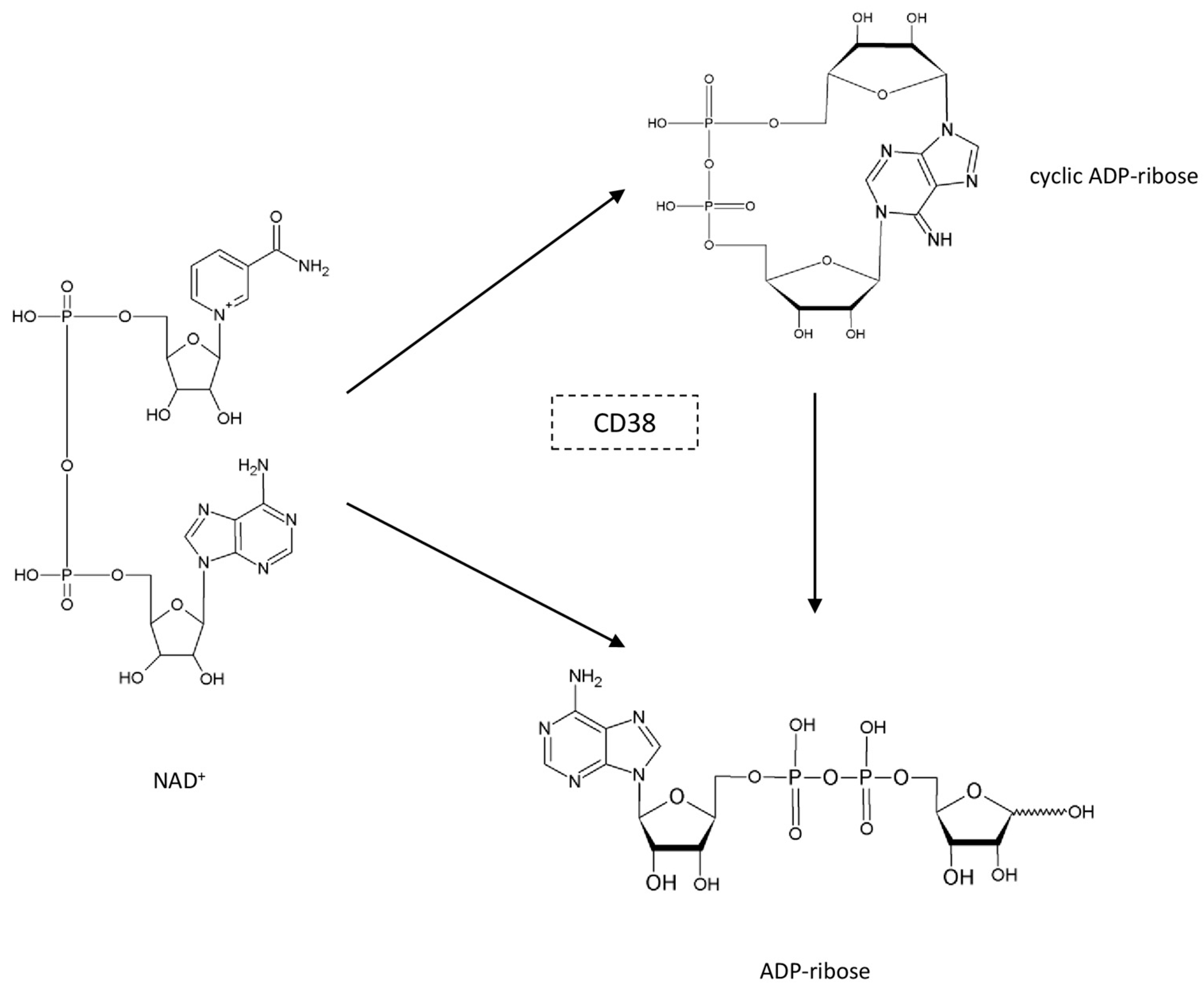
3. The Role of Adipose PARPs and CD38 as NAD+ Consumers
3.1. PARPs in Adipose Tissue
3.2. CD38 in Adipose Tissue
4. Strategies for NAD+ Boosting in Adipose Tissue
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Abdelaal, M.; le Roux, C.W.; Docherty, N.G. Morbidity and mortality associated with obesity. Ann. Transl. Med. 2017, 5, 161. [Google Scholar] [CrossRef] [PubMed]
- Ghanta, R.K.; LaPar, D.J.; Zhang, Q.; Devarkonda, V.; Isbell, J.M.; Yarboro, L.T.; Kern, J.A.; Kron, I.L.; Speir, A.M.; Fonner, C.E.; et al. Obesity Increases Risk-Adjusted Morbidity, Mortality, and Cost Following Cardiac Surgery. J. Am. Heart Assoc. 2017, 6, e003831. [Google Scholar] [CrossRef] [PubMed]
- Tamara, A.; Tahapary, D.L. Obesity as a predictor for a poor prognosis of COVID-19: A systematic review. Diabetes Metab. Syndr. 2020, 14, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Di Pino, A.; DeFronzo, R.A. Insulin Resistance and Atherosclerosis: Implications for Insulin-Sensitizing Agents. Endocr. Rev. 2019, 40, 1447–1467. [Google Scholar] [CrossRef]
- Finck, B.N. Targeting Metabolism, Insulin Resistance, and Diabetes to Treat Nonalcoholic Steatohepatitis. Diabetes 2018, 67, 2485–2493. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Ruskovska, T.; Bernlohr, D.A. Oxidative stress and protein carbonylation in adipose tissue-implications for insulin resistance and diabetes mellitus. J. Proteom. 2013, 92, 323–334. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 2012, 8, 287–296. [Google Scholar] [CrossRef]
- Jukarainen, S.; Heinonen, S.; Rämö, J.T.; Rinnankoski-Tuikka, R.; Rappou, E.; Tummers, M.; Muniandy, M.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; et al. Obesity Is Associated with Low NAD(+)/SIRT Pathway Expression in Adipose Tissue of BMI-Discordant Monozygotic Twins. J. Clin. Endocrinol. Metab. 2016, 101, 275–283. [Google Scholar] [CrossRef]
- Li, P.; Ge, J.; Li, H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 96–115. [Google Scholar] [CrossRef] [PubMed]
- Poulose, N.; Raju, R. Sirtuin regulation in aging and injury. Biochim. Biophys. Acta 2015, 1852, 2442–2455. [Google Scholar] [CrossRef] [PubMed]
- Klar, A.J.; Fogel, S.; Macleod, K. MAR1-a Regulator of the HMa and HMalpha Loci in Saccharomyces cerevisiae. Genetics 1979, 93, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes. Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef]
- Kupis, W.; Pałyga, J.; Tomal, E.; Niewiadomska, E. The role of sirtuins in cellular homeostasis. J. Physiol. Biochem. 2016, 72, 371–380. [Google Scholar] [CrossRef]
- Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, Y.; Wang, S.; Zhang, Y. Sirtuin Deacetylation Mechanism and Catalytic Role of the Dynamic Cofactor Binding Loop. J. Phys. Chem. Lett. 2013, 4, 491–495. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Song, J.; Ke, S.F.; Zhou, C.C.; Zhang, S.L.; Guan, Y.F.; Xu, T.Y.; Sheng, C.Q.; Wang, P.; Miao, C.Y. Nicotinamide phosphoribosyltransferase is required for the calorie restriction-mediated improvements in oxidative stress, mitochondrial biogenesis, and metabolic adaptation. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 44–57. [Google Scholar] [CrossRef]
- Drew, J.E.; Farquharson, A.J.; Horgan, G.W.; Williams, L.M. Tissue-specific regulation of sirtuin and nicotinamide adenine dinucleotide biosynthetic pathways identified in C57Bl/6 mice in response to high-fat feeding. J. Nutr. Biochem. 2016, 37, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Jia, R.; Wang, G.; Hong, S.; Song, L.; Sun, B.; Chen, K.; Wang, N.; Wang, Q.; Luo, X.; et al. Depot-specific regulation of NAD(+)/SIRTs metabolism identified in adipose tissue of mice in response to high-fat diet feeding or calorie restriction. J. Nutr. Biochem. 2020, 80, 108377. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Yoshida, M.; Johnson, S.; Takikawa, A.; Usui, I.; Tobe, K.; Nakagawa, T.; Yoshino, J.; Imai, S. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD+ and Function in Mice. Cell Metab. 2015, 21, 706–717. [Google Scholar] [CrossRef]
- Stromsdorfer, K.L.; Yamaguchi, S.; Yoon, M.J.; Moseley, A.C.; Franczyk, M.P.; Kelly, S.C.; Qi, N.; Imai, S.; Yoshino, J. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep. 2016, 16, 1851–1860. [Google Scholar] [CrossRef]
- Shi, W.; Hegeman, M.A.; van Dartel, D.A.M.; Tang, J.; Suarez, M.; Swarts, H.; van der Hee, B.; Arola, L.; Keijer, J. Effects of a wide range of dietary nicotinamide riboside (NR) concentrations on metabolic flexibility and white adipose tissue (WAT) of mice fed a mildly obesogenic diet. Mol. Nutr. Food Res. 2017, 61, 1600878. [Google Scholar] [CrossRef]
- Gouranton, E.; Romier, B.; Marcotorchino, J.; Tourniaire, F.; Astier, J.; Peiretti, F.; Landrier, J.F. Visfatin is involved in TNFα-mediated insulin resistance via an NAD(+)/Sirt1/PTP1B pathway in 3T3-L1 adipocytes. Adipocyte 2014, 3, 180–189. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Engin, A.B. Adipocyte-Macrophage Cross-Talk in Obesity. Adv. Exp. Med. Biol. 2017, 960, 327–343. [Google Scholar] [CrossRef]
- Hertzel, A.V.; Yong, J.; Chen, X.; Bernlohr, D.A. Immune Modulation of Adipocyte Mitochondrial Metabolism. Endocrinology 2022, 163, bqac094. [Google Scholar] [CrossRef]
- Ruskovska, T.; Bernlohr, D.A. Effects of TNFα, IL6 and IL1β on 3T3-L1 Adipocytes in Culture. Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota-Twin Cities, Minneapolis, MN, USA. 2015; Unpublished work. [Google Scholar]
- Zhang, T.; Liu, J.; Tong, Q.; Lin, L. SIRT3 Acts as a Positive Autophagy Regulator to Promote Lipid Mobilization in Adipocytes via Activating AMPK. Int. J. Mol. Sci. 2020, 21, 372. [Google Scholar] [CrossRef] [PubMed]
- Kranendonk, M.E.; van Herwaarden, J.A.; Stupkova, T.; de Jager, W.; Vink, A.; Moll, F.L.; Kalkhoven, E.; Visseren, F.L. Inflammatory characteristics of distinct abdominal adipose tissue depots relate differently to metabolic risk factors for cardiovascular disease: Distinct fat depots and vascular risk factors. Atherosclerosis 2015, 239, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Heiker, J.T.; Gärtner, D.; Björnson, E.; Schön, M.R.; Flehmig, G.; Klöting, N.; Krohn, K.; Fasshauer, M.; Stumvoll, M.; et al. Extensive weight loss reveals distinct gene expression changes in human subcutaneous and visceral adipose tissue. Sci. Rep. 2015, 5, 14841. [Google Scholar] [CrossRef] [PubMed]
- Gaddipati, R.; Sasikala, M.; Padaki, N.; Mukherjee, R.M.; Sekaran, A.; Jayaraj-Mansard, M.; Rabella, P.; Rao-Guduru, V.; Reddy-Duvvuru, N. Visceral adipose tissue visfatin in nonalcoholic fatty liver disease. Ann. Hepatol. 2010, 9, 266–270. [Google Scholar] [CrossRef]
- Rappou, E.; Jukarainen, S.; Rinnankoski-Tuikka, R.; Kaye, S.; Heinonen, S.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Saunavaara, V.; Rissanen, A.; et al. Weight Loss Is Associated with Increased NAD(+)/SIRT1 Expression but Reduced PARP Activity in White Adipose Tissue. J. Clin. Endocrinol. Metab. 2016, 101, 1263–1273. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef]
- Heeren, J.; Münzberg, H. Novel aspects of brown adipose tissue biology. Endocrinol. Metab. Clin. N. Am. 2013, 42, 89–107. [Google Scholar] [CrossRef][Green Version]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef]
- Tamucci, K.A.; Namwanje, M.; Fan, L.; Qiang, L. The dark side of browning. Protein Cell 2018, 9, 152–163. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Elattar, S.; Satyanarayana, A. Can Brown Fat Win the Battle Against White Fat? J. Cell Physiol. 2015, 230, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Franczyk, M.P.; Chondronikola, M.; Qi, N.; Gunawardana, S.C.; Stromsdorfer, K.L.; Porter, L.C.; Wozniak, D.F.; Sasaki, Y.; Rensing, N.; et al. Adipose tissue NAD(+) biosynthesis is required for regulating adaptive thermogenesis and whole-body energy homeostasis in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 23822–23828. [Google Scholar] [CrossRef] [PubMed]
- Crisol, B.M.; Veiga, C.B.; Lenhare, L.; Braga, R.R.; Silva, V.R.R.; da Silva, A.S.R.; Cintra, D.E.; Moura, L.P.; Pauli, J.R.; Ropelle, E.R. Nicotinamide riboside induces a thermogenic response in lean mice. Life Sci. 2018, 211, 1–7. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Yi, D.; Lin, F.; Viscarra, J.A.; Tabuchi, C.; Ngo, K.; Shin, G.; Lee, A.Y.; Wang, Y.; Sul, H.S. Aifm2, a NADH Oxidase, Supports Robust Glycolysis and Is Required for Cold- and Diet-Induced Thermogenesis. Mol. Cell 2020, 77, 600–617.e604. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Teng, R.; Di, L.; Rogers, H.; Wu, H.; Kopp, J.B.; Noguchi, C.T. PPARα and Sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes 2013, 62, 4122–4131. [Google Scholar] [CrossRef]
- Teng, R.; Gavrilova, O.; Suzuki, N.; Chanturiya, T.; Schimel, D.; Hugendubler, L.; Mammen, S.; Yver, D.R.; Cushman, S.W.; Mueller, E.; et al. Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nat. Commun. 2011, 2, 520. [Google Scholar] [CrossRef]
- Szántó, M.; Bai, P. The role of ADP-ribose metabolism in metabolic regulation, adipose tissue differentiation, and metabolism. Genes. Dev. 2020, 34, 321–340. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Lettieri-Barbato, D.; D’Angelo, F.; Sciarretta, F.; Tatulli, G.; Tortolici, F.; Ciriolo, M.R.; Aquilano, K. Maternal high calorie diet induces mitochondrial dysfunction and senescence phenotype in subcutaneous fat of newborn mice. Oncotarget 2017, 8, 83407–83418. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Aarhus, R. ADP-ribosyl cyclase: An enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef]
- Wei, W.; Graeff, R.; Yue, J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca(2+) signaling pathway. World J. Biol. Chem. 2014, 5, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- McEllistrim, C.; Krawczyk, J.; O’Dwyer, M.E. New developments in the treatment of multiple myeloma-clinical utility of daratumumab. Biologics 2017, 11, 31–43. [Google Scholar] [CrossRef]
- Burgler, S. Role of CD38 Expression in Diagnosis and Pathogenesis of Chronic Lymphocytic Leukemia and Its Potential as Therapeutic Target. Crit. Rev. Immunol. 2015, 35, 417–432. [Google Scholar] [CrossRef]
- Ding, Y.; Gao, H.; Zhang, Q. The biomarkers of leukemia stem cells in acute myeloid leukemia. Stem Cell Investig. 2017, 4, 19. [Google Scholar] [CrossRef]
- Higashida, H.; Yuhi, T.; Akther, S.; Amina, S.; Zhong, J.; Liang, M.; Nishimura, T.; Liu, H.X.; Lopatina, O. Oxytocin release via activation of TRPM2 and CD38 in the hypothalamus during hyperthermia in mice: Implication for autism spectrum disorder. Neurochem. Int. 2018, 119, 42–48. [Google Scholar] [CrossRef]
- Deshpande, D.A.; Guedes, A.G.P.; Lund, F.E.; Subramanian, S.; Walseth, T.F.; Kannan, M.S. CD38 in the pathogenesis of allergic airway disease: Potential therapeutic targets. Pharmacol. Ther. 2017, 172, 116–126. [Google Scholar] [CrossRef]
- Cai, G.; Cole, S.A.; Freeland-Graves, J.H.; MacCluer, J.W.; Blangero, J.; Comuzzie, A.G. Principal component for metabolic syndrome risk maps to chromosome 4p in Mexican Americans: The San Antonio Family Heart Study. Hum. Biol. 2004, 76, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. Faseb J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.H.; Harrington, W.W.; Luo, G.; Milliken, N.O.; Ulrich, J.C.; Chen, J.; Rajpal, D.K.; Qian, Y.; Carpenter, T.; Murray, R.; et al. Genetic Ablation of CD38 Protects against Western Diet-Induced Exercise Intolerance and Metabolic Inflexibility. PLoS ONE 2015, 10, e0134927. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Cheng, R.; Zhou, X.Y.; Zhu, J.G.; Zhu, C.; Qin, D.N.; Kou, C.Z.; Guo, X.R. Gene expression profiles of adipose tissue of high-fat diet-induced obese rats by cDNA microarrays. Mol. Biol. Rep. 2010, 37, 3691–3695. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Miao, L.J.; Wang, X.N.; Huang, C.C.; Qian, Y.S.; Huang, X.; Wang, X.L.; Jin, W.Z.; Ji, G.J.; Fu, M.; et al. CD38 deficiency suppresses adipogenesis and lipogenesis in adipose tissues through activating Sirt1/PPARγ signaling pathway. J. Cell Mol. Med. 2018, 22, 101–110. [Google Scholar] [CrossRef]
- Benzi, A.; Sturla, L.; Heine, M.; Fischer, A.W.; Spinelli, S.; Magnone, M.; Sociali, G.; Parodi, A.; Fenoglio, D.; Emionite, L.; et al. CD38 downregulation modulates NAD(+) and NADP(H) levels in thermogenic adipose tissues. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158819. [Google Scholar] [CrossRef]
- Méndez-Lara, K.A.; Rodríguez-Millán, E.; Sebastián, D.; Blanco-Soto, R.; Camacho, M.; Nan, M.N.; Diarte-Añazco, E.M.G.; Mato, E.; Lope-Piedrafita, S.; Roglans, N.; et al. Nicotinamide Protects Against Diet-Induced Body Weight Gain, Increases Energy Expenditure, and Induces White Adipose Tissue Beiging. Mol. Nutr. Food Res. 2021, 65, e2100111. [Google Scholar] [CrossRef]
- Luo, C.; Yang, C.; Wang, X.; Chen, Y.; Liu, X.; Deng, H. Nicotinamide reprograms adipose cellular metabolism and increases mitochondrial biogenesis to ameliorate obesity. J. Nutr. Biochem. 2022, 107, 109056. [Google Scholar] [CrossRef]
- Nascimento, E.B.M.; Moonen, M.P.B.; Remie, C.M.E.; Gariani, K.; Jörgensen, J.A.; Schaart, G.; Hoeks, J.; Auwerx, J.; van Marken Lichtenbelt, W.D.; Schrauwen, P. Nicotinamide Riboside Enhances In Vitro Beta-adrenergic Brown Adipose Tissue Activity in Humans. J. Clin. Endocrinol. Metab. 2021, 106, 1437–1447. [Google Scholar] [CrossRef]
- Poljšak, B.; Kovač, V.; Milisav, I. Current Uncertainties and Future Challenges Regarding NAD+ Boosting Strategies. Antioxidants 2022, 11, 1637. [Google Scholar] [CrossRef]
- Sabbagh, F.; Kim, B.S. Ex Vivo Transdermal Delivery of Nicotinamide Mononucleotide Using Polyvinyl Alcohol Microneedles. Polymers 2023, 15, 2031. [Google Scholar] [CrossRef] [PubMed]
- Bañales-Luna, M.; Figueroa-Vega, N.; Marín-Aragón, C.I.; Perez-Luque, E.; Ibarra-Reynoso, L.; Gallardo-Blanco, H.L.; López-Aguilar, I.; Malacara, J.M. Associations of nicotidamide-N-methyltransferase, FTO, and IRX3 genetic variants with body mass index and resting energy expenditure in Mexican subjects. Sci. Rep. 2020, 10, 11478. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.R.; Aird, T.P.; Farquharson, A.J.; Horgan, G.W.; Fisher, E.; Wilson, J.; Hopkins, G.E.; Anderson, B.; Ahmad, S.A.; Davis, S.R.; et al. Inter-individual responses to sprint interval training, a pilot study investigating interactions with the sirtuin system. Appl. Physiol. Nutr. Metab. 2018, 43, 84–93. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Type of Adipose Tissue | Major Metabolic Outcome/s |
|---|---|---|
| High-fat diet (HFD) | White adipose tissue (WAT) | Type 2 diabetes Decreased NAD+ levels [19] Obesity Glucose intolerance Altered regulation of the SIRT/NAD+ system [21] |
| Calorie restriction (CR) | Visceral adipose tissue (VAT) | Increased NAD+ Decreased oxidative stress Increased mitogenesis [20] |
| Nnmt knockdown and high-fat diet | White adipose tissue (WAT) | Reduced diet-induced obesity Enhanced insulin sensitivity Increased energy expenditure Increased NAD+ [22] |
| Adipocyte-specific Nampt knockout (ANKO) | Visceral adipose tissue (VAT) Subcutaneous adipose tissue (SubQ) | Multi-organ insulin resistance Adipose tissue inflammation Decrease of plasma adiponectin and adipsin [25] |
| Adipocyte-specific Nampt knockout (ANKO) | Brown adipose tissue (BAT) | Cold intolerance Impaired thermogenic response Decreased NAD+ in BAT Hypertrophy and whitening of BAT [44] |
| High-fat diet supplemented with nicotinamide riboside (NR) | Epididymal white adipose tissue (eWAT) | Improved metabolic flexibility with NR addition [26] |
| Regular diet supplement with nicotinamide riboside (NR) | Brown adipose tissue (BAT) | Reduced abdominal visceral fat Increased heat production [45] |
| Whole body Aifm2-knockout | Brown adipose tissue (BAT) Inguinal white adipose tissue (iWAT) | Impaired thermogenesis Decreased BAT and iWAT NAD+/NADH ratio Increased body weight and adiposity [46] |
| Aifm2-overexpression in UCP1+ cells | Brown adipose tissue (BAT) Inguinal white adipose tissue (iWAT) | Increased thermogenesis Increased BAT and iWAT NAD+/NADH ratio Decreased body weight and adiposity [46] |
| Adipocyte-specific deletion of erythropoietin receptor | Subcutaneous WAT (SubQ) | Obesity Glucose intolerance Insulin resistance Decreased oxygen consumption Decreased NAD+ in SubQ [47] |
| PARP1 knockout mouse | Brown adipose tissue (BAT) | Reduced fat accumulation Higher energy expenditure Higher NAD+ in BAT Higher mitochondrial content in BAT [51] |
| Maternal high-calorie diet | Subcutaneous WAT (SubQ) of newborn mice at the end of lactation | Increased SubQ mass Increased adipocyte size Decreased NAD+/NADH ratio Increased oxidative damage to mitochondrial proteins Impaired mitochondrial bioenergetics [52] |
| Maternal high-calorie diet supplemented with niacin | Subcutaneous WAT (SubQ) of newborn mice at the end of lactation | Increased NAD+/NADH ratio in SubQ [52] |
| CD38 knockout mice on high-fat high-sucrose diet | White adipose tissue (WAT) Brown adipose tissue (BAT) | Higher levels of NAD+ in WAT and BAT Improved metabolic flexibility [64] |
| CD38 knockout mice on high-fat diet | White adipose tissue (WAT) | Less body weight gain [66] |
| Cold exposure | Interscapular brown adipose tissue (iBAT) | Down-regulation of CD38 at both mRNA and protein levels in iBAT Increased NAD+ in iBAT [67] |
| Cold exposure, CD38 knockout mice | Interscapular brown adipose tissue (iBAT) | Increased NAD+ in iBAT [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruskovska, T.; Bernlohr, D.A. The Role of NAD+ in Metabolic Regulation of Adipose Tissue: Implications for Obesity-Induced Insulin Resistance. Biomedicines 2023, 11, 2560. https://doi.org/10.3390/biomedicines11092560
Ruskovska T, Bernlohr DA. The Role of NAD+ in Metabolic Regulation of Adipose Tissue: Implications for Obesity-Induced Insulin Resistance. Biomedicines. 2023; 11(9):2560. https://doi.org/10.3390/biomedicines11092560
Chicago/Turabian StyleRuskovska, Tatjana, and David A. Bernlohr. 2023. "The Role of NAD+ in Metabolic Regulation of Adipose Tissue: Implications for Obesity-Induced Insulin Resistance" Biomedicines 11, no. 9: 2560. https://doi.org/10.3390/biomedicines11092560
APA StyleRuskovska, T., & Bernlohr, D. A. (2023). The Role of NAD+ in Metabolic Regulation of Adipose Tissue: Implications for Obesity-Induced Insulin Resistance. Biomedicines, 11(9), 2560. https://doi.org/10.3390/biomedicines11092560